


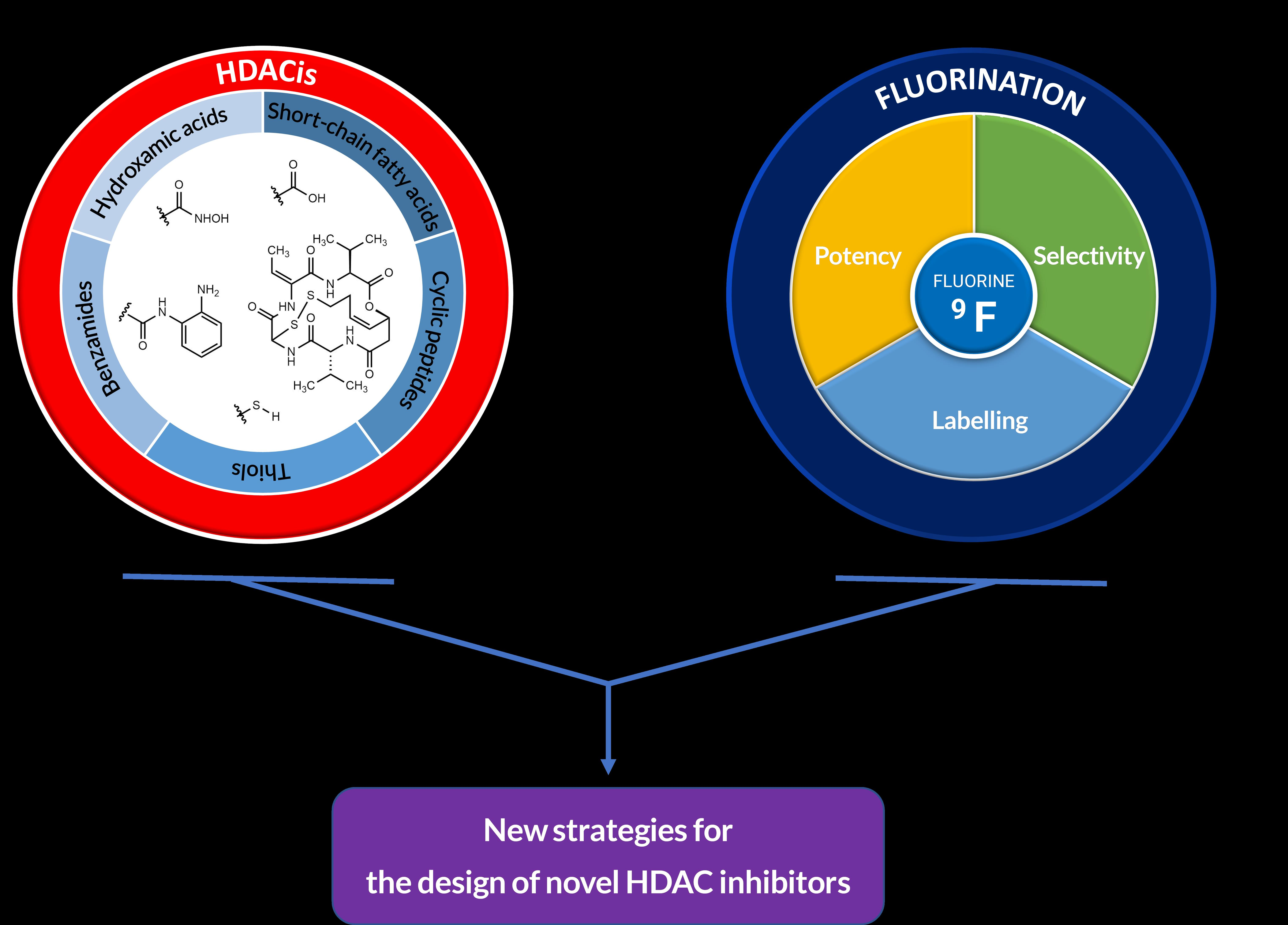
The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors



Abstract

