
Synthesis and Application Dichalcogenides as Radical Reagents with Photochemical Technology


Abstract
1. Introduction
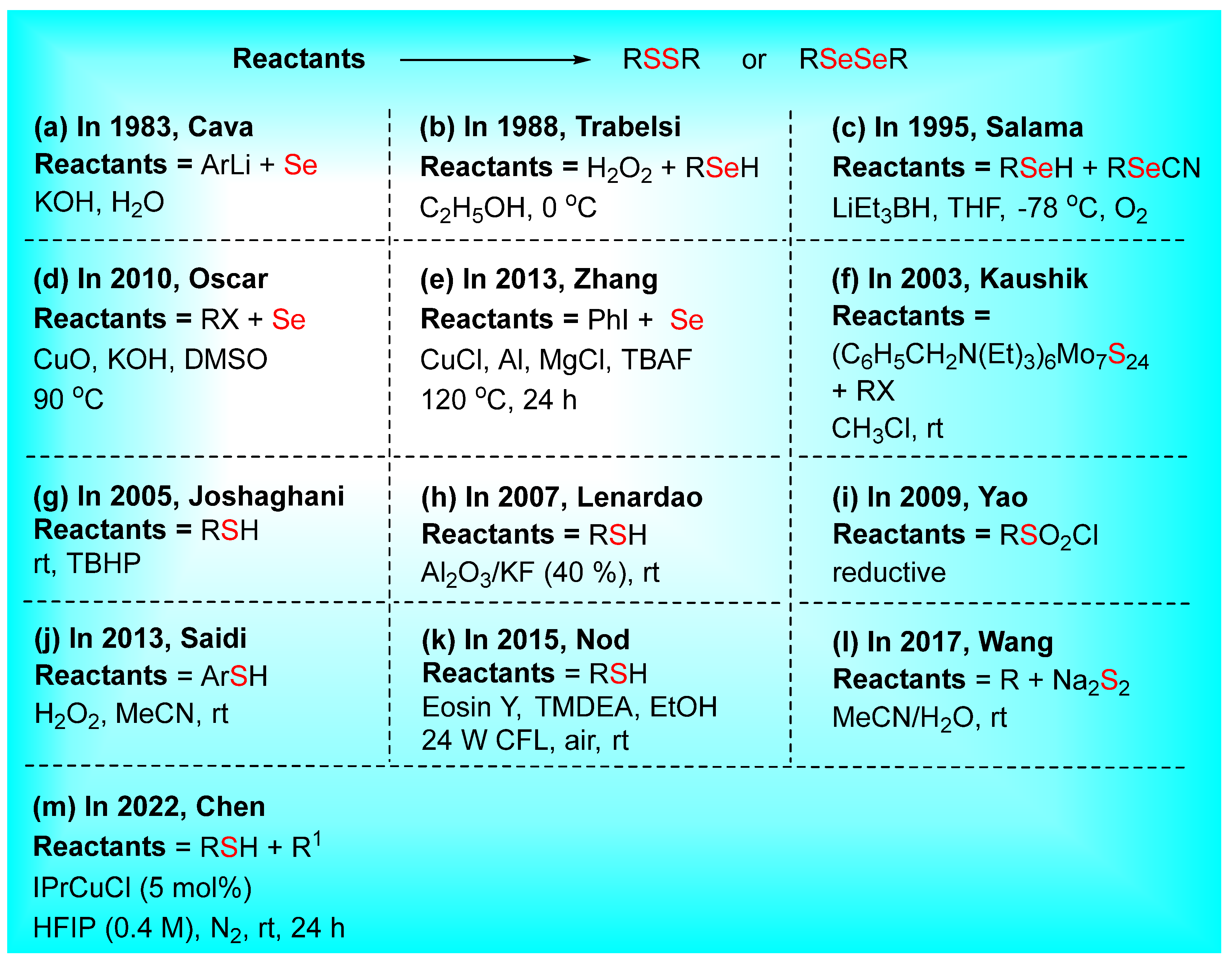
2. Synthesis of Dichalcogenides (Disulfides and Diselenides)
3. Application of Dichalcogenides under Visible-Light Irradiation
3.1. Cyclization of Diselenides/Disulfides with Alkenes and Alkynes
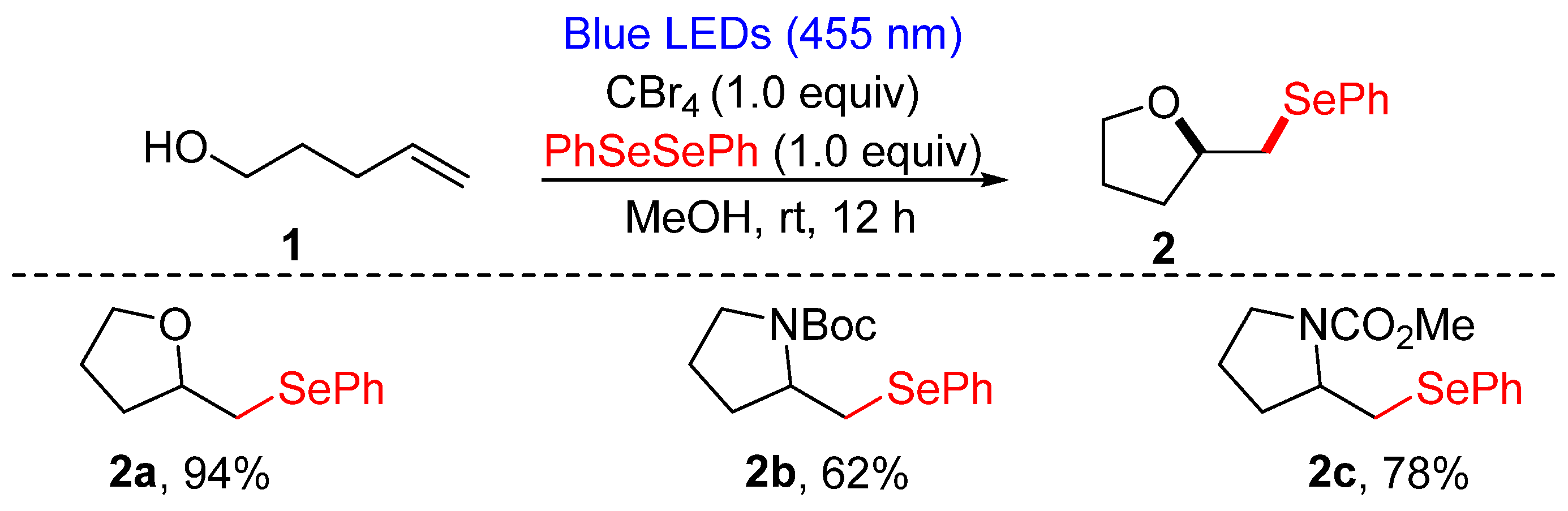
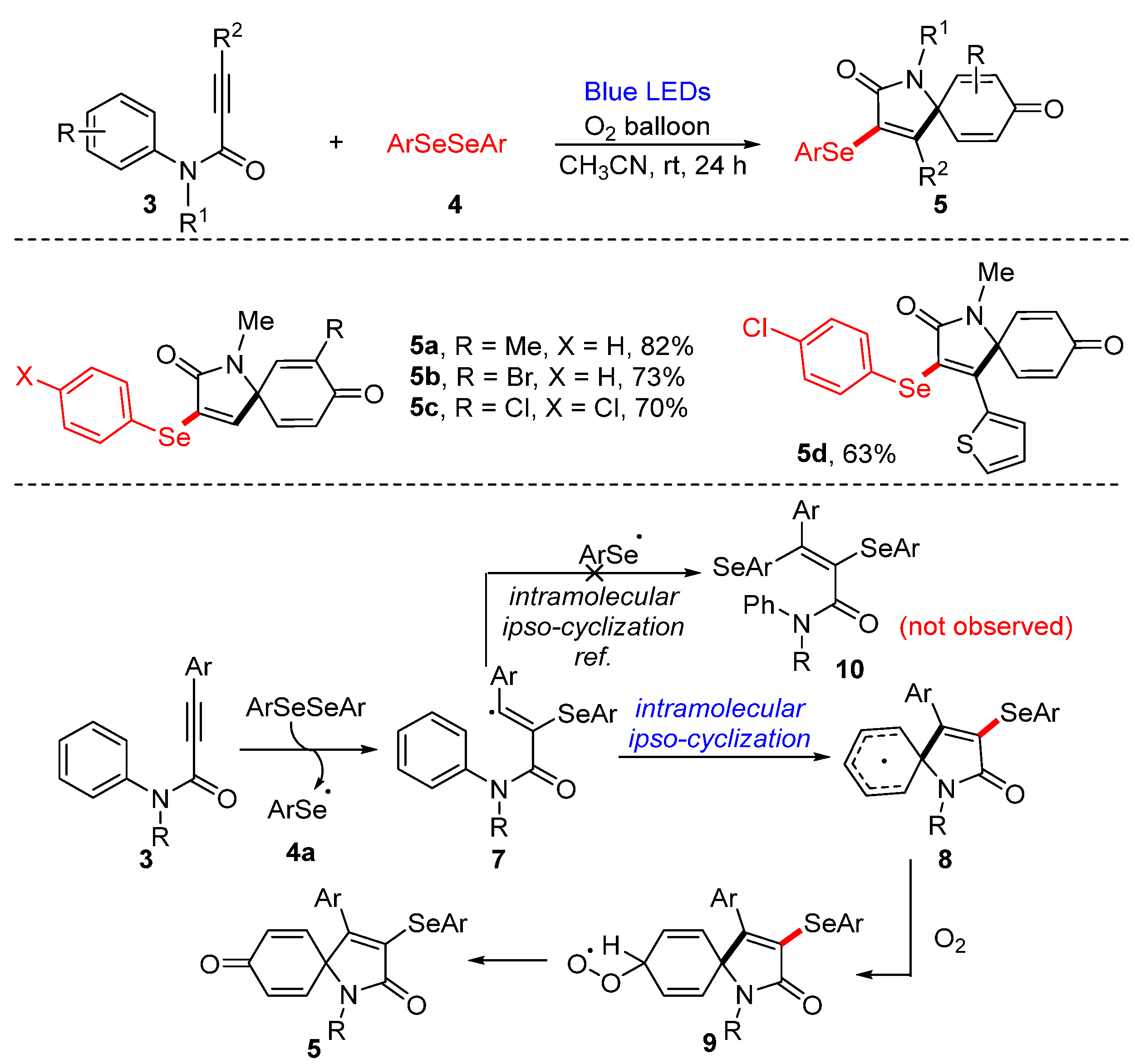
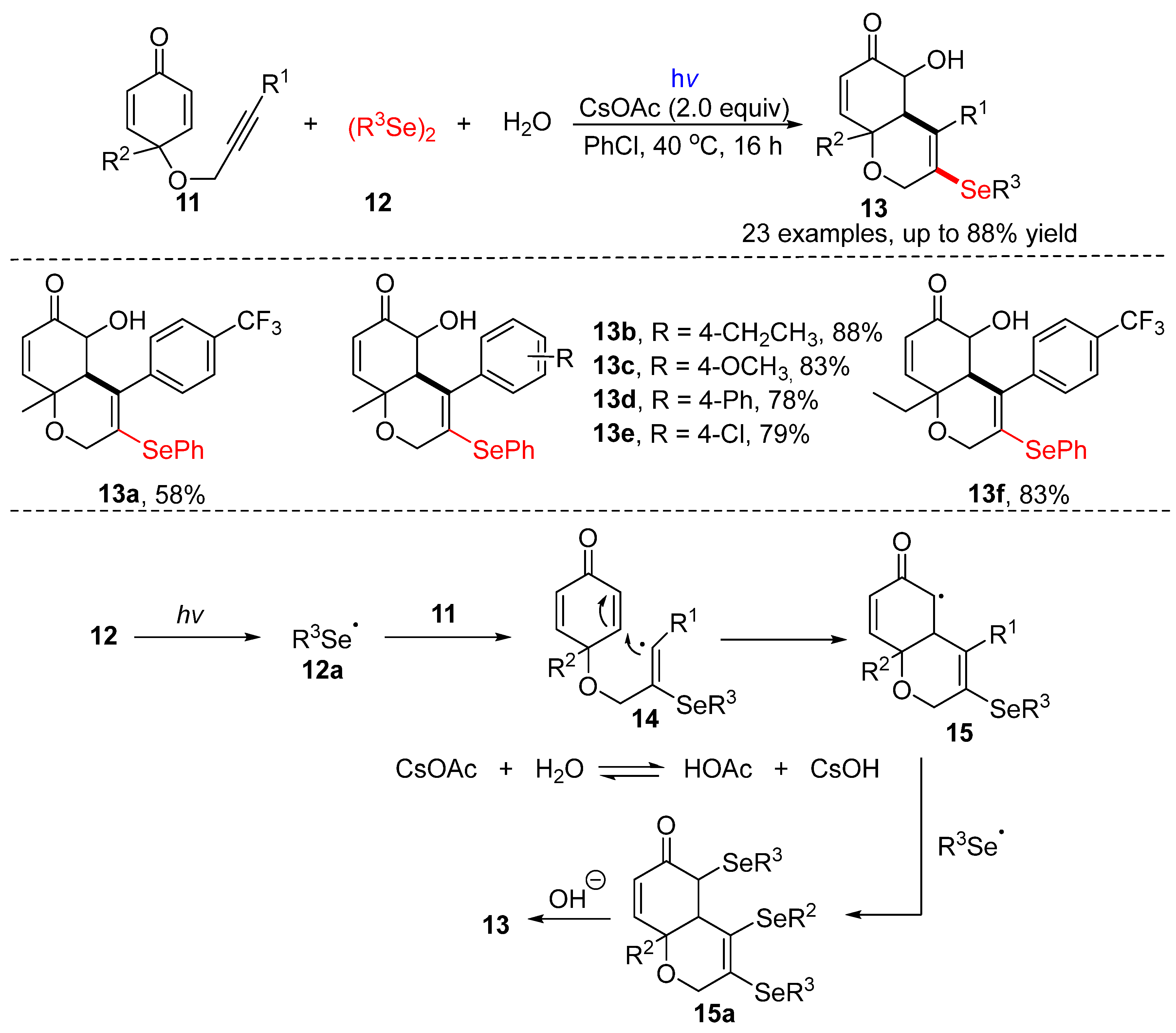
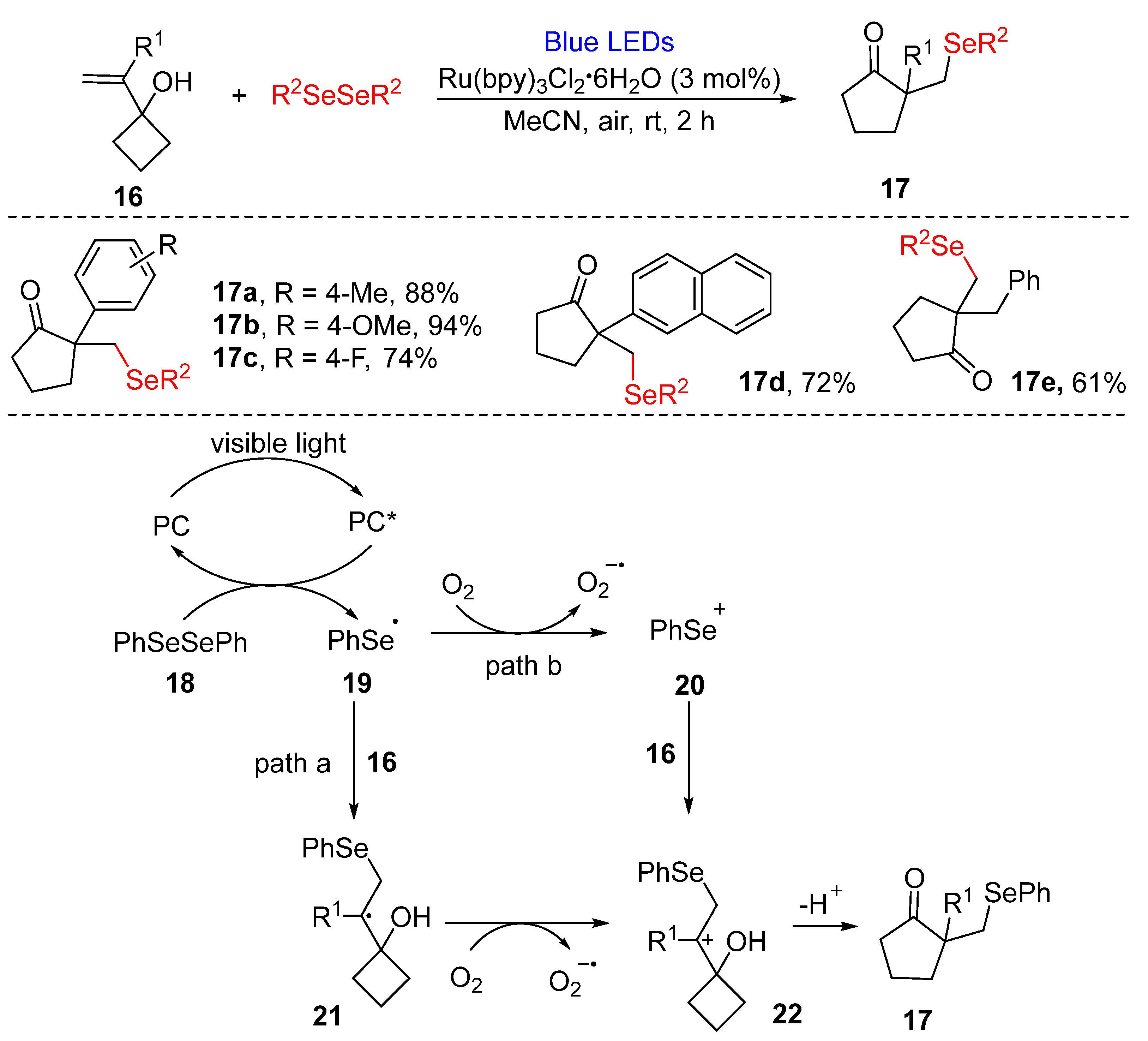
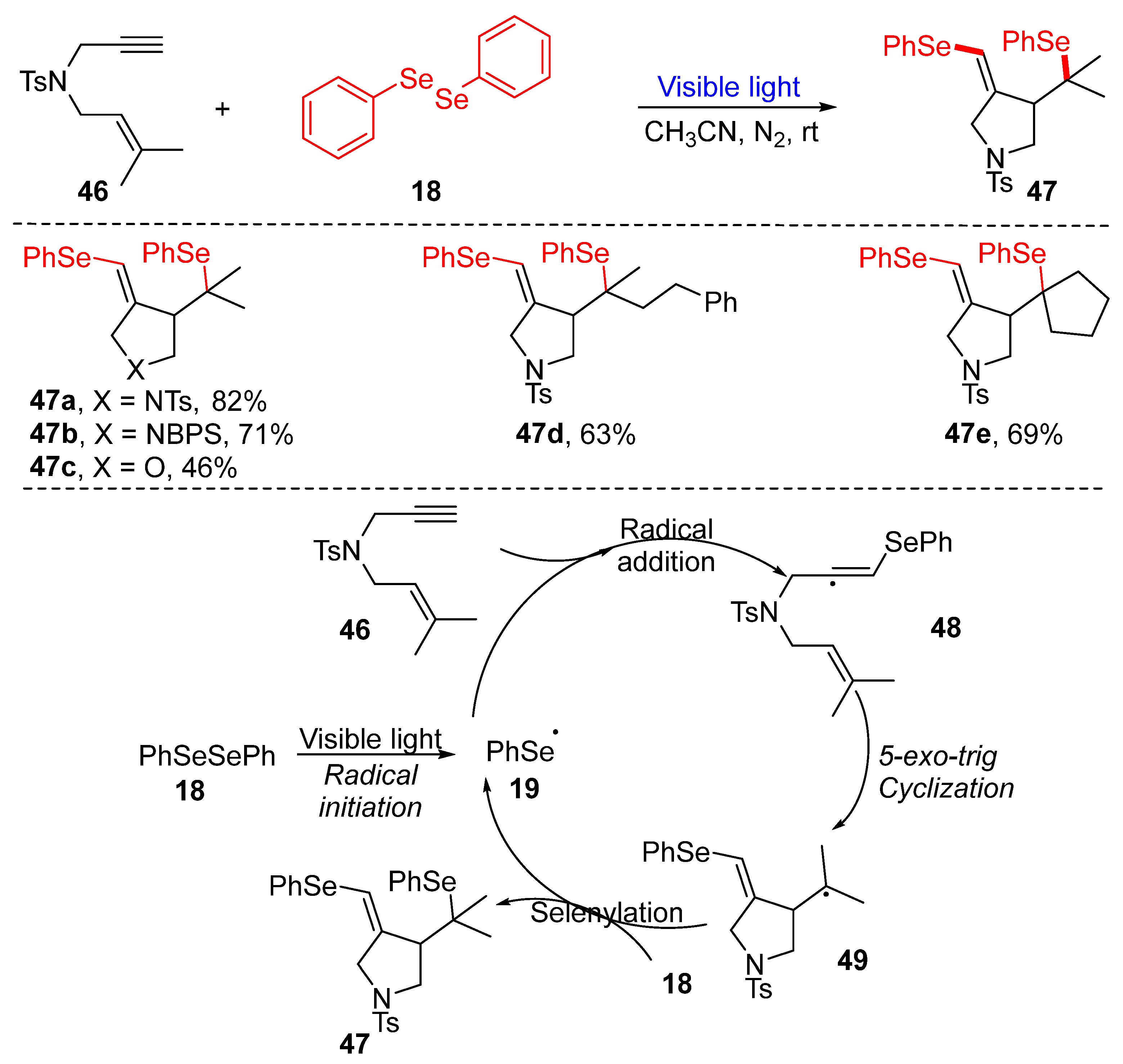
3.1.1. Cyclization of Diselenides with Alkenes and Alkynes
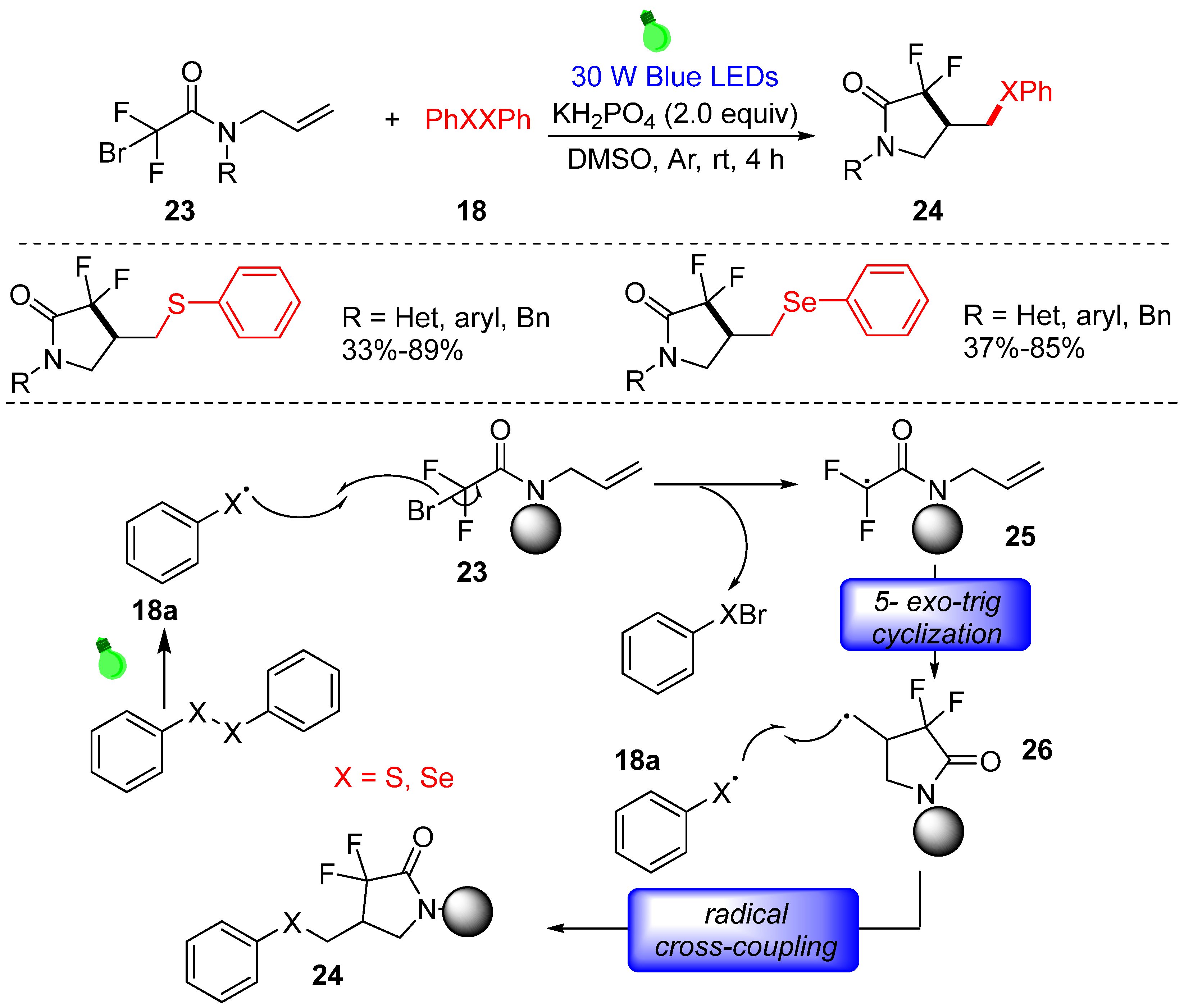
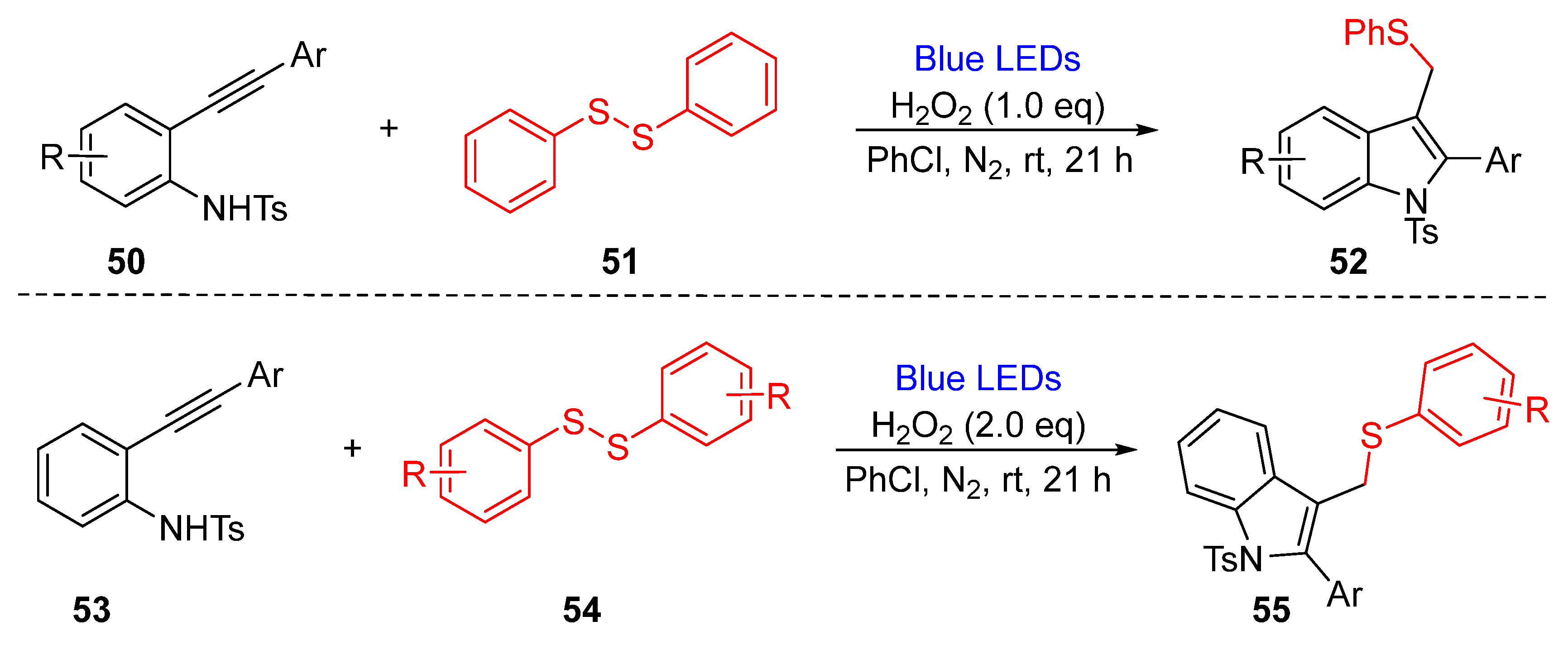
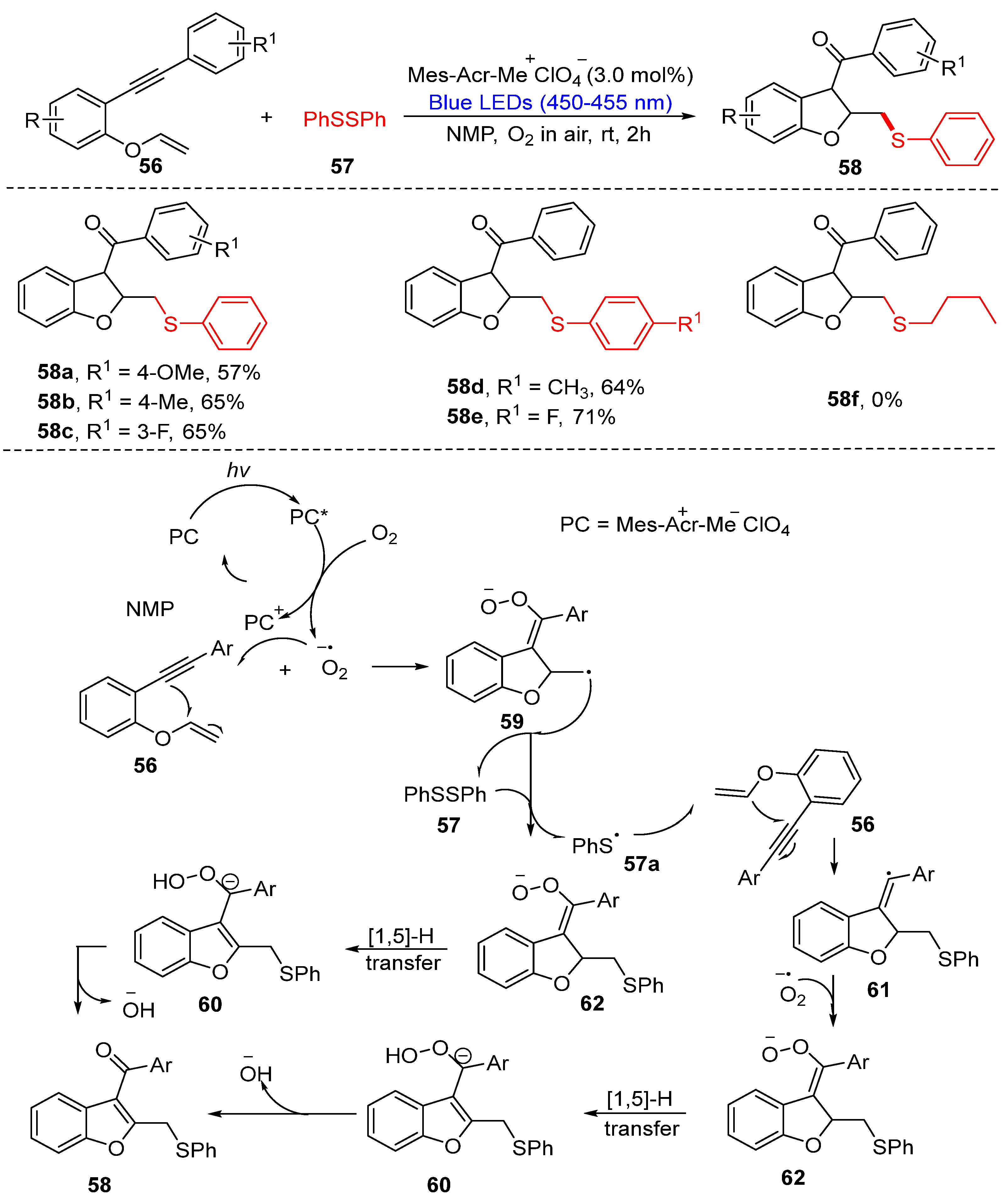
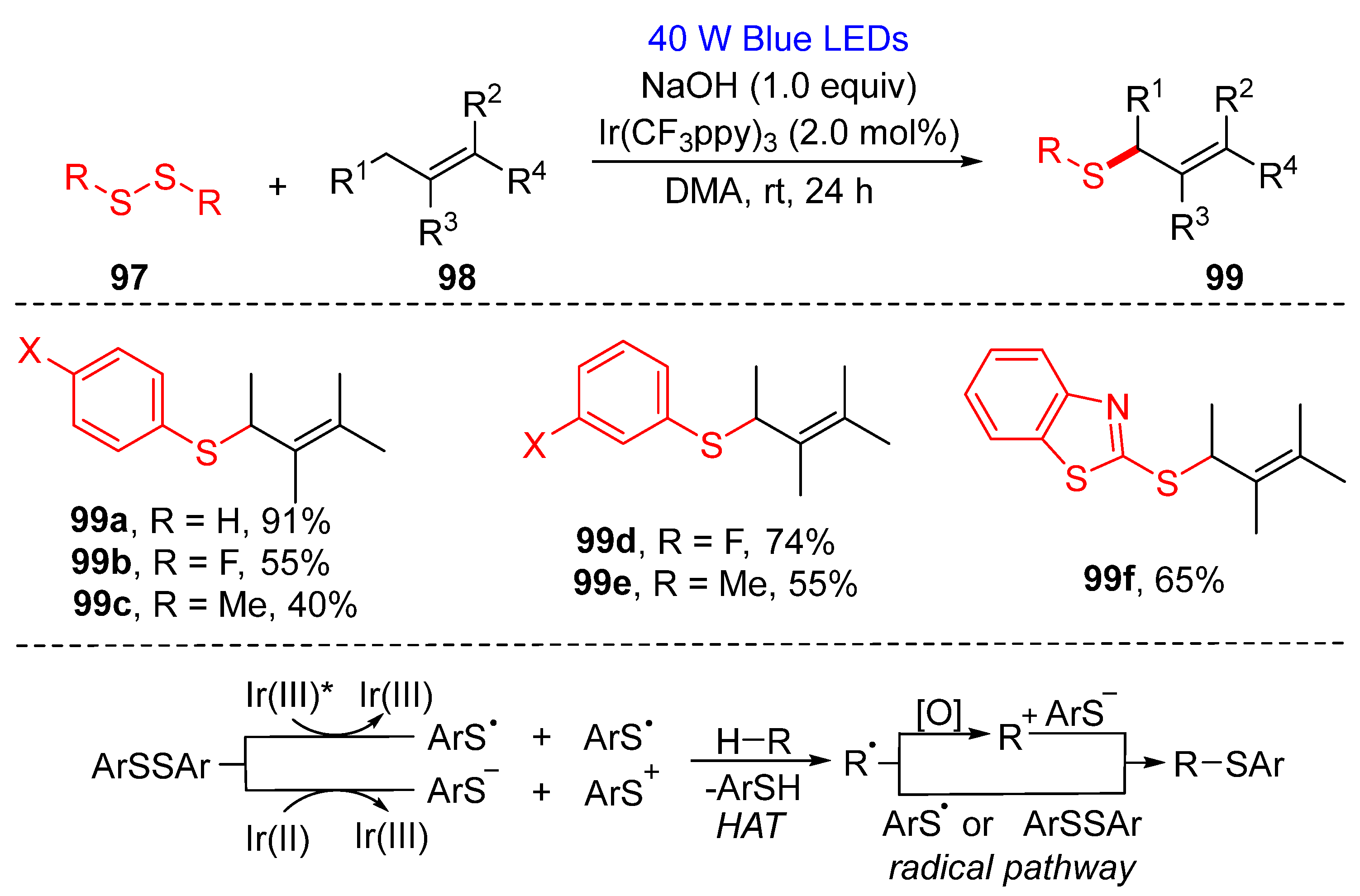
3.1.2. Cyclization of Disulfides with Alkenes and Alkynes
3.2. Direct Selenylation/Sulfuration of C−H/C−C/C−N Bonds
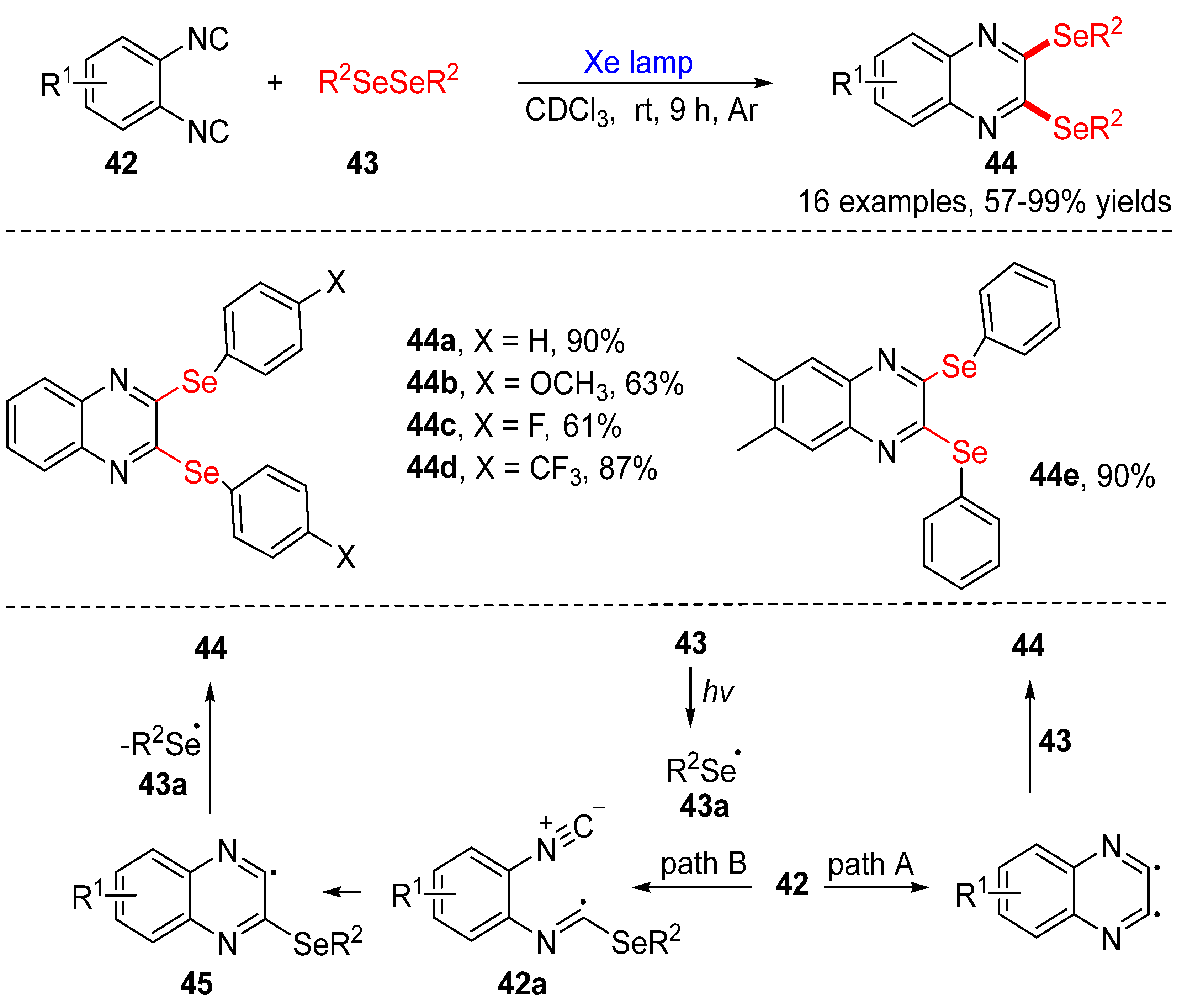
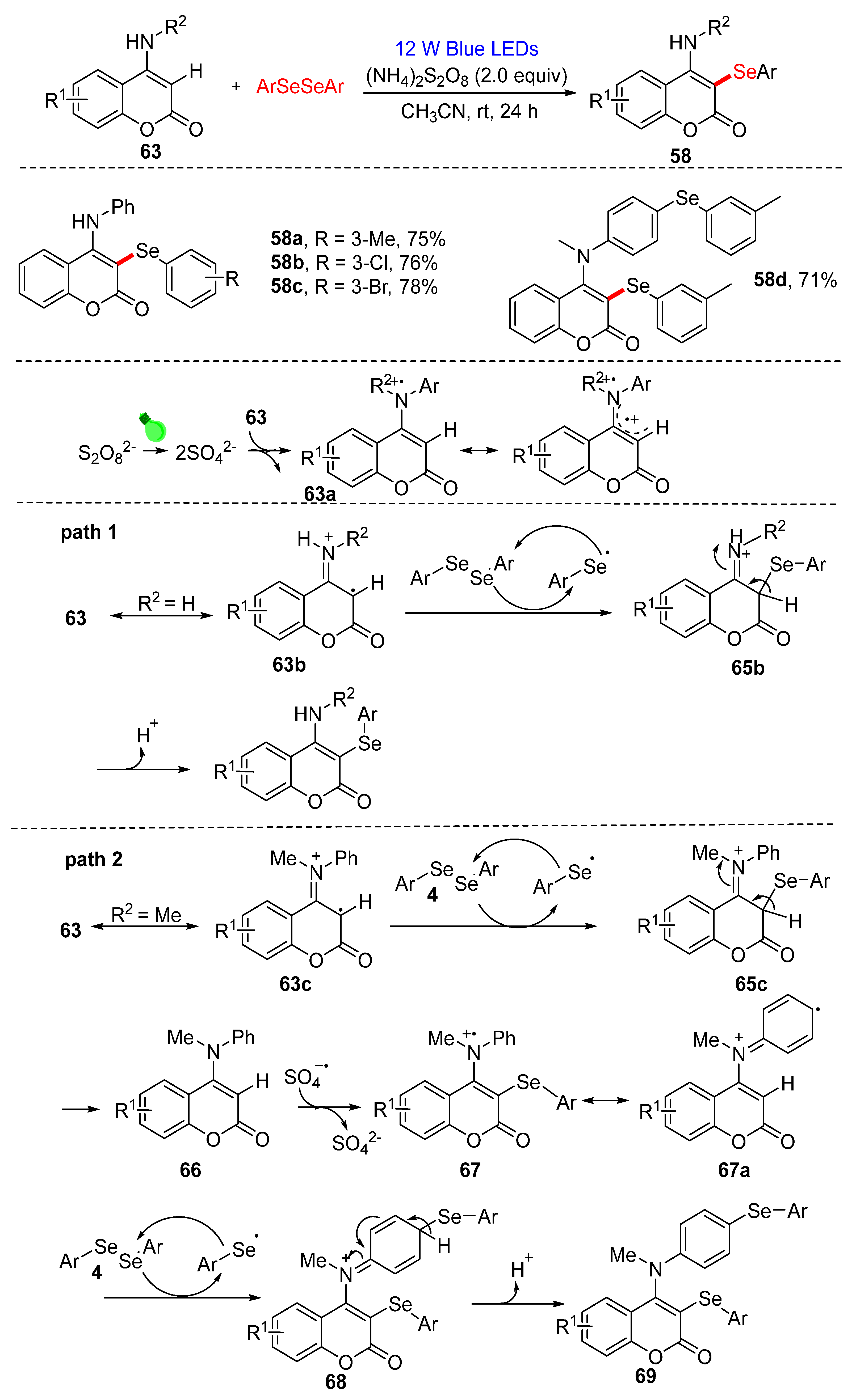
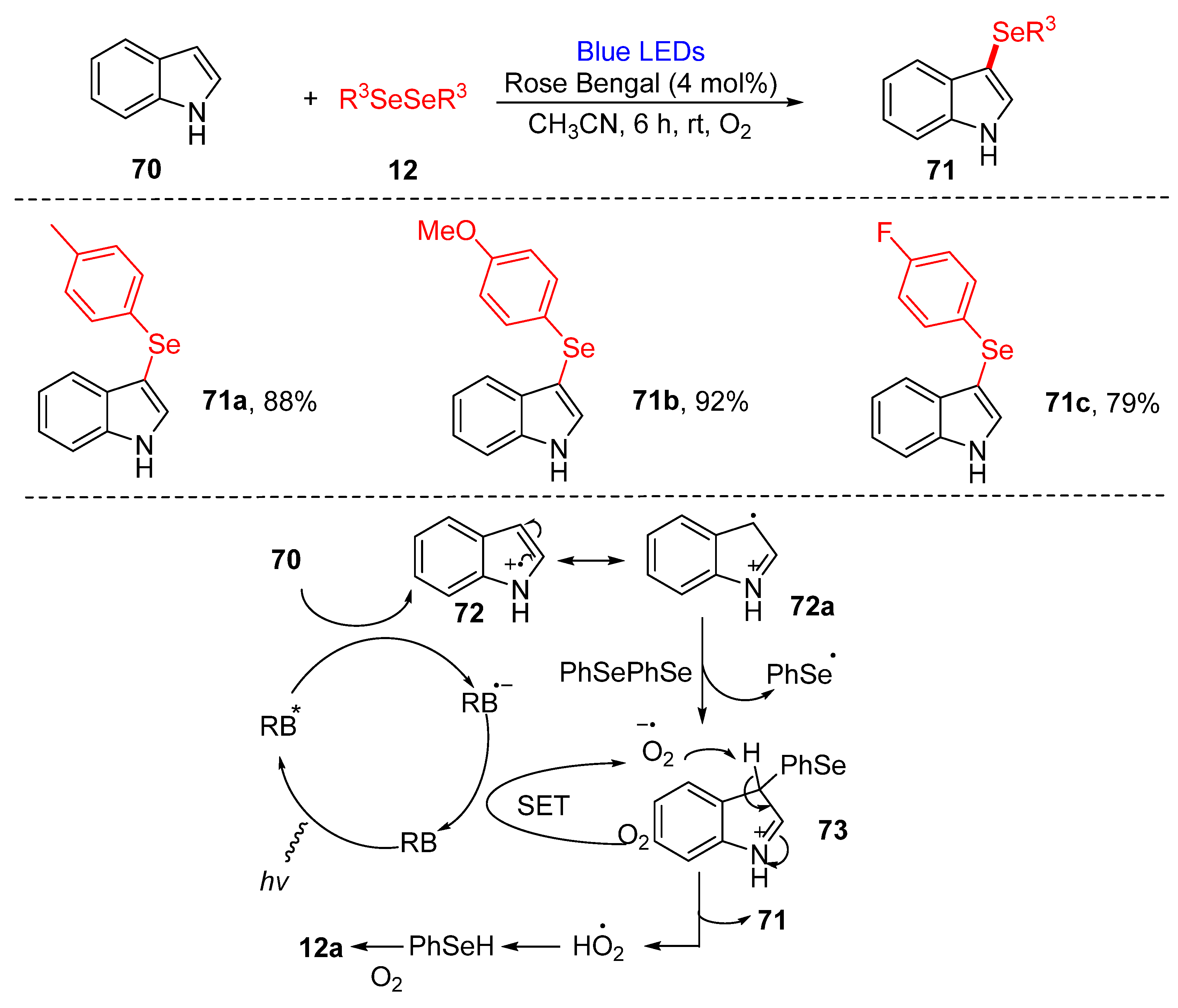
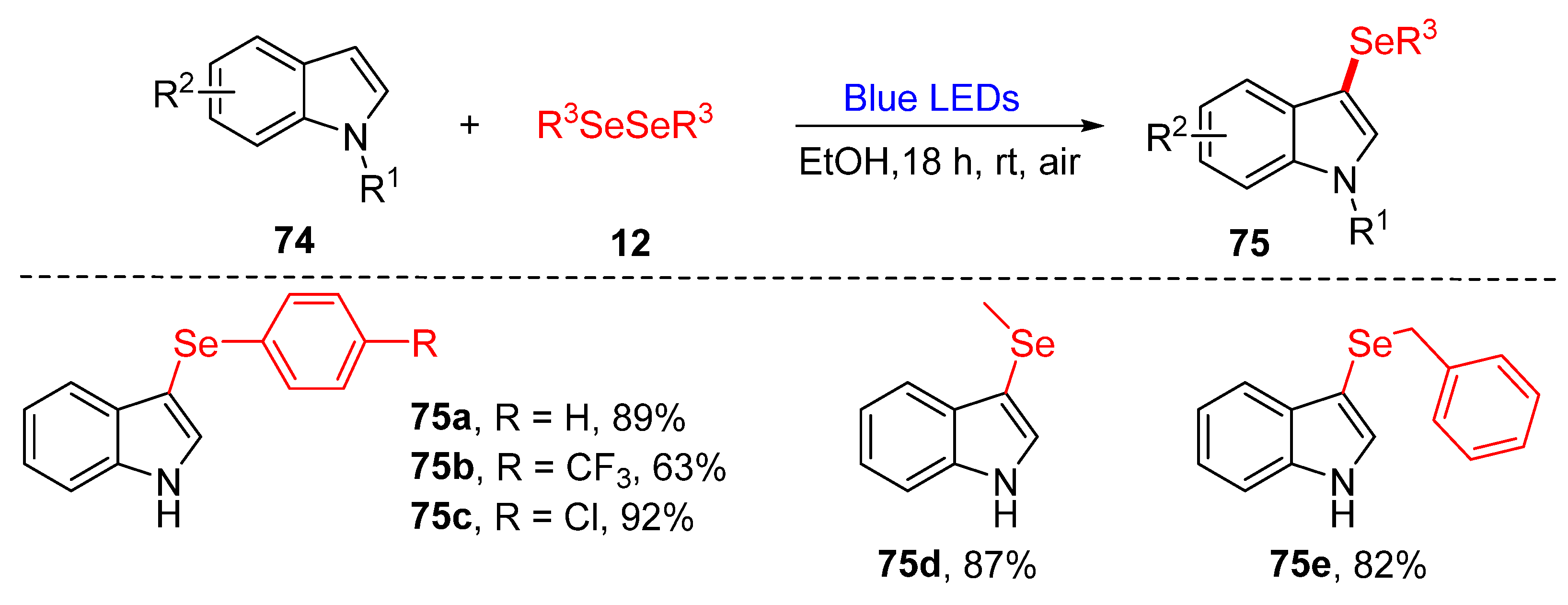
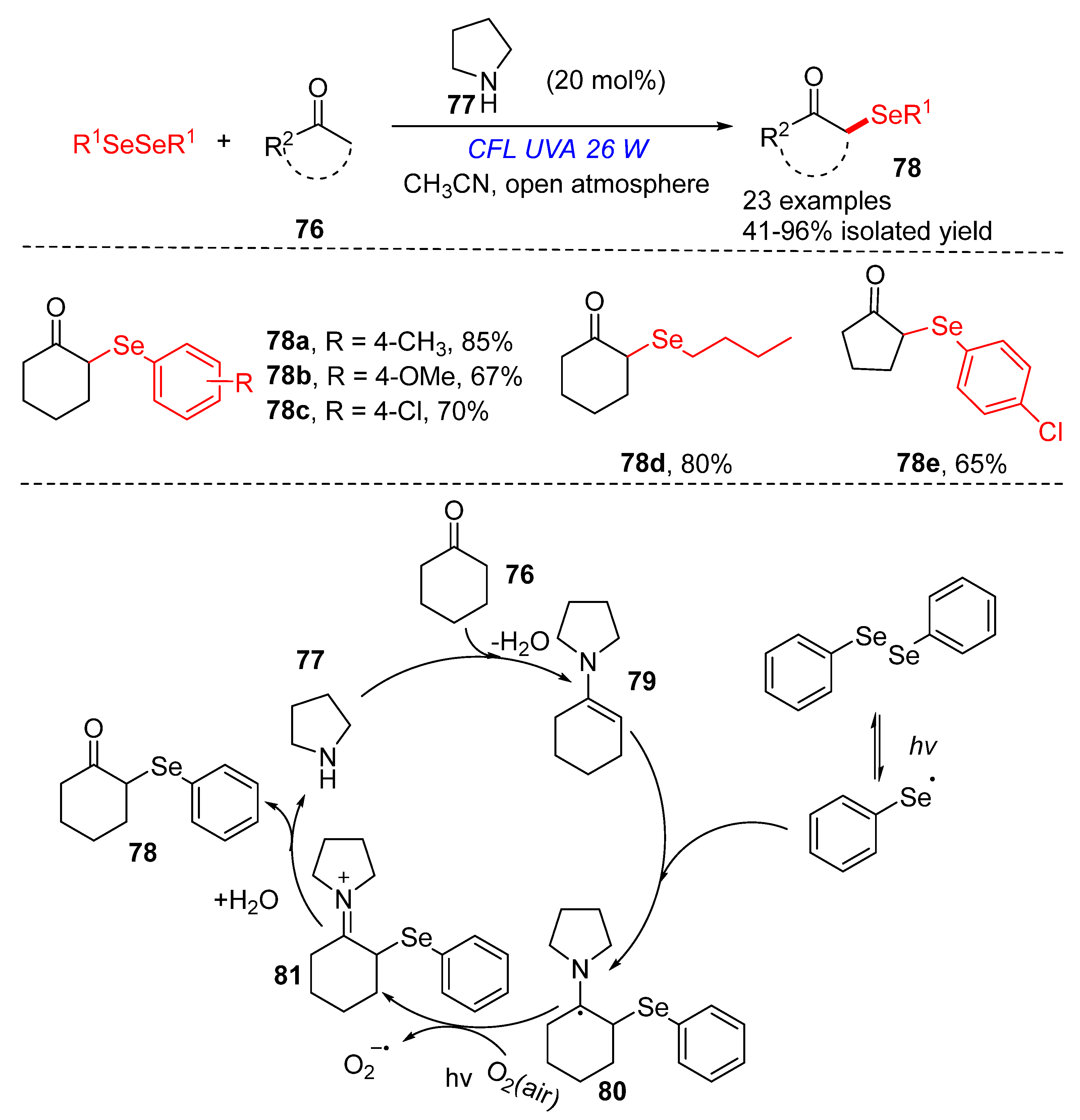
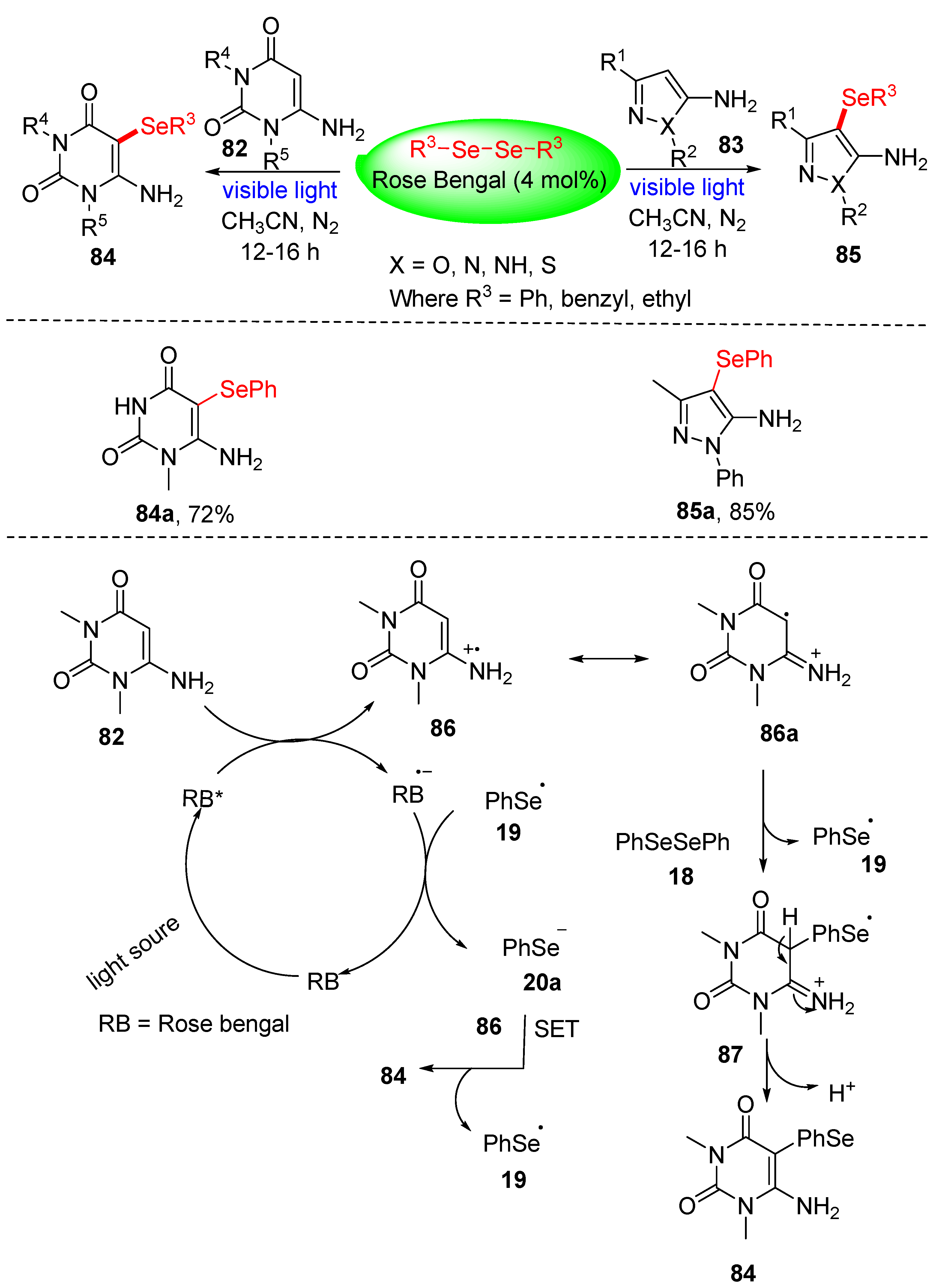
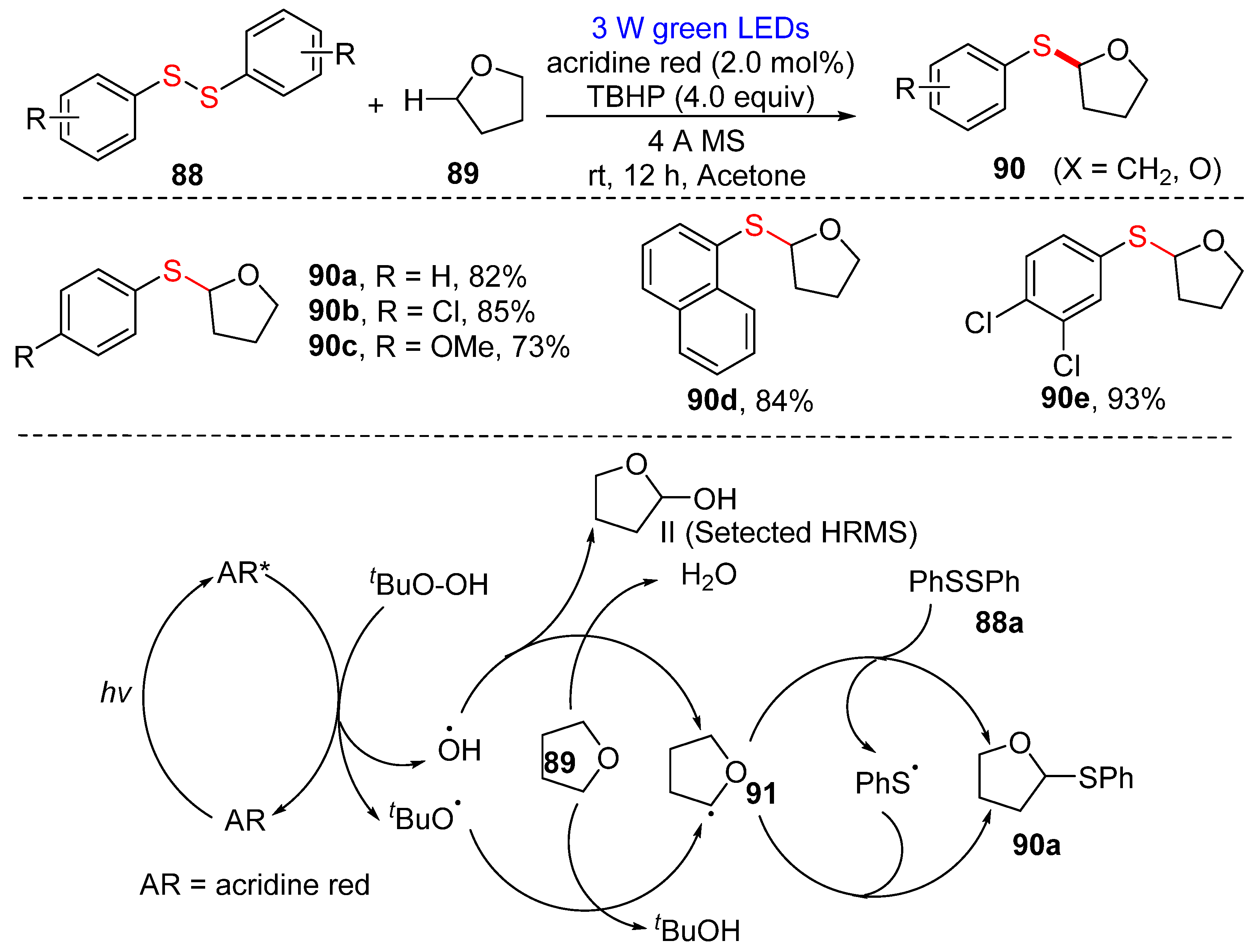
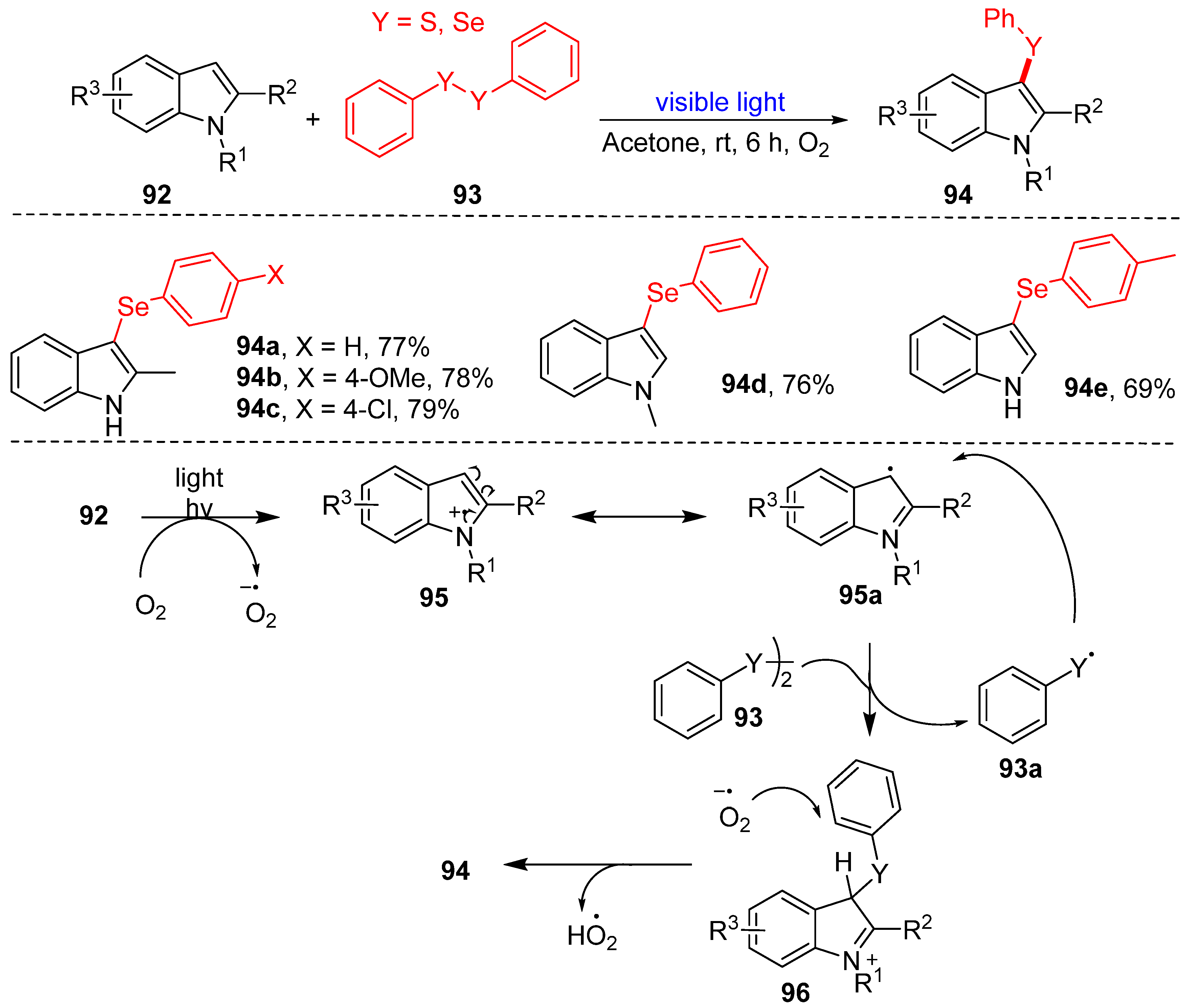
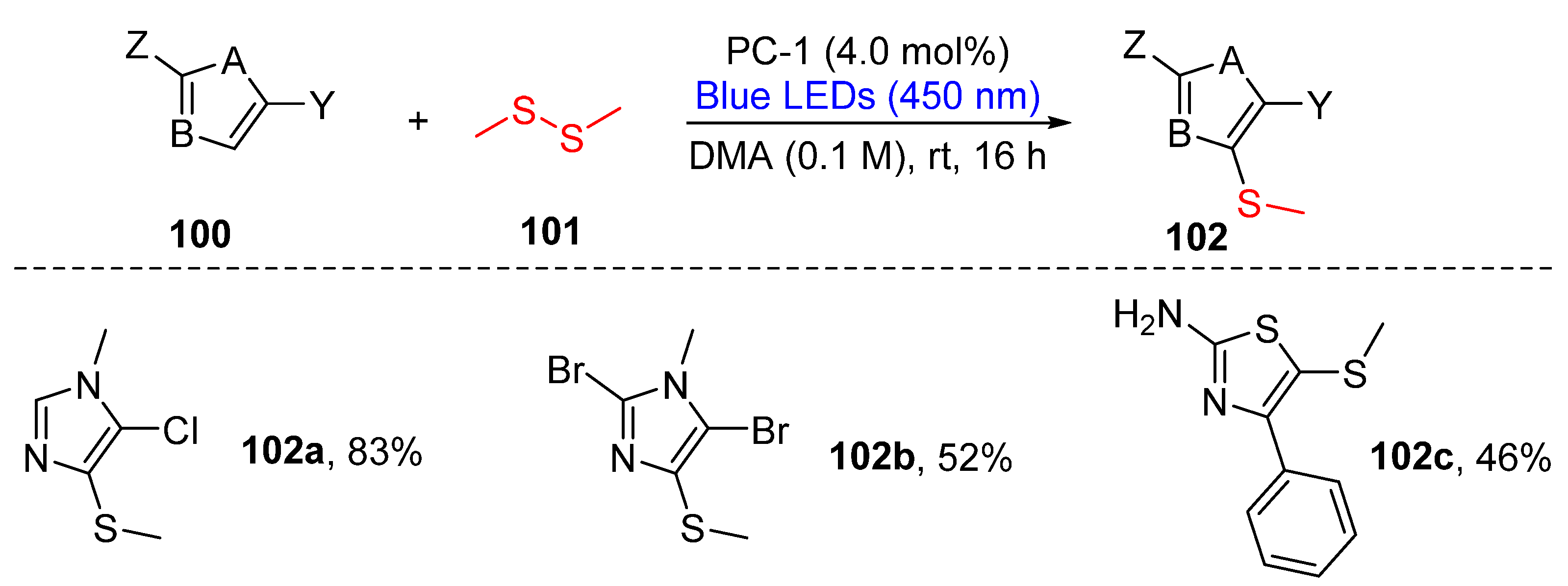
3.2.1. Direct Selenylation/Sulfuration of C−H Bonds
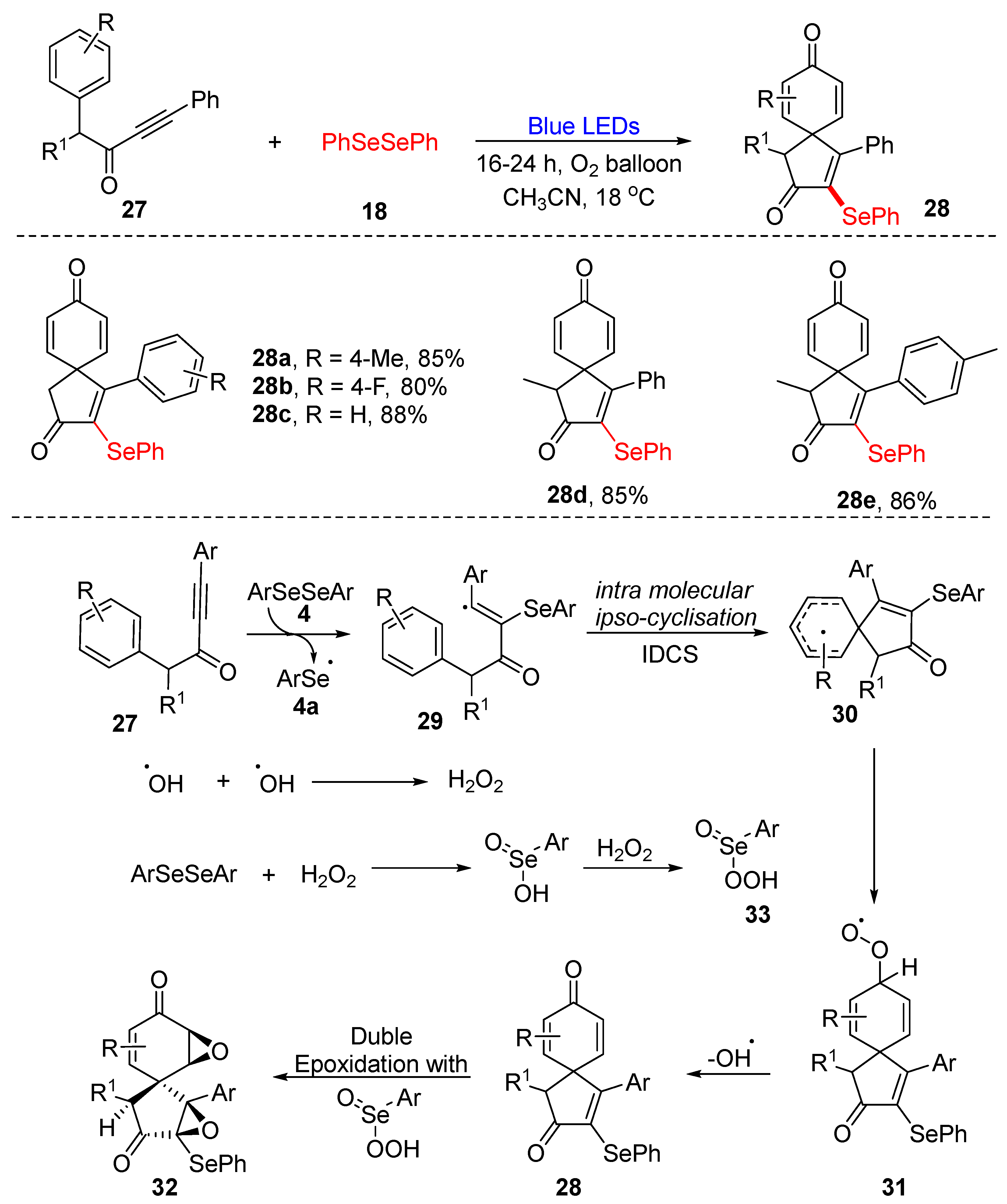
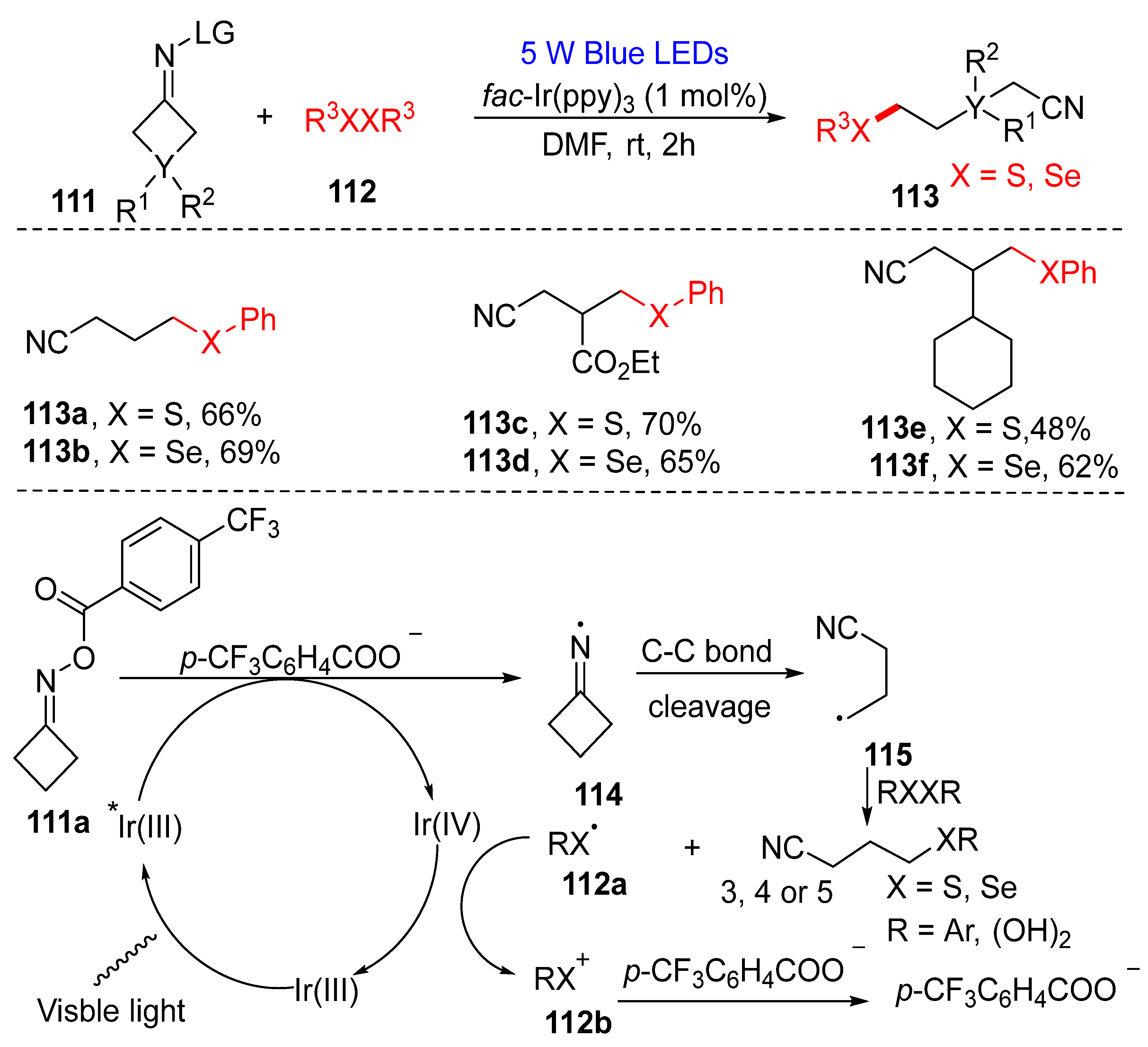
3.2.2. Direct Selenylation of C−C Bonds
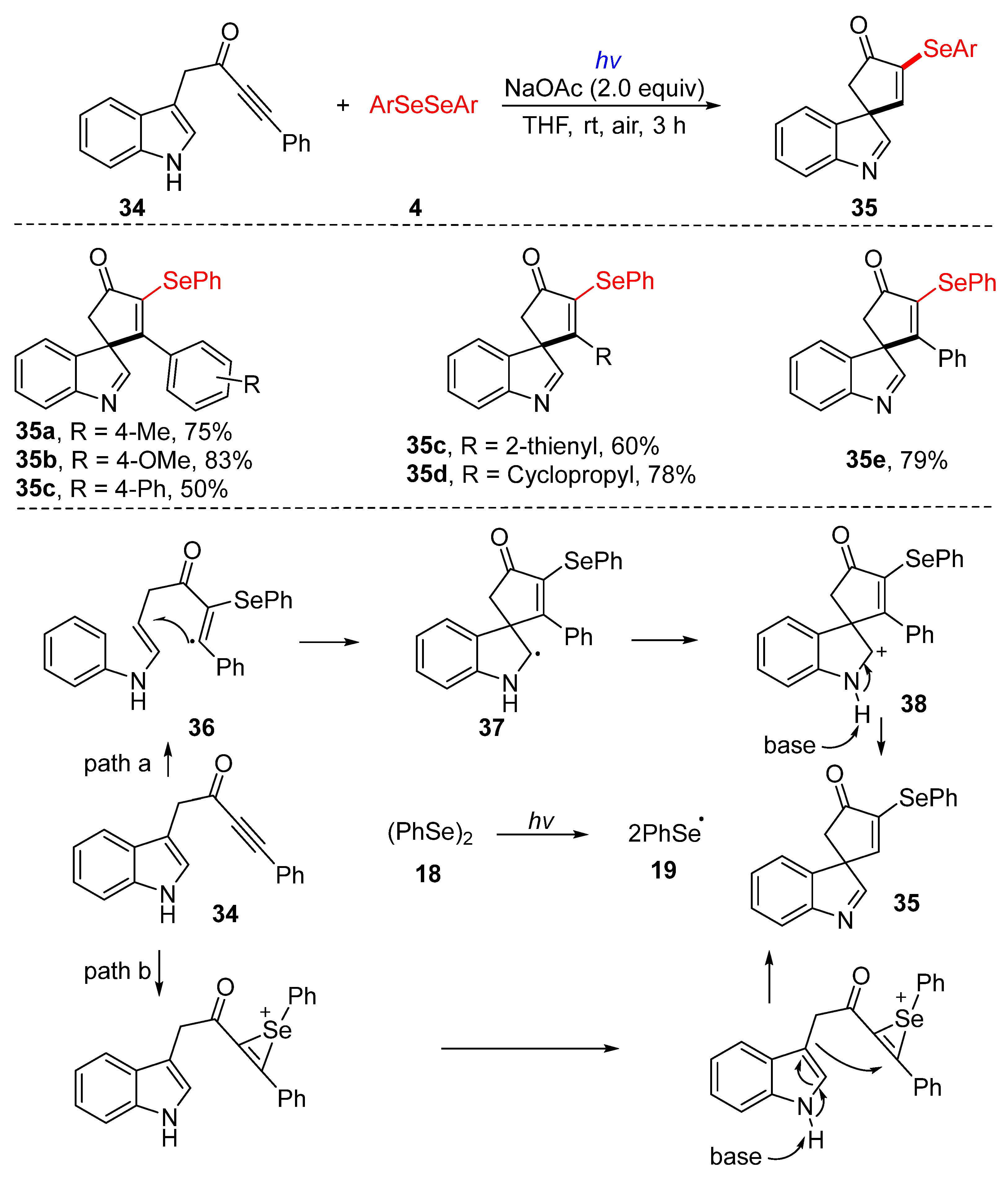
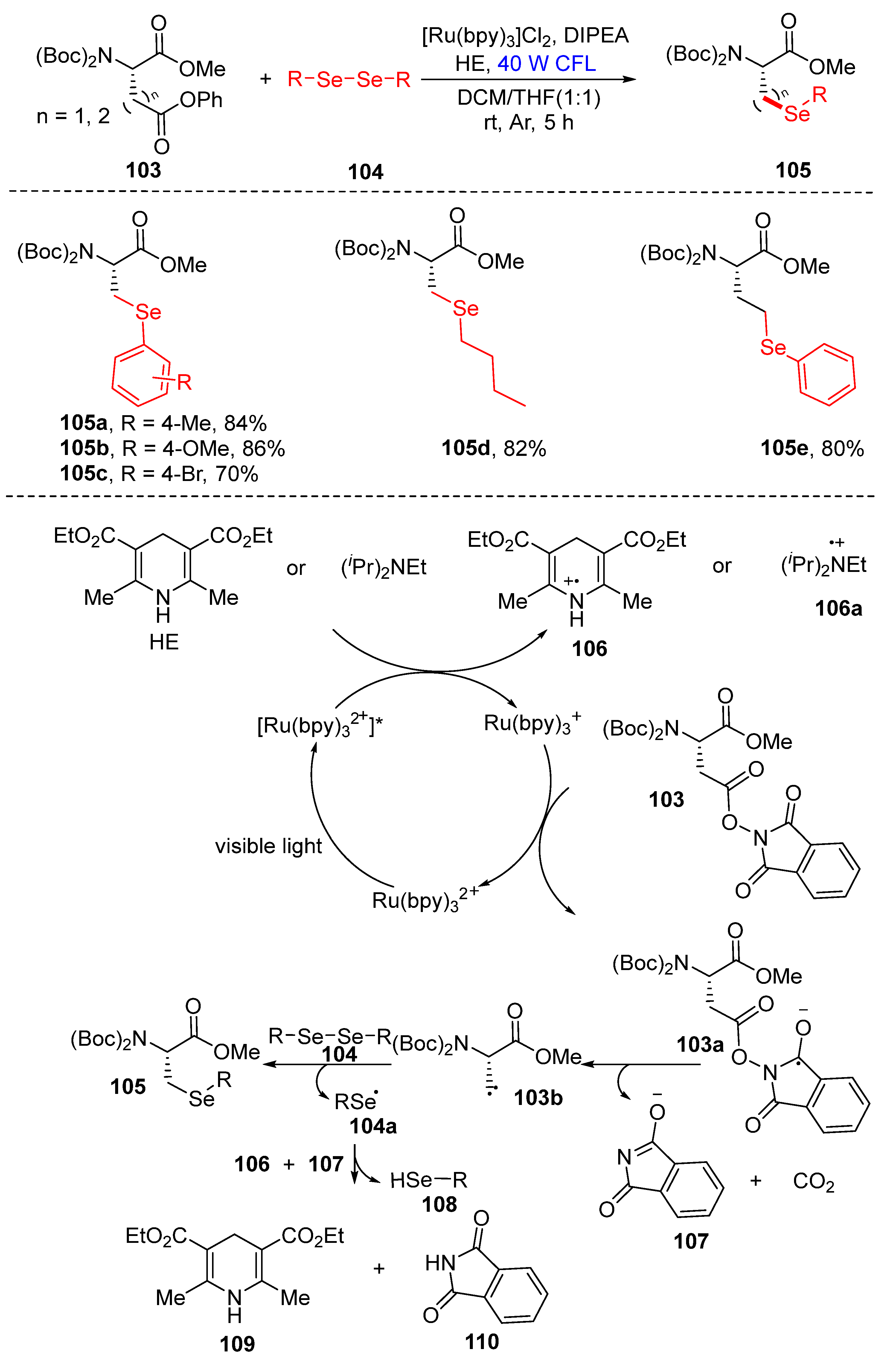
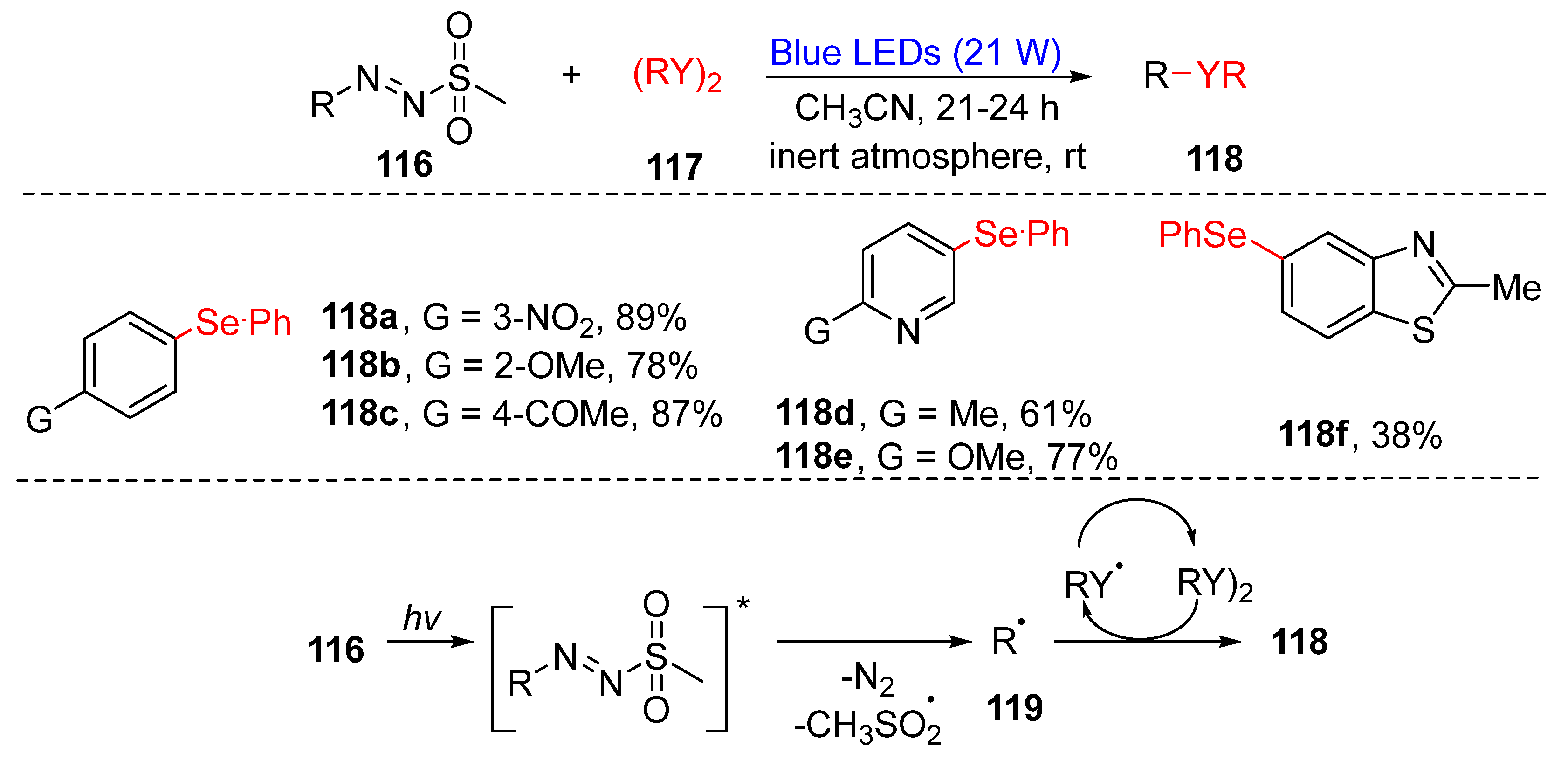
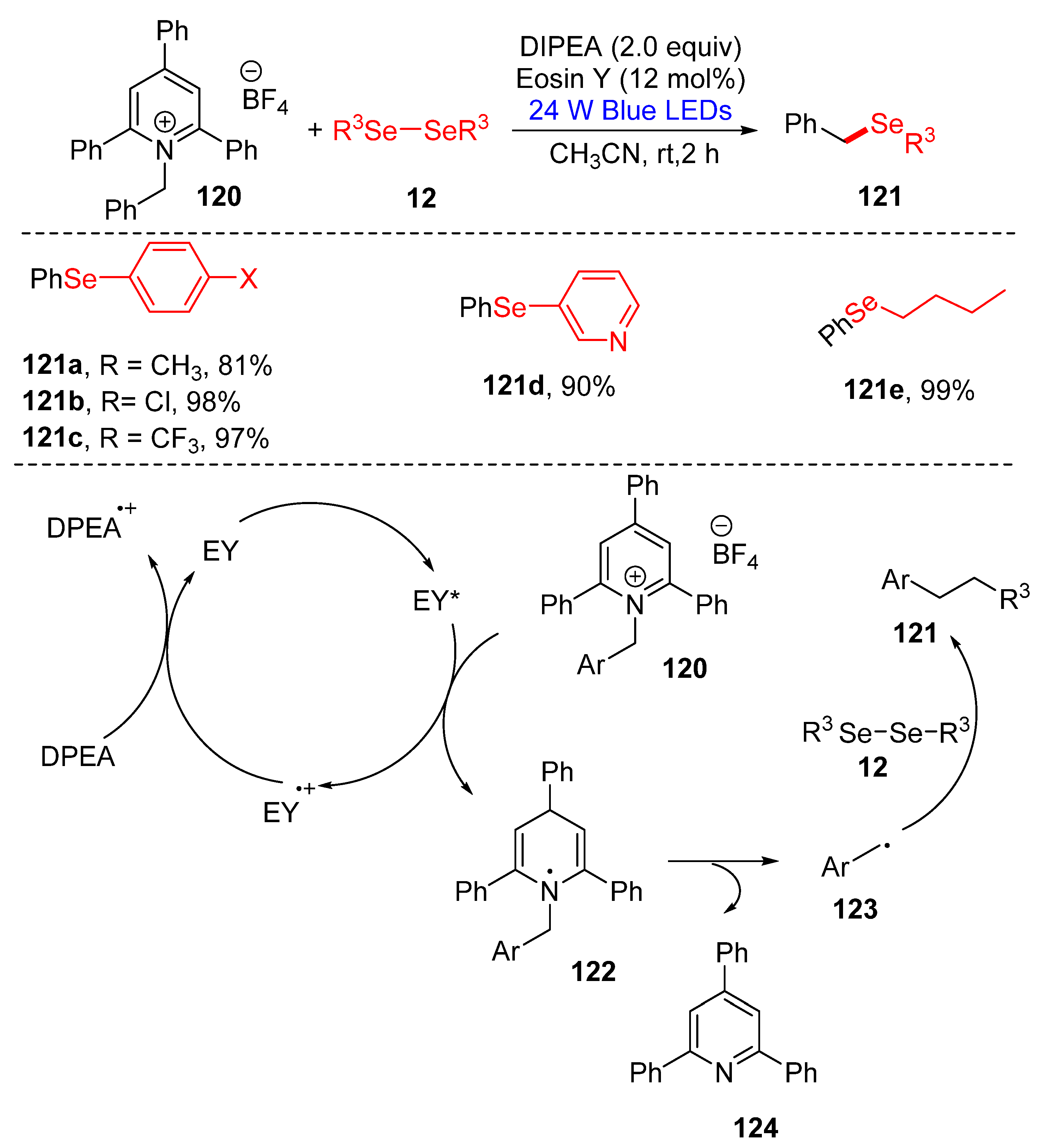
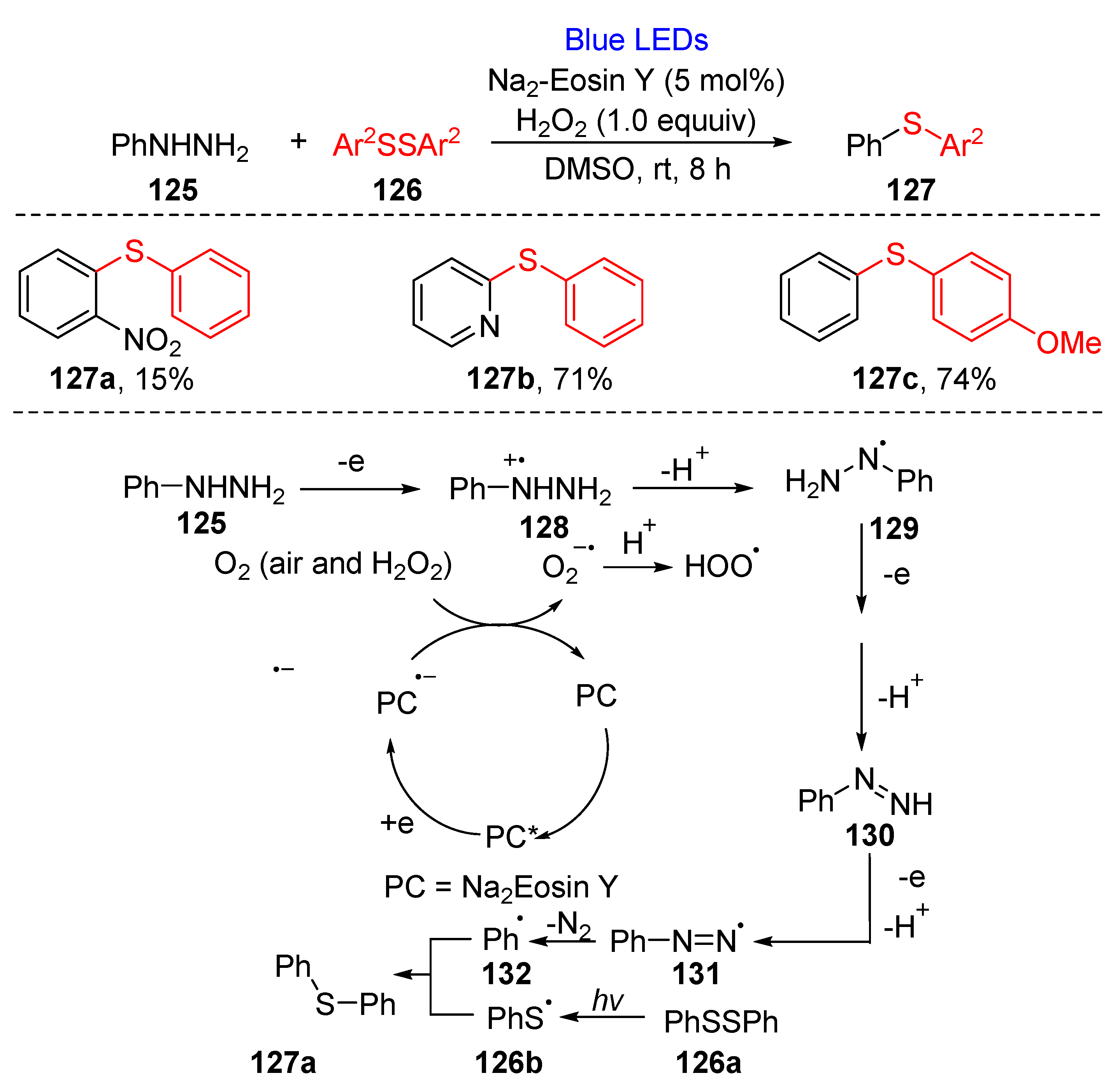
3.2.3. Direct Selenylation/Sulfuration of C−N Bonds
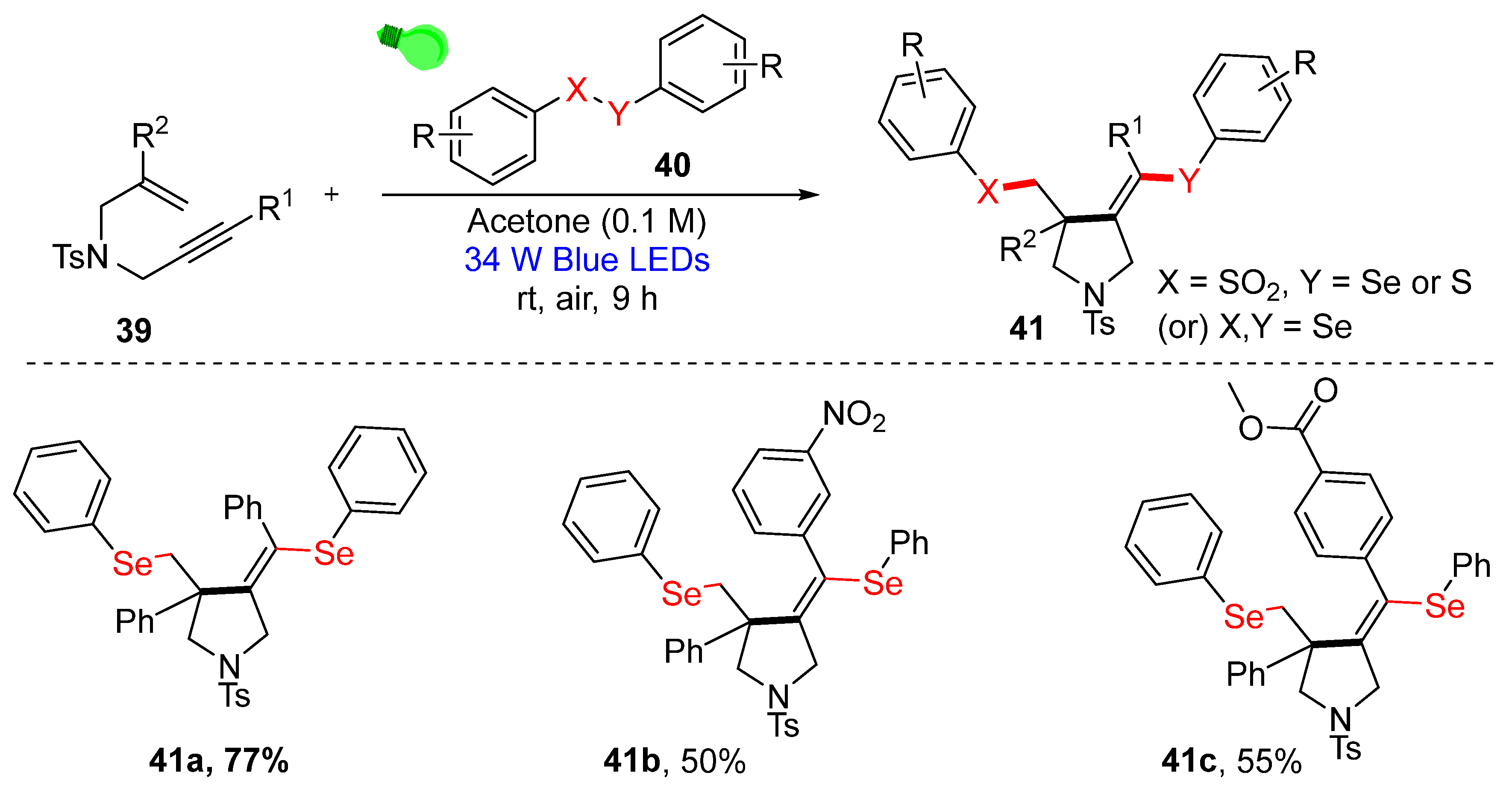
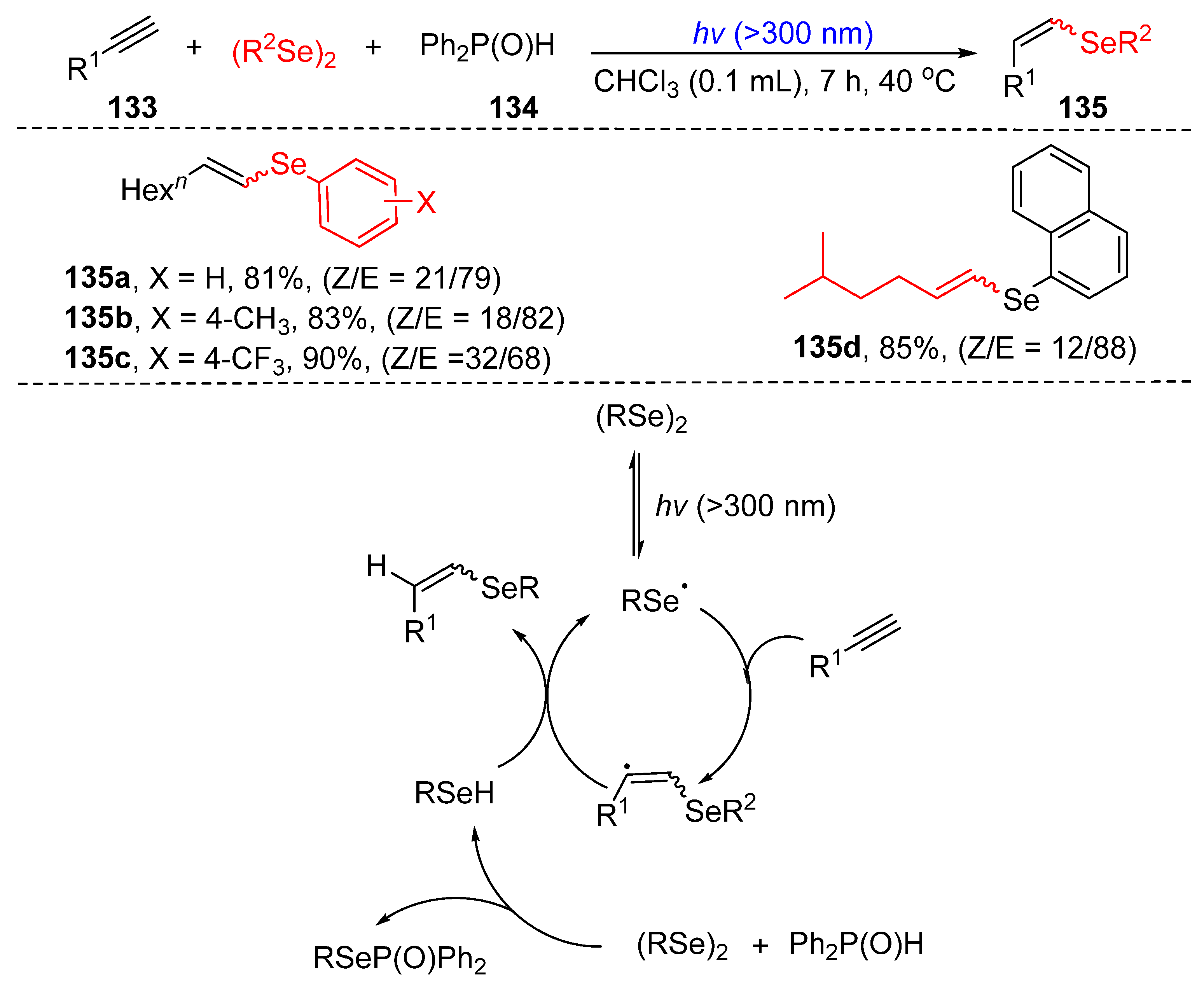
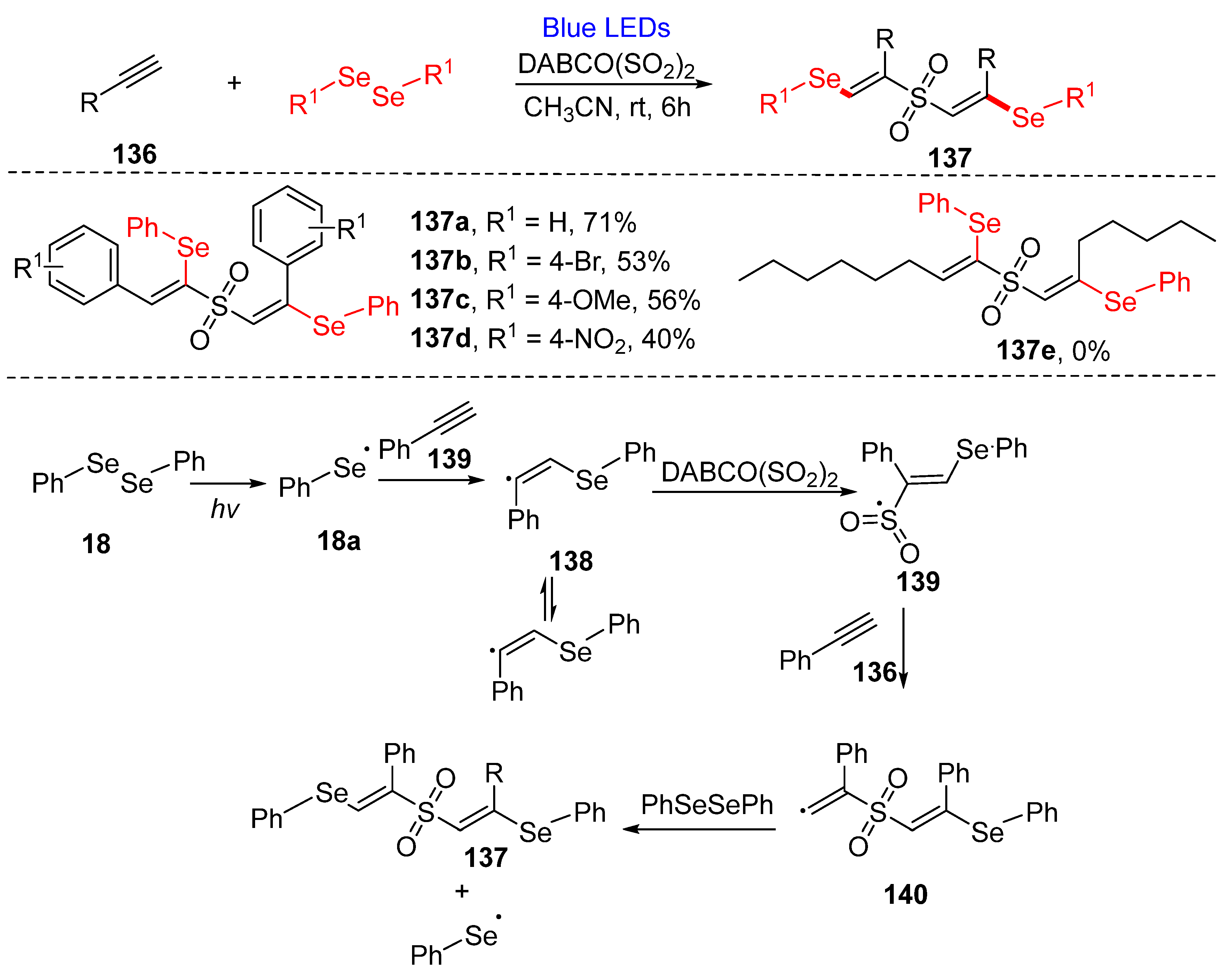
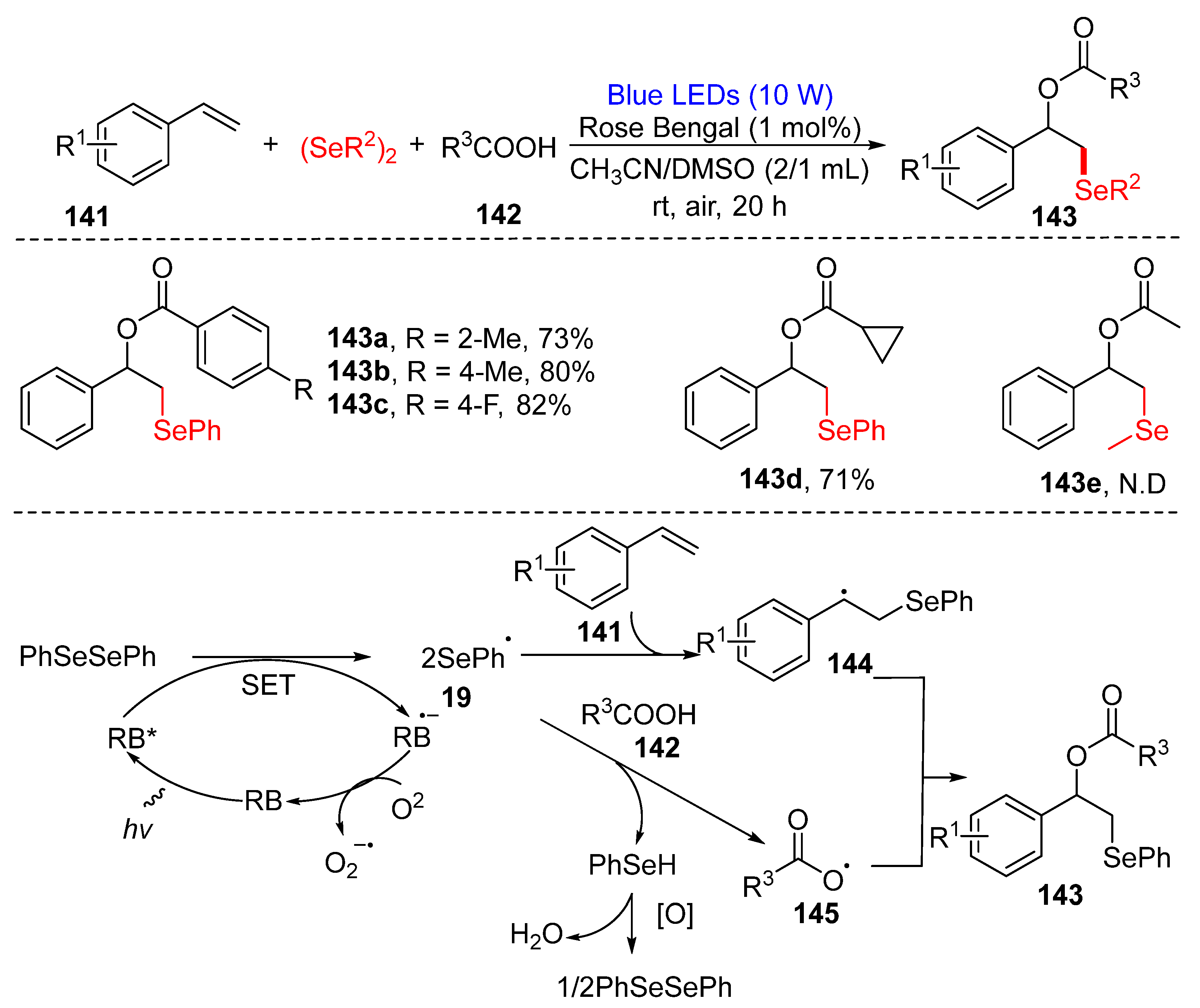
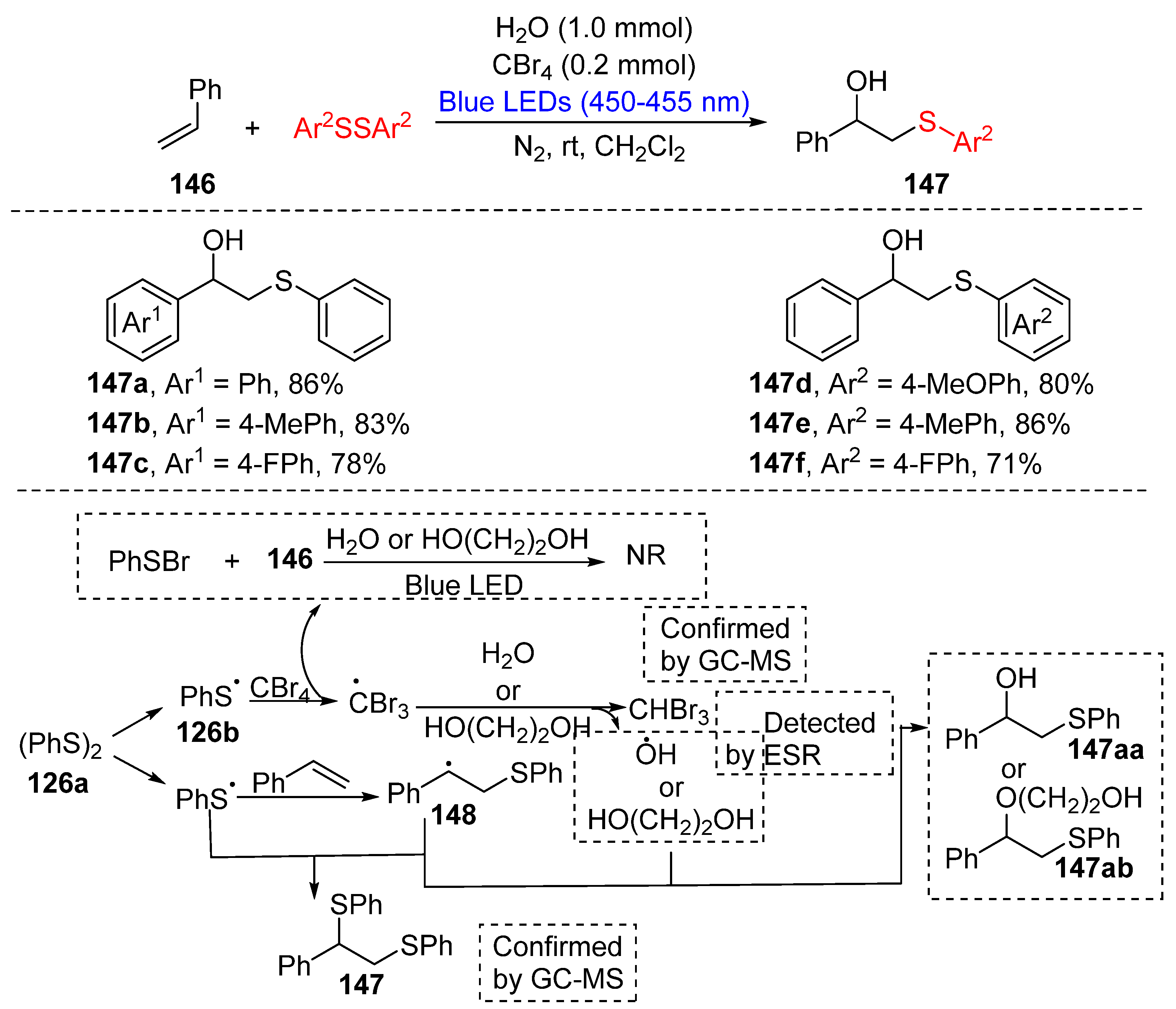
3.3. Visible-Light-Enabled Seleno-and Sulfur-Bifunctionalization of Alkenes/Alkynes
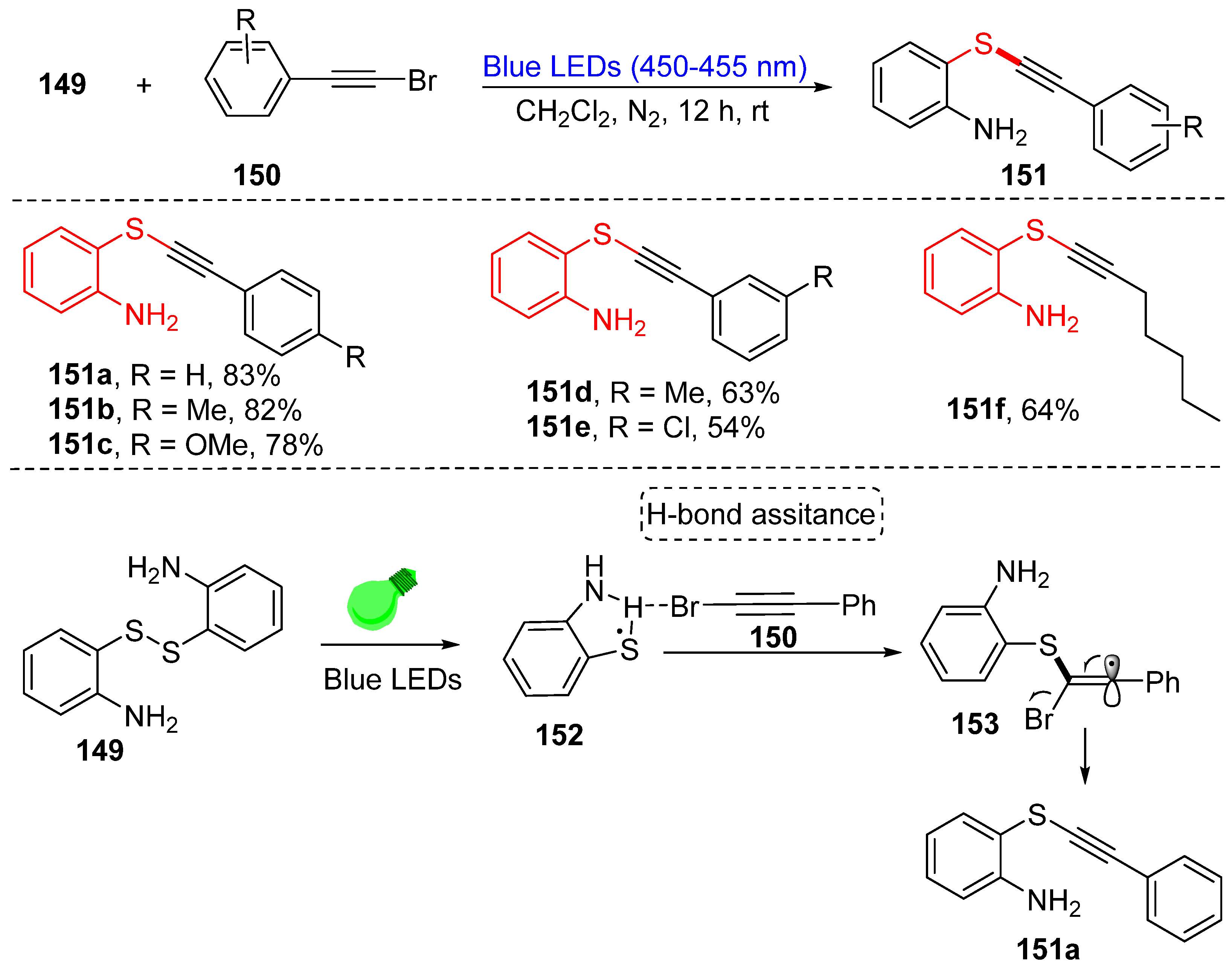
4. Construction of the C(sp)-S Bond
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Meng, J.P.; Zhao, D.Y.; Tang, S.; Sun, K. Synthesis and applications of thiosulfonates and selenosulfonates as free-radical reagents. Chin. Chem. Lett. 2023, 34, 107736. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Sun, K.; Meng, J.P.; Zhang, B. Study on the photoelectric technology in the synthesis of selenium-containing heterocycles. Chin. J. Org. Chem. 2021, 41, 4588–4609. [Google Scholar] [CrossRef]
- Chen, Z.; Lai, H.Q.; Hou, L.Y.; Chen, T.F. Rational design and action mechanisms of chemically innovative organoselenium in cancer therapy. Chem. Commun. 2020, 56, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Ortgies, S.; Breder, A. Oxidative alkene functionalizations via selenium-π-acid catalysis. ACS Catal. 2017, 7, 5828–5840. [Google Scholar] [CrossRef]
- Lomaestro, B.M.; Malone, M. Glutathione in health and disease: Pharmacotherapeutic issues. Ann. Pharmacother. 1995, 29, 1263–1273. [Google Scholar] [CrossRef]
- Ma, N.; Li, Y.; Ren, H.F.; Xu, H.P.; Li, Z.B.; Zhang, X. Selenium-containing block copolymers and their oxidation-responsive aggregates. Polym. Chem. 2010, 1, 1609–1614. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic Diselenide Bonds: Exchange Reaction Induced by Visible Light without Catalysis. Angew. Chem. Int. Ed. 2014, 53, 6781–6785. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Nivsarkar, M.; Acharya, J. A new reagent for the efficient synthesis of disulfides from alkyl halides. Tetrahedron Lett. 2003, 44, 887–889. [Google Scholar] [CrossRef]
- Sun, B.; Luo, C.; Zhang, X.; Guo, M.; Sun, M.; Yu, H.; Chen, Q.; Yang, W.; Wang, M.; Zuo, S. Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat. Commun. 2019, 10, 3211–3219. [Google Scholar] [CrossRef]
- Jen, K.Y.; Cava, M.P. Improved synthesis of aromatic diselenides. J. Org. Chem. 1983, 48, 1449–1451. [Google Scholar] [CrossRef]
- Krief, A.; Mahieu, A.F.D.; Dumont, W.; Trabelsi, M. Efficient syntheses of diselenides from selenols. Synthesis 1988, 2, 131–133. [Google Scholar] [CrossRef]
- Zhong, P.; Guo, M.P. Preparation of diselenides by the oxidation of selenols using trichloroisocyanuric acid. Synth. Commun. 2001, 31, 1507–1510. [Google Scholar] [CrossRef]
- De, R.M.; Rong, Z.H.; Qiang, F.W.; Zhou, X.J. First example of selenium transfer reaction of primary selenoamides and selenourea. Novel synthesis of dialkyl diselenides from alkyl halides. J. Organomet. Chem. 1995, 485, 19–24. [Google Scholar] [CrossRef]
- Singh, D.; Deobald, A.M.; Camargo, L.R.S.; Tabarelli, G.; Rodrigues, O.E.D.; Braga, A.L. An efficient one-pot synthesis of symmetrical diselenides or ditellurides from halides with CuO nanopowder/Seo or Teo/base. Org. Lett. 2010, 12, 3288–3291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Ran, L.; Luo, J.; Xiong, F.; Jing, L.; Qin, D. Synthesis of diaryl selenyl ethers by copper-catalyzed one-pot method. Chem. Res. Appl. 2013, 25, 1632–1638. [Google Scholar]
- Chen, X.; Cui, M.F.; Zhang, J. Phase transfer catalytic synthesis of bicyclohexyl disulfide. Fine Chem. 2005, 22, 874–877. [Google Scholar]
- Joshaghani, M.; Khosropour, A.; Jafary, H. Mild and highly efficient preparation of symmetrical disulfides and diselenides using bipyridinum hydrobromide perbromide as a new oxidative reagent. Phosphorus Sulfur Silicon 2005, 180, 117–123. [Google Scholar] [CrossRef]