1. Introduction
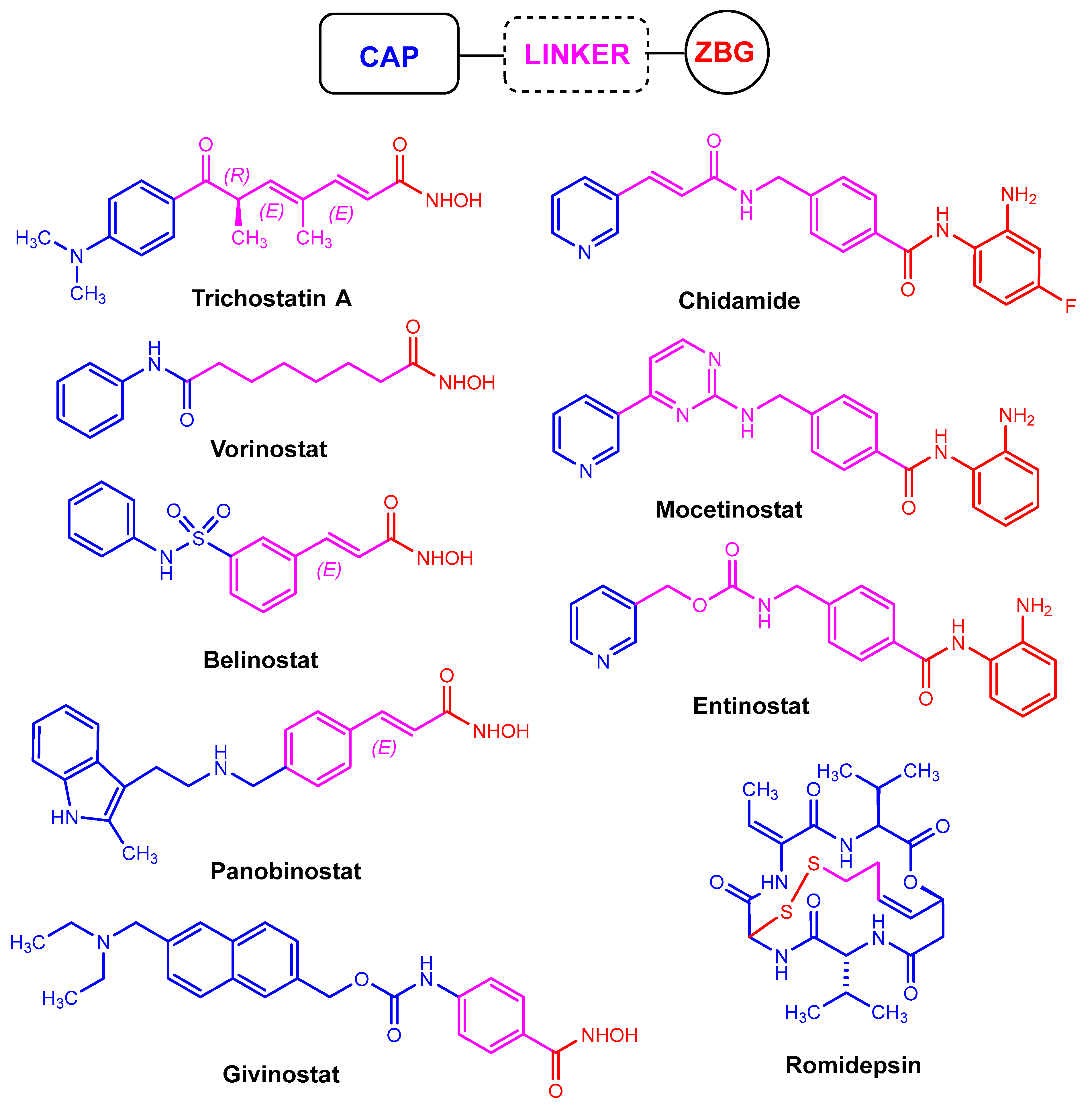
2. Introduction of the Chemical Modification Generating Fluorination in HDACis
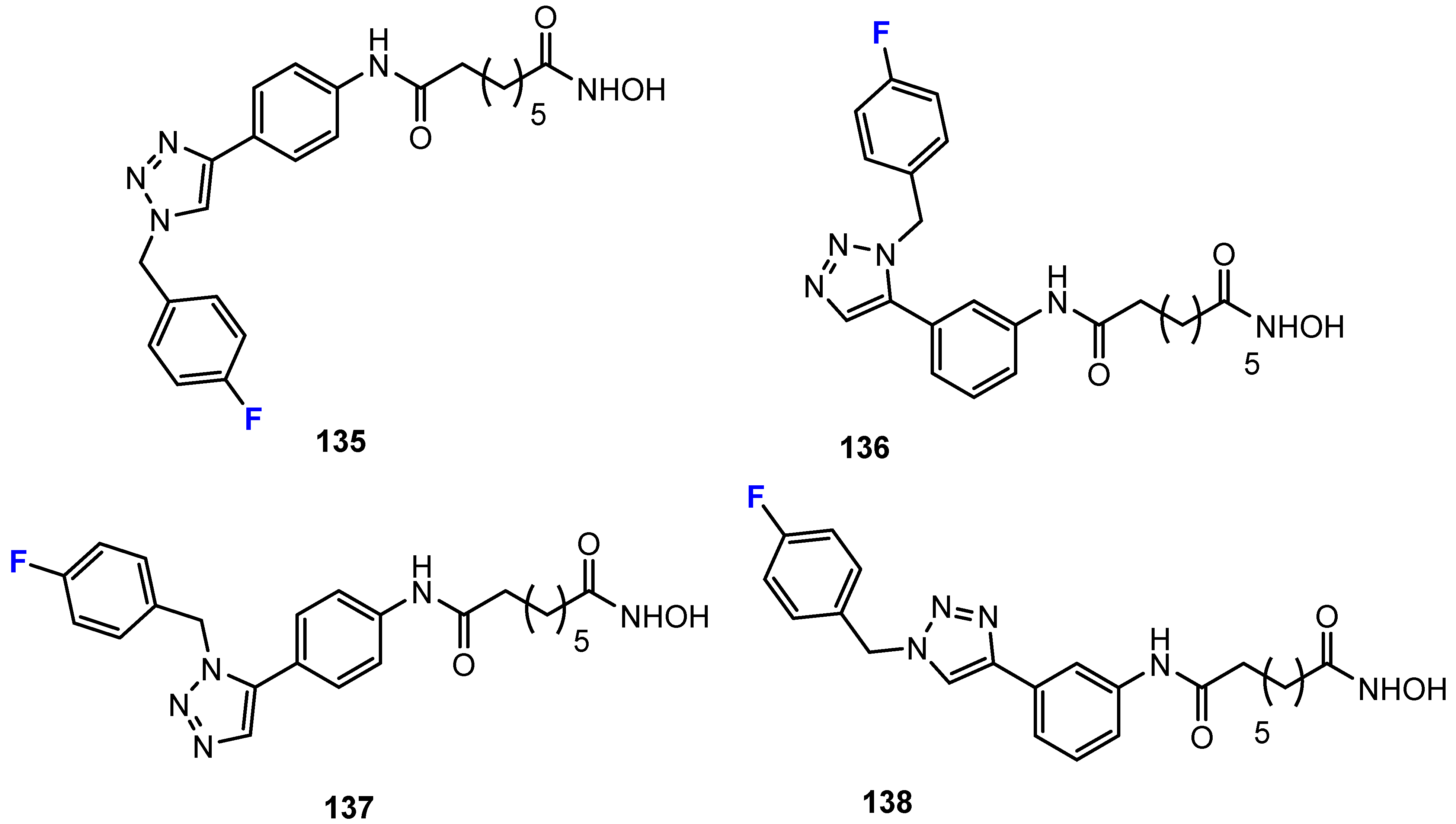
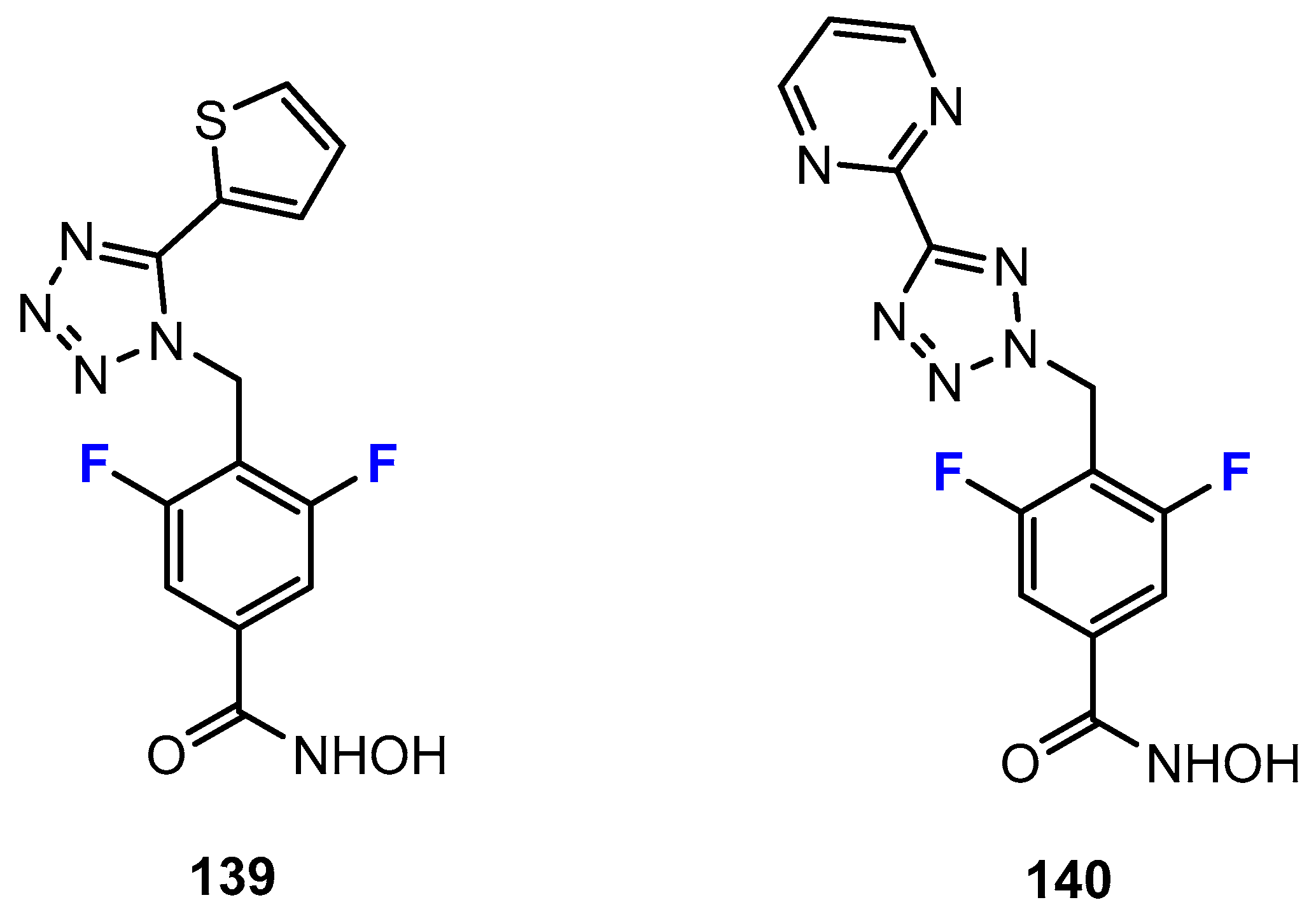