- Lenardao, E.J.; Lara, R.G.; Silva, M.S. Clean and fast oxidative transformation of thiolsto disulfides under solvent-free conditions. Tetrahedron Lett. 2007, 39, 7668–7670. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Reddy, M.S.; Yao, M.L. Synthesis of diaryl disulfides via the reductive coupling of arylsulfonyl chlorides. Tetrahedron Lett. 2009, 50, 7340–7342. [Google Scholar] [CrossRef]
- Rajabi, F.; Kakeshpool, T.; Saidi, M.R. Supported iron oxide nanoparticles: Recoverable and efficient catalyst for oxidative S−S coupling of thiolsto disulfides. Catal. Commun. 2013, 40, 13–17. [Google Scholar] [CrossRef]
- Talla, A.; Driessen, B.; Straathof, N.J.W.; Milroy, L.G.; Brunsveld, L.; Noel, T. Metal-free photocatalytic aerobic oxidation of thiols to disulfides in batch and continuous-flow. Adv. Synth. Catal. 2015, 357, 2180–2186. [Google Scholar] [CrossRef]
- Yu, B.; Zheng, Y.; Yuan, Z. Toward direct protein S-persulfidation: A prodrug approach that directly delivers hydrogen persulfide. J. Am. Chem. Soc. 2018, 140, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Zhu, S.; Hu, Y.; Yang, P.; Chu, X.; He, G.; Wang, H.; Chen, G. Synthesis of N-acyl sulfenamides via copper catalysis and their use as S-sulfenylating reagents of thiols. Nat. Commun. 2022, 13, 6445. [Google Scholar] [CrossRef] [PubMed]
- Conner, E.S.; Crocker, K.E.; Fernando, R.G.; Fronczek, F.R.; Stanley, G.G.; Ragains, J.R. Visible-light-promoted selenofunctionalization of alkenes. Org. Lett. 2013, 15, 5558–5561. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, H.; Mandal, A.; Dana, S.; Baidyaa, M. Visible-light-induced synthetic approach for selenylative spirocyclization of N-aryl alkynamides with molecular oxygen as oxidant. Adv. Synth. Catal. 2018, 360, 1099–1103. [Google Scholar] [CrossRef]
- Ma, X.L.; Wang, Q.; Feng, X.Y.; Mo, Z.Y.; Pan, Y.M.; Chen, Y.Y.; Xin, M.; Xu, Y.L. Metal-free visible-light induced cyclization/substitution cascade reaction of alkyne-tethered cyclohexadienones and diselenides: Access to 5-hydroxy-3-selenyl-4a,8a-dihydro-2H-chromen-6(5H)-ones. Green Chem. 2019, 21, 3547–3551. [Google Scholar] [CrossRef]
- Jung, H.I.; Kim, D.Y. Synthesis of β-selenylated cyclopentanones via photoredox-catalyzed selenylation ring-expansion cascades of alkenyl cyclobutanols. Synlett 2019, 30, 1361–1365. [Google Scholar] [CrossRef]
- Ye, Z.P.; Xia, P.; Liu, F.; Hu, Y.Z.; Song, D.; Xiao, J.A.; Huang, P.; Xiang, H.Y.; Chen, X.Q.; Yang, H. Visible-light-induced, catalyst-free radical cross-coupling cyclization of N-allylbromodifluoroacetamides with disulfides or diselenides. J. Org. Chem. 2020, 85, 5670–5682. [Google Scholar] [CrossRef]
- Sahoo, S.R.; Das, B.; Sarkar, D.; Henkel, F.; Reuter, H. Visible-light assisted selenylative intramolecular dearomative carbo-spirocyclisation (IDCS) of homologated-ynones. Eur. J. Org. Chem. 2020, 7, 891–896. [Google Scholar] [CrossRef]