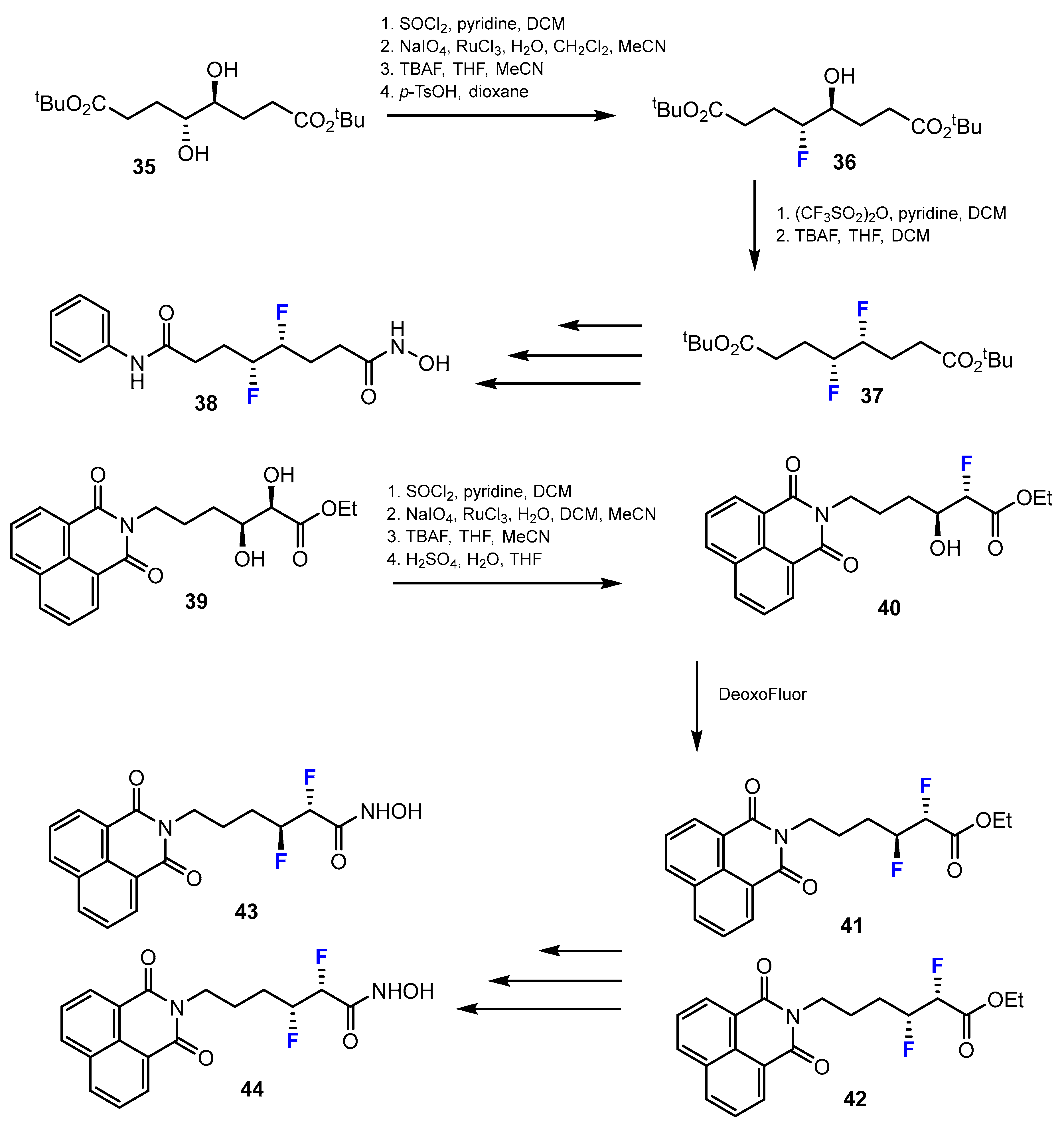
2.1. Hydroxamic Acids
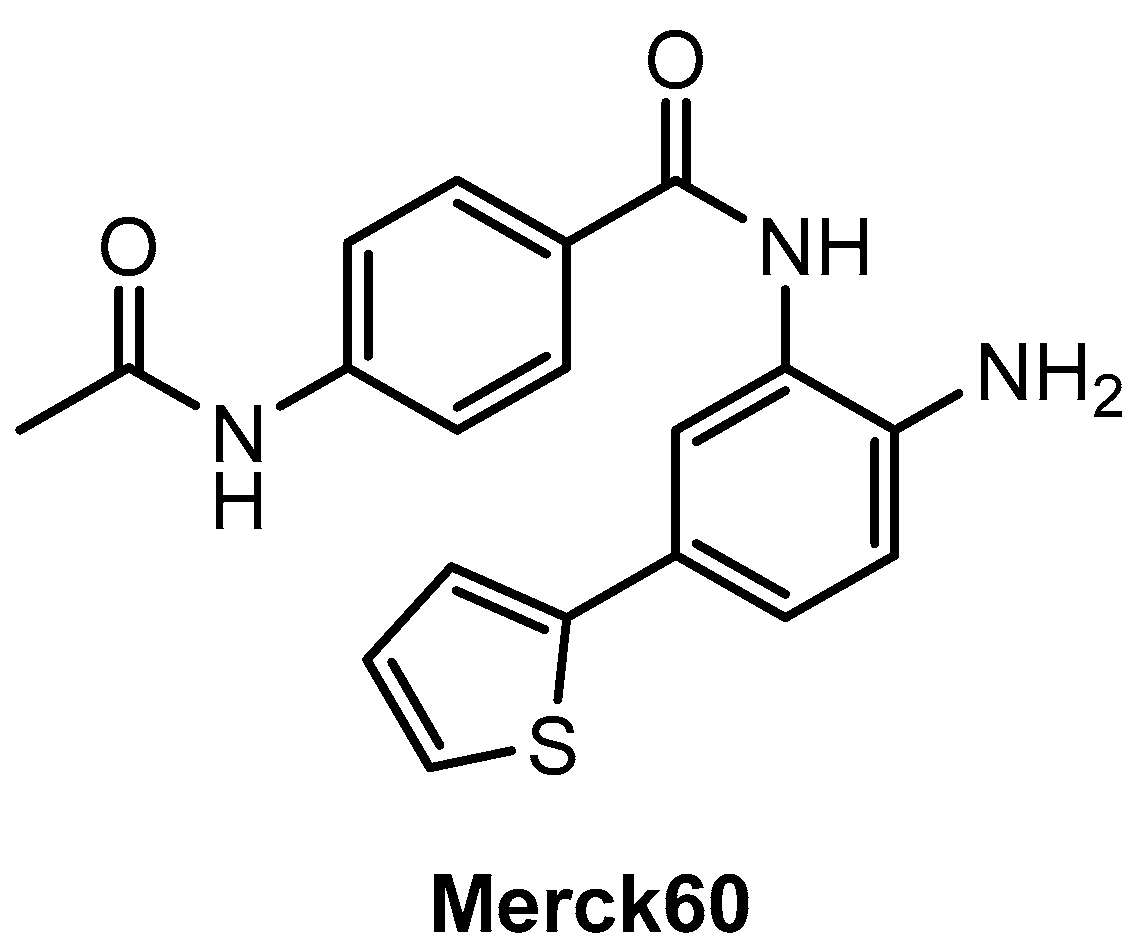
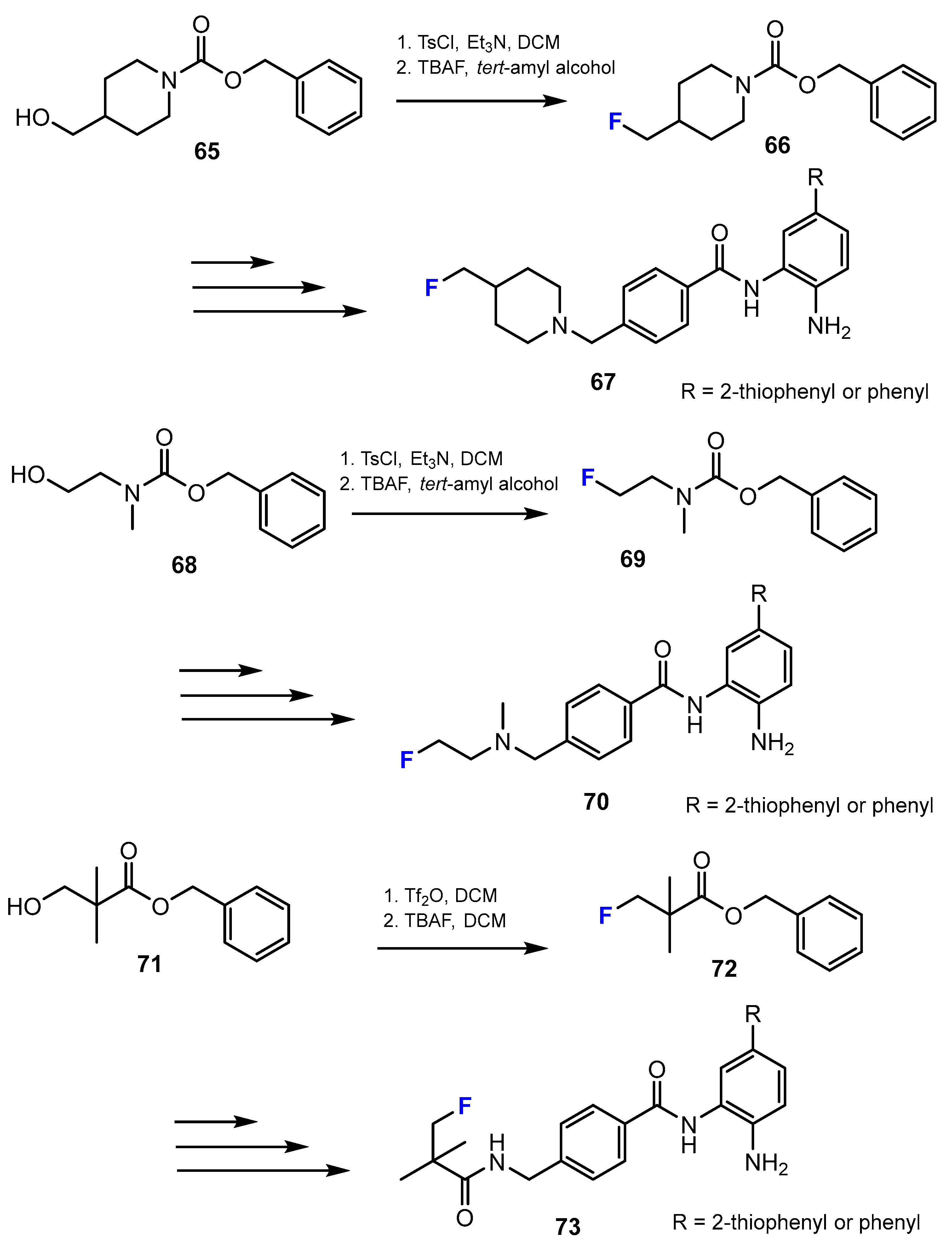