- Zhou, X.J.; Liu, H.Y.; Mo, Z.Y.; Ma, X.L.; Chen, Y.Y.; Tang, H.T.; Pan, Y.M.; Xu, Y.L. Visible-light-promoted selenylative spirocyclization of indolyl-ynones toward the formation of 3-selenospiroindolenine anticancer agents. Chem. Asian J. 2020, 15, 1536–1539. [Google Scholar] [CrossRef]
- Mutra, M.R.; Kudale, V.S.; Li, J.; Tsaia, W.H.; Wang, J.J. Alkene versus alkyne reactivity in unactivated 1,6-enynes: Regio- and chemoselective radical cyclization with chalcogens under metal- and oxidant-free conditions. Green Chem. 2020, 22, 2288–2300. [Google Scholar] [CrossRef]
- Tran, C.C.; Kawaguchi, S.; Sato, F.; Nomoto, A.; Ogawa, A. Photoinduced cyclizations of o-diisocyanoarenes with organic diselenides and thiols that afford chalcogenated quinoxalines. J. Org. Chem. 2020, 85, 7258–7266. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Sun, Y.; Pan, Y.J.; Yu, H.G.; Han, Y.; Shi, Y.C.; Yan, C.G.; Zhu, S.Q. Visible-light mediated diaryl selenylative cyclization of 1,6-Enynes. J. Org. Chem. 2021, 86, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Li, P.; Zhang, Y.; Wang, L. Visible-light-induced a tandem oxidative cyclization of 2-alkynylanilines with disulfides(diselenides) to 3-sulfenyl and 3-selenylindoles under transition-metal-free and photocatalyst-free conditions. Org. Chem. Front. 2017, 4, 1322–1330. [Google Scholar] [CrossRef]
- Li, X.; Xu, Z.; Wang, L.; Wang, F.; Yang, J.; Li, P. A facile synthesis of functionalized benzofurans via visible-light-induced tandem cyclization of 1,6-enynes with disulfides. ChemPhotoChem 2021, 5, 142–148. [Google Scholar] [CrossRef]
- Romero, N.A.; Nicewicz, D.A. Organic Photoredox Catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef]
- Li, H.; Yao, Y.; Li, L. Coumarins as potential antidiabetic agents. J. Pharmacol. 2017, 69, 1253–1264. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Seoane-Rivero, R.; Navarro, R.; Marcos-Fernandez, A. Coumarins into polyurethanes for smart and functional materials. Polymers 2020, 12, 630. [Google Scholar] [CrossRef]
- Yang, D.; Li, G.; Xing, C.; Cui, W.; Li, K.; Wei, W. Metal- and photocatalyst-free visible-light-promoted regioselective selenylation of coumarin derivatives via oxidation-induced C−H functionalization. Org. Chem. Front. 2018, 5, 2974–2979. [Google Scholar] [CrossRef]
- Jin, J.; MacMillan, D.W.C. Direct α-arylation of ethers through the combination of photoredox-mediated C−H functionalization and the minisci reaction. Angew. Chem. Int. Ed. 2015, 54, 1565–1569. [Google Scholar] [CrossRef]
- Devari, S.; Shah, B.A. Visible-light-promoted C−H functionalization of ethers and electron-deficient arenes. Chem. Commun. 2016, 52, 1490–1493. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, B.; Yang, C.; Xia, W. Visible-light-promoted direct amination of phenols via oxidative cross-dehydrogenative coupling reaction. Org. Lett. 2016, 18, 3326–3329. [Google Scholar] [CrossRef] [PubMed]