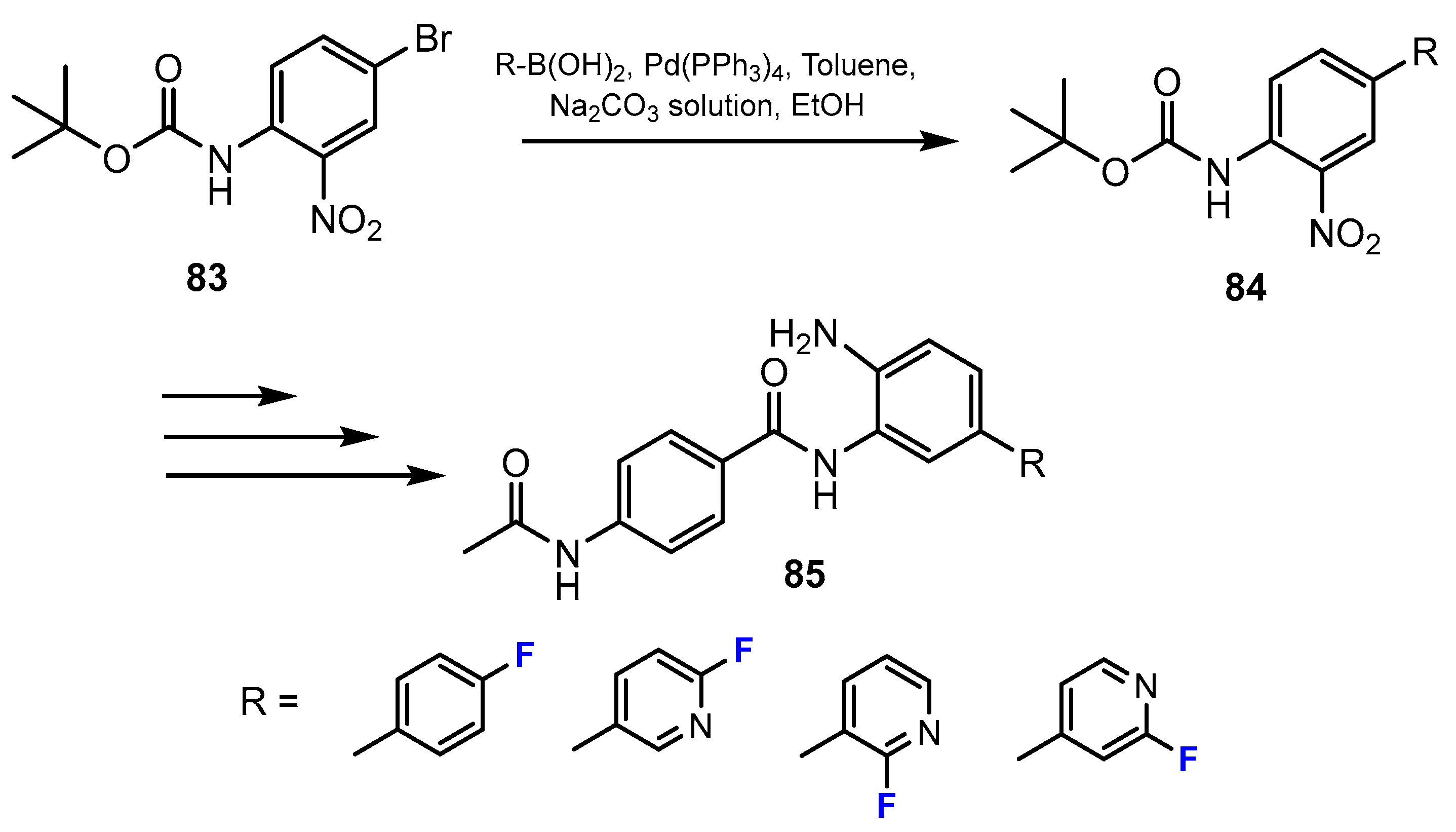
2.2. Benzamides
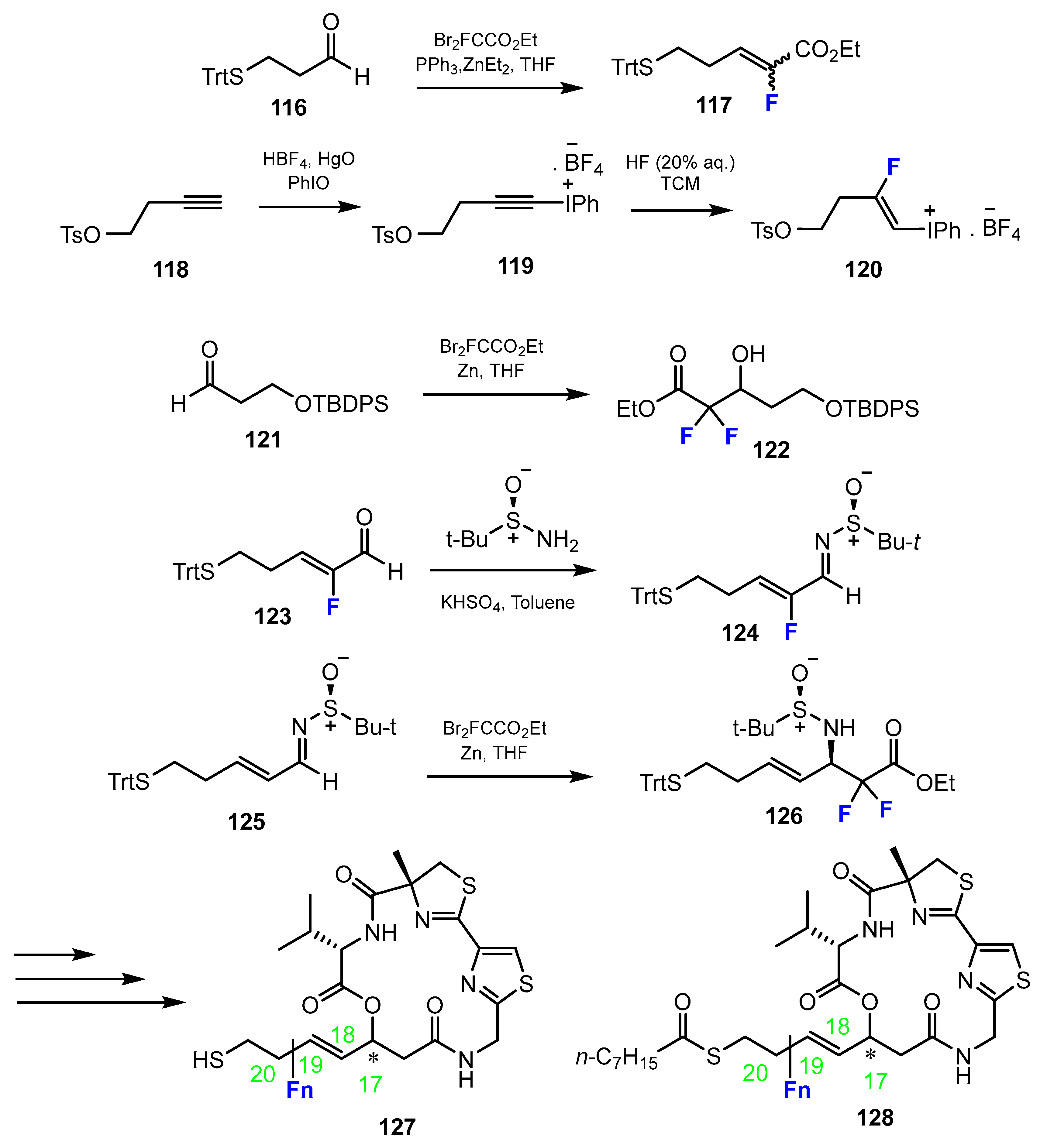
2.3. Thiols
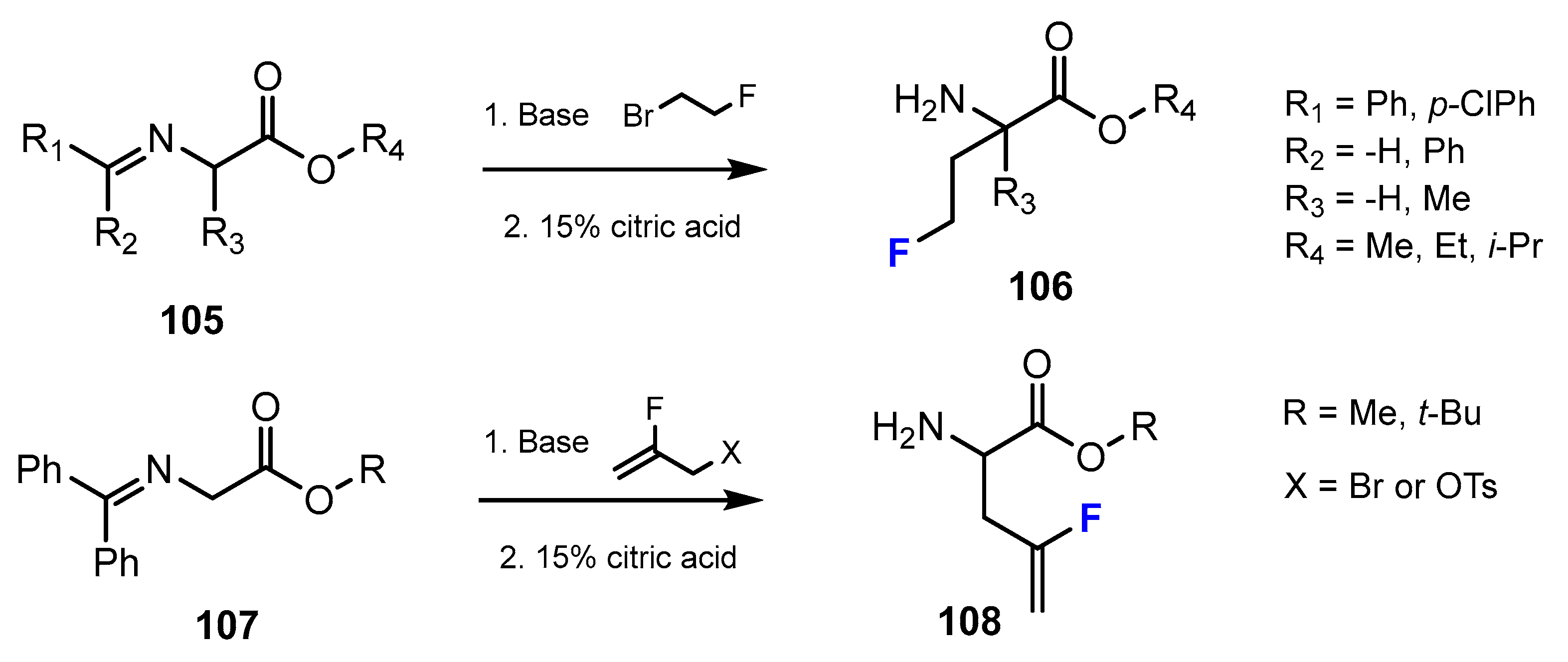
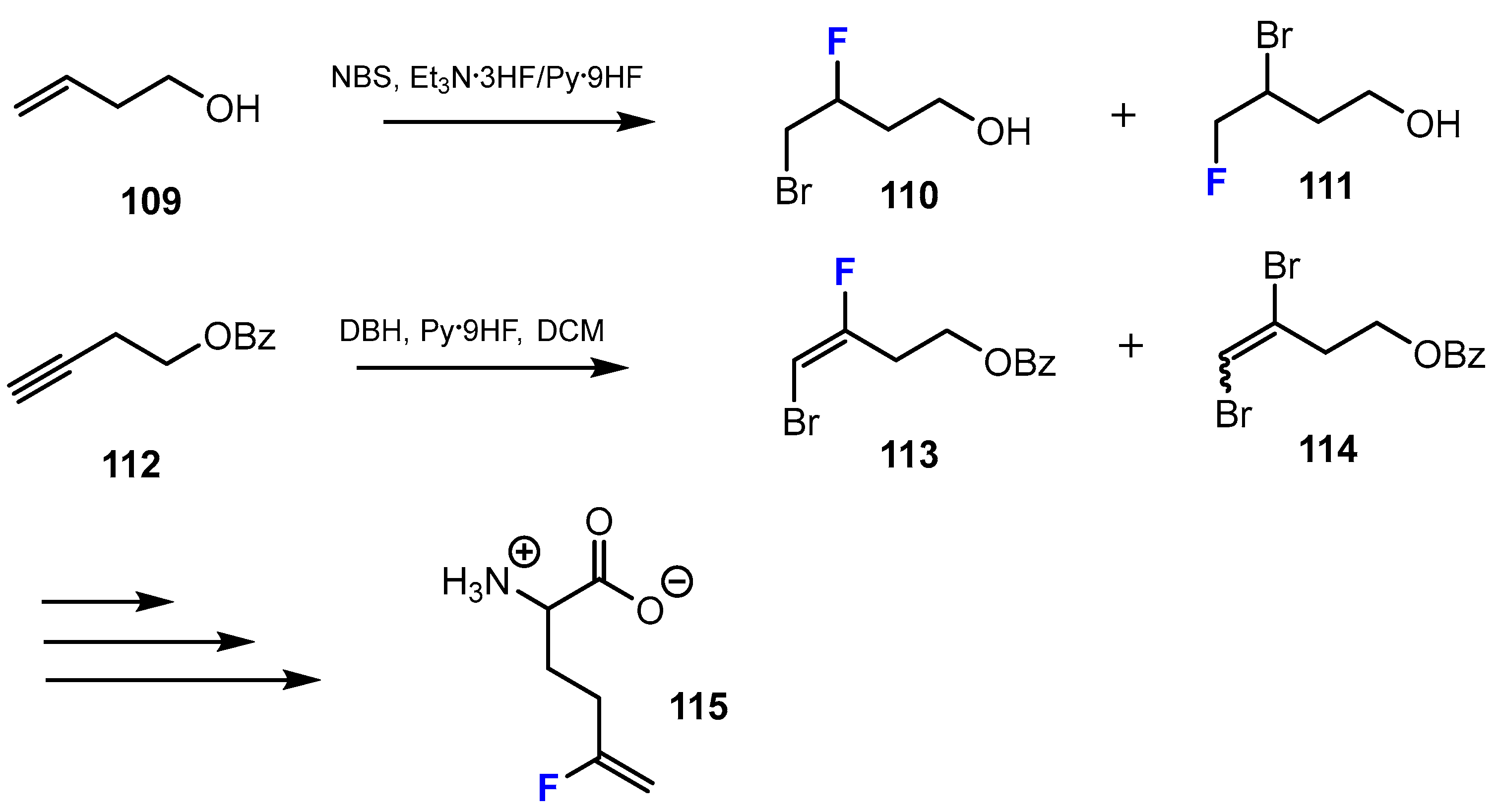
2.4. Short-Chain Fatty Acids