- Saba, S.; Rafifique, J.; Franco, M.S.; Schneider, A.R.; Espíndola, L.; Silva, D.O.; Braga, A.L. Rose Bengal catalysed photo-induced selenylation of indoles, imidazoles and arenes: A metal free approach. Org. Biomol. Chem. 2018, 16, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Lemir, I.D.; Castro-Godoy, W.D.; Heredia, A.A.; Schmidt, L.C.; Arguello, J.E. Metal- and photocatalyst-free synthesis of 3-selenylindoles and asymmetric diaryl selenides promoted by visible-light. RSC Adv. 2019, 9, 22685–22694. [Google Scholar] [CrossRef]
- Sousa, F.S.S.; Birmann, P.T.; Baldinotti, R.; Fronza, M.G.; Balaguez, R.; Alves, D.; Bruning, C.A.; Savegnago, L. α-(phenylselanyl) acetophenone mitigates reserpine-induced pain-depression dyad: Behavioral, biochemical and molecular docking evidences. Brain Res. Bull. 2018, 142, 129–137. [Google Scholar] [CrossRef]
- Dalberto, B.T.; Schneider, P.H. Photoinduced metal-free α-selenylation of ketones. RSC Adv. 2020, 10, 10502–10509. [Google Scholar] [CrossRef]
- Ali, D.; Parvin, T.; Choudhury, L.H. Visible-light-mediated C(sp2)−H selenylation of amino pyrazole and amino uracils in the presence of rose bengal as an organophotocatalyst. J. Org. Chem. 2022, 87, 1230–1239. [Google Scholar] [CrossRef]
- Zhu, X.; Xie, X.; Li, P.; Guo, J.; Wang, L. Visible-light-induced direct thiolation at α-C(sp3)−H of ethers with disulfides using acridine red as photocatalyst. Org. Lett. 2016, 18, 1546–1549. [Google Scholar] [CrossRef]
- Rathore, V.; Kuma, S. Visible-light-induced metal and reagent-free oxidative coupling of C(sp2)−H bonds with organo-dichalcogenides: Synthesis of 3-organochalcogenyl indoles. Green Chem. 2019, 21, 2670–2676. [Google Scholar] [CrossRef]
- Kim, J.; Kang, B.; Hong, S.H. Direct allylic C(sp3)−H thiolation with disulfides via visible-light photoredox catalysis. ACS Catal. 2020, 10, 6013–6022. [Google Scholar] [CrossRef]
- Sandfort, F.; Knecht, T.; Pinkert, T.; Daniliuc, C.G.; Glorius, F. Site-selective thiolation of (multi)halogenated heteroarenes. J. Am. Chem. Soc. 2020, 142, 6913–6919. [Google Scholar] [CrossRef]
- Tyson, E.L.; Ament, M.S.; Yoon, T.P. Transition metal photoredox catalysis of radical thiol-ene reactions. J. Org. Chem. 2013, 78, 2046–2050. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M.L.; Weragoda, G.K.; Monaghan, R.; Connell, T.U.; Brzozowski, M.A.; Scully, D.; Burton, J.; Lupton, D.W.; Polyzos, A.A. Visible-light photocatalytic thiolation of aryl, heteroaryl and vinyl iodides. Org. Biomol. Chem. 2018, 16, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Yang, H.J.; Fu, H. Visible-light photoredox synthesis of chiral α-selenoamino acids. Org. Lett. 2016, 18, 1968–1971. [Google Scholar] [CrossRef] [PubMed]
- Anand, D.; He, Y.; Li, L.; Zhou, L. A photocatalytic sp3 C−S, C−Se and C−B bond formation through C−C bond cleavage of cycloketone oxime esters. Org. Biomol. Chem. 2019, 17, 533–540. [Google Scholar] [CrossRef]
- He, B.Q.; Yu, X.Y.; Wang, P.Z.; Chen, J.R.; Xiao, W.J. A photoredox catalyzed iminyl radical-triggered C−C bond cleavage/addition/Kornblum oxidation cascade of oxime esters and styrenes: Synthesis of ketonitriles. Chem. Commun. 2018, 54, 12262–12265. [Google Scholar] [CrossRef]
- Li, L.; Chen, H.; Mei, M.; Zhou, L. Visible-light promoted γ-cyanoalkyl radical generation: Three-component cyanopropylation etherification of unactivated alkenes. Chem. Commun. 2017, 53, 11544–11547. [Google Scholar] [CrossRef]
- Zhang, J.J.; Duan, X.H.; Wu, Y.; Yang, J.C.; Guo, L.N. Transition-metal free C−C bond cleavage/borylation of cycloketone oxime esters. Chem. Sci. 2018, 10, 161–166. [Google Scholar] [CrossRef]
- Liu, J.J.; Tian, M.M.; Li, Y.X.; Shan, X.W.; Li, A.K.; Lu, K.; Fagnoni, M.; Protti, S.; Zhao, X. Metal-free synthesis of unsymmetrical aryl selenides and tellurides via visible-light-driven activation of arylazo sulfones. Eur. J. Org. Chem. 2020, 47, 7358–7367. [Google Scholar] [CrossRef]
- He, F.S.; Ye, S.Q.; Wu, J. Recent advances in pyridinium salts as radical reservoirs in organic synthesis. ACS Catal. 2019, 9, 8943–8960. [Google Scholar] [CrossRef]
- Basch, C.H.; Liao, J.N.; Xu, J.Y.; Piane, J.J.; Watson, M.P. Harnessing alkyl amines as electrophiles for nickel-catalyzed cross couplings via C−N Bond Activation. J. Am. Chem. Soc. 2017, 139, 5313–5316. [Google Scholar] [CrossRef] [PubMed]
- Klauck, F.J.R.; Jame, M.J.; Glorius, F. Deaminative strategy for the visible-light-mediated generation of alkyl radicals. Angew. Chem. Int. Ed. 2017, 56, 12336–12339. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.P.; Wang, G.Q.; Li, S.H.; Shi, Z.Z. Selective C−N borylation of alkyl amines promoted by lewis base. Angew. Chem. Int. Ed. 2018, 57, 15227–15232. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.S.; Qiao, J.M.; Li, A.K.; Jiang, Z.Y.; Lu, K.; Zhao, X. Metal-free synthesis of unsymmetrical selenides from pyridinium salts and diselenides catalysed by visible-light. Tetrahedron Lett. 2021, 72, 153071. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, W.; Li, H.; Meng, Y.; Zhang, T.; Chen, Q.Y.; Zhu, C. Metal-free synthesis of ketones by visible-light induced aerobic oxidative radical addition of aryl hydrazines to alkenes. Green Chem. 2017, 19, 2941–2944. [Google Scholar] [CrossRef]
- Ding, Y.; Li, H.; Meng, Y.; Zhang, T.; Li, J.; Chen, Q.Y.; Zhu, C. Direct synthesis of hydrazones by visible-light mediated aerobic oxidative cleavage of the C[double bond, length as m-dash]C bond. Org. Chem. Front. 2017, 4, 1611–1614. [Google Scholar] [CrossRef]