2.5. Cyclic Peptides
3. Biological Significance and Purpose of Fluorination in HDACis
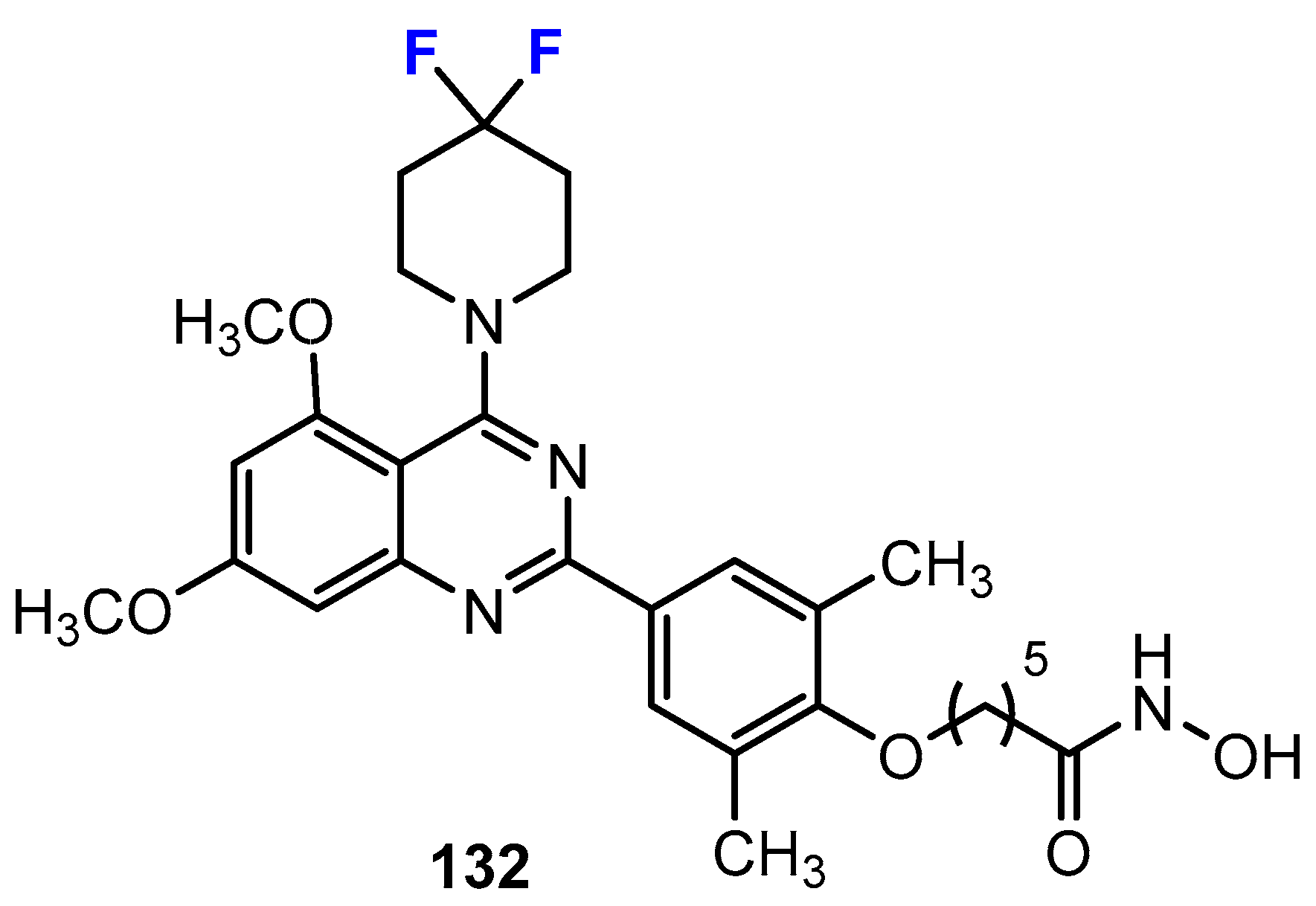