- Taniguchi, T.; Naka, T.; Imoto, M.; Takeda, M.; Nakai, T.; Mihara, M.; Mizuno, T.; Nomoto, A.; Ogawa, A. Transition-metal-free and oxidant-free cross-coupling of arylhydrazines with disulfides: Base-promoted synthesis of unsymmetrical aryl sulfides. J. Org. Chem. 2017, 82, 6647–6655. [Google Scholar] [CrossRef]
- Taniguchi, T.; Murata, A.; Takeda, M.; Mizuno, T.; Nomoto, A.; Ogawa, A. Atom-economical synthesis of unsymmetrical diaryl selenides from arylhydrazines and diaryl diselenides. Eur. J. Org. Chem. 2017, 2017, 4928–4934. [Google Scholar] [CrossRef]
- Hosseini-Sarvari, M.; Jafari, F.; Mohajeri, A.; Hassani, N. Cu2O/TiO2 nanoparticles as visible-light photocatalysts concerning C(sp2)−P bond formation. Catal. Sci. Technol. 2018, 8, 4044–4051. [Google Scholar] [CrossRef]
- Li, R.; Shi, T.; Chen, X.L.; Lv, Q.Y.; Zhang, Y.L.; Peng, Y.Y.; Qu, L.B.; Yu, B. Visible-light-promoted organic dye-catalyzed sulfidation and phosphorylation of arylhydrazines toward aromatic sulfides and diarylphosphoryl hydrazides. N. J. Chem. 2019, 43, 13642–13646. [Google Scholar] [CrossRef]
- Hu, H.; Chen, X.; Sun, K.; Wang, J.; Liu, Y.; Liu, H.; Yu, B.; Sun, Y.; Qu, L.; Zhao, Y. Silver-catalyzed decarboxylative cascade radical cyclization of tert-carboxylic acids and o-(allyloxy)arylaldehydes towards chroman-4-one derivatives. Org. Chem. Front. 2018, 5, 2925–2929. [Google Scholar] [CrossRef]
- Kobiki, Y.; Kawaguchi, S.; Ogawa, A. Highly regioselective hydroselenation of inactivated terminal alkynes using diselenide-Ph2P(O)H mixed systems under visible-light irradiation. Tetrahedron Lett. 2013, 54, 5453–5456. [Google Scholar] [CrossRef]
- Chen, H.; Ding, R.; Tang, H.; Pan, Y.; Xu, Y.; Chen, Y. Simultaneous construction of C−Se and C−S bonds via the visible-light-mediated multicomponent cascade reaction of diselenides, alkynes, and SO2. Chem. Asian J. 2019, 14, 3264–3268. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Doi, M.; Ogawa, I.; Hirao, T. Highly Selective Three-Component Coupling of Ethyl Propiolate, Alkenes, and Diphenyl Diselenide under Visible-Light Irradiation. Angew. Chem. Int. Ed. 1999, 38, 2027–2029. [Google Scholar] [CrossRef]
- Ogawa, A.; Ogawa, I.; Sonoda, N. A Novel Three-Component Coupling of Alkynes, Vinylcyclopropanes, and Diphenyl Diselenide under Visible-Light Irradiation. J. Org. Chem. 2000, 65, 7682–7685. [Google Scholar] [CrossRef]
- Tsuchii, K.; Doi, M.; Hirao, T.; Ogawa, A. Highly selective sequential addition and cyclization reactions involving diphenyl diselenide, an alkyne, and alkenes under visible-light irradiation. Angew. Chem. Int. Ed. 2003, 42, 3490–3493. [Google Scholar] [CrossRef]
- Chen, J.; Chen, R.; Mei, L.; Yan, S.; Wu, Y.; Li, Q.; Yuan, B. Visible-light-induced difunctionalization of styrenes: An efficient and green protocol for the synthesis of β-acyloxyselenides. Asian J. Org. Chem. 2020, 9, 181–184. [Google Scholar] [CrossRef]
- Srivastava, V.; Singh, P.K.; Singh, P.P. Visible-light photoredox catalysed amidation of carboxylic acids with amines. Tetrahedron Lett. 2019, 60, 40–43. [Google Scholar] [CrossRef]
- Luly, J.R.; Yi, N.; Soderquist, J.; Stein, H.; Cohen, J.; Perun, T.J.; Plattner, J.J. New inhibitors of human renin that contain novel Leu-Val replacements. J. Med. Chem. 1987, 30, 1609–1616. [Google Scholar] [CrossRef]
- Alvarez-Ibarra, C.; Cuervo-Rodrìguez, R.; Fernìnedz-Monreal, M.C.; Ruiz, M.P. Synthesis, configurational assignment and conformational analysis of beta-hydroxy sulfoxides, bioisosteres of oxisuran metabolites, and their o-methyl derivatives. J. Org. Chem. 1994, 59, 7284–7291. [Google Scholar] [CrossRef]
- Corey, E.J.; Clark, D.A.; Goto, G.; Marfat, A.; Mioskowski, C.; Samuelsson, B.; Hammarstrom, S. Stereospecific total synthesis of a "slow reacting substance" of anaphylaxis, leukotriene C-1. J. Am. Chem. Soc. 1980, 102, 1436–1439. [Google Scholar] [CrossRef]
- Ruan, H.; Meng, L.G.; Zhu, L.; Wang, L. Visible-light-induced hydroxysulfurization and alkoxysulfurization of styrenes in the absence of photocatalyst: Synthesis of β-Hydroxysulfides and β-alkoxysulfides. Adv. Synth. Catal. 2019, 361, 3217–3222. [Google Scholar] [CrossRef]
- Wang, H.; Gao, X.; Lv, Z.; Abdelilah, T.; Lei, A. Recent advances in oxidative R1-H/R2-H cross-coupling with hydrogen evolution via photo-electrochemistry. Chem. Rev. 2019, 119, 6769–6787. [Google Scholar] [CrossRef]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic carbon-sulfur bond formation in natural product biosynthesis. Chem. Rev. 2017, 117, 5521–5577. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Ruan, H.; Xu, H.; Li, Z.; Meng, L.G.; Wang, L. Amino-assisted synthesis of alkynylthioethers via a visible-light-induced C(sp)–SII coupling between bromoalkynes and 2,2′-diaminodiaryldisulfides. Org. Chem. Front. 2021, 8, 5345–5351. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Zhang, Y.; Sun, K.; Yu, T.; Liu, F.; Wang, X. Synthesis and Application Dichalcogenides as Radical Reagents with Photochemical Technology. Molecules 2023, 28, 1998. https://doi.org/10.3390/molecules28041998
Wang C, Zhang Y, Sun K, Yu T, Liu F, Wang X. Synthesis and Application Dichalcogenides as Radical Reagents with Photochemical Technology. Molecules. 2023; 28(4):1998. https://doi.org/10.3390/molecules28041998
Chicago/Turabian StyleWang, Cairong, Yan Zhang, Kai Sun, Tingting Yu, Fei Liu, and Xin Wang. 2023. "Synthesis and Application Dichalcogenides as Radical Reagents with Photochemical Technology" Molecules 28, no. 4: 1998. https://doi.org/10.3390/molecules28041998
APA StyleWang, C., Zhang, Y., Sun, K., Yu, T., Liu, F., & Wang, X. (2023). Synthesis and Application Dichalcogenides as Radical Reagents with Photochemical Technology. Molecules, 28(4), 1998. https://doi.org/10.3390/molecules28041998