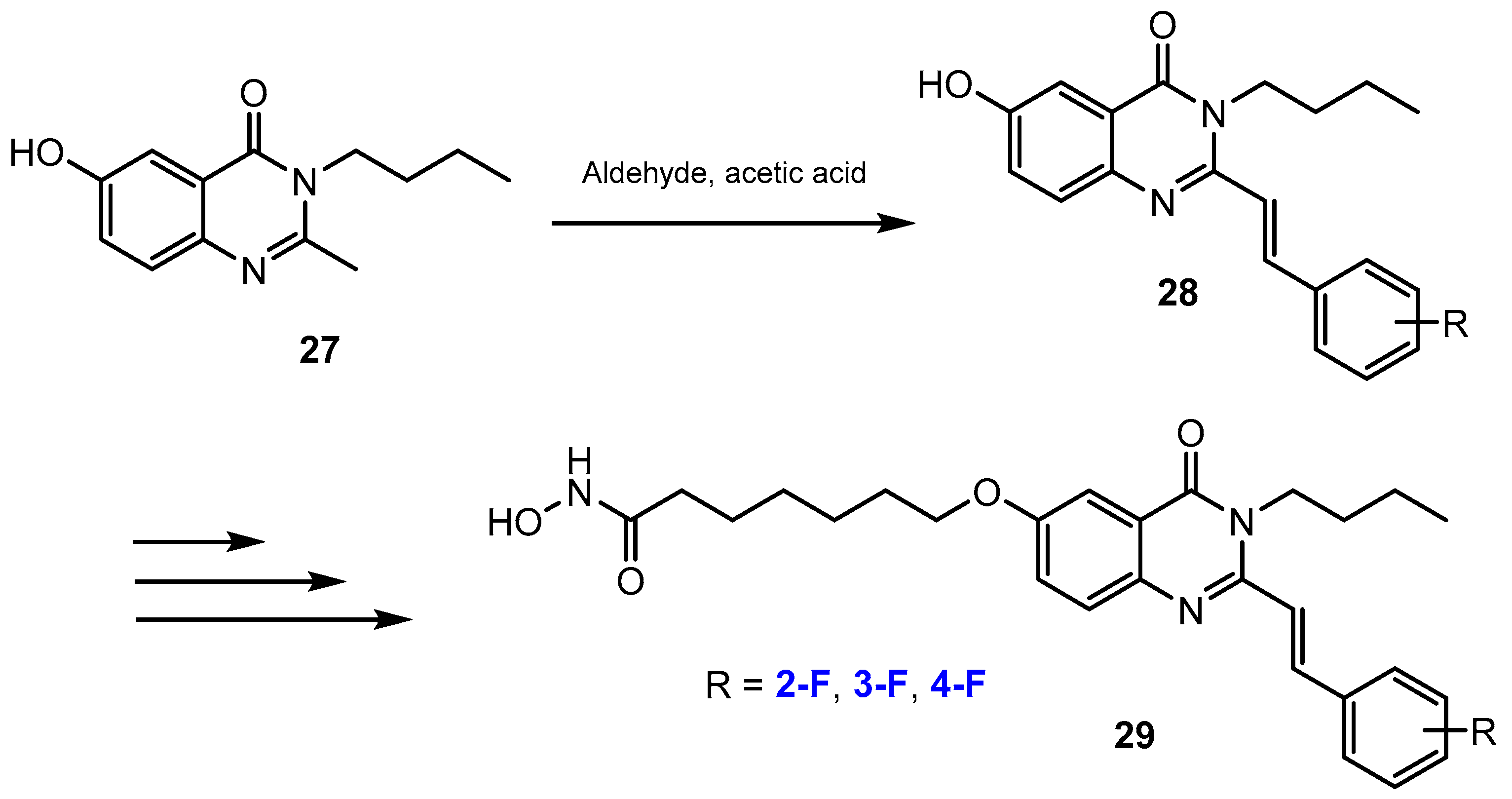
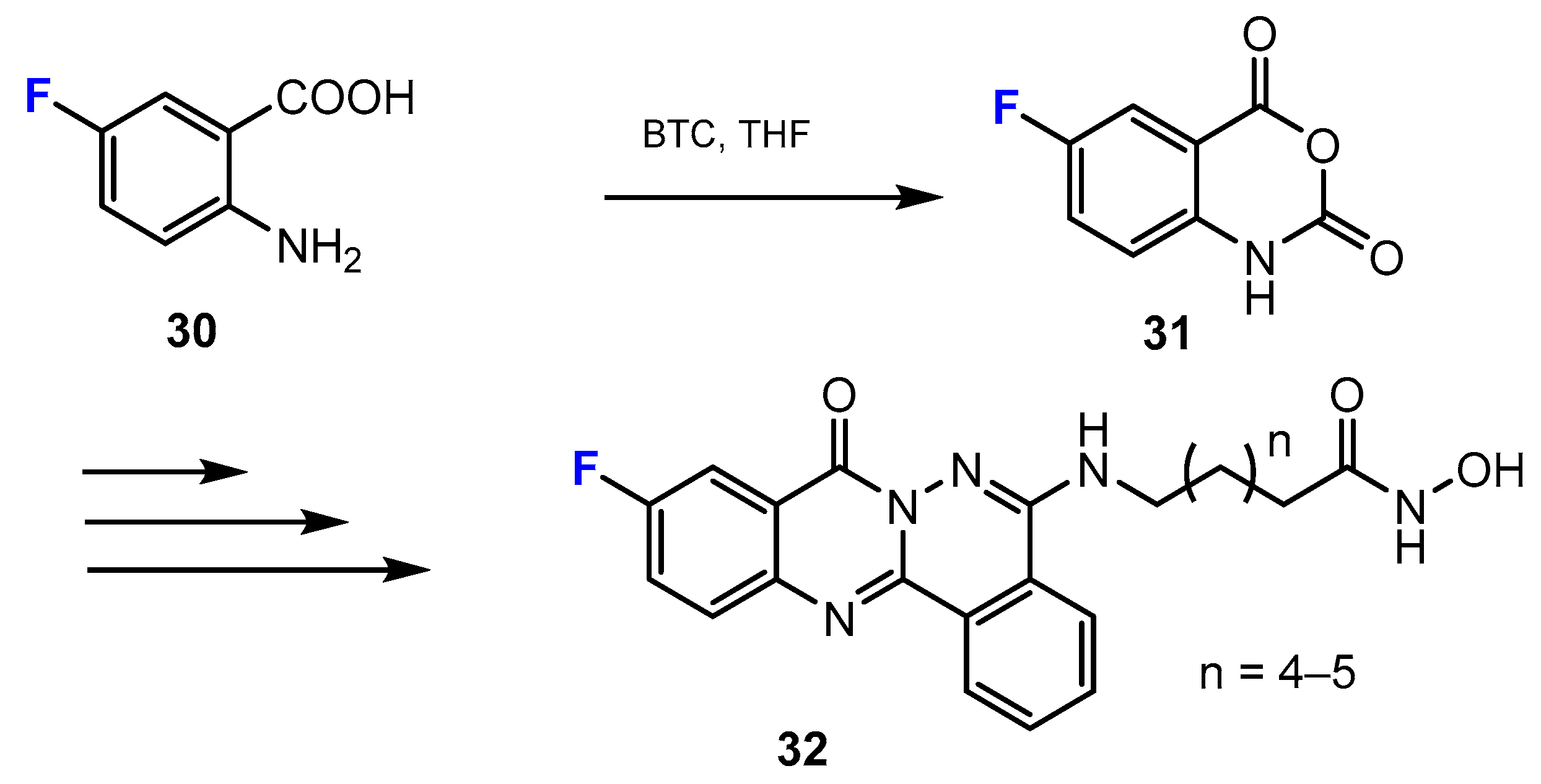
3.1. Potency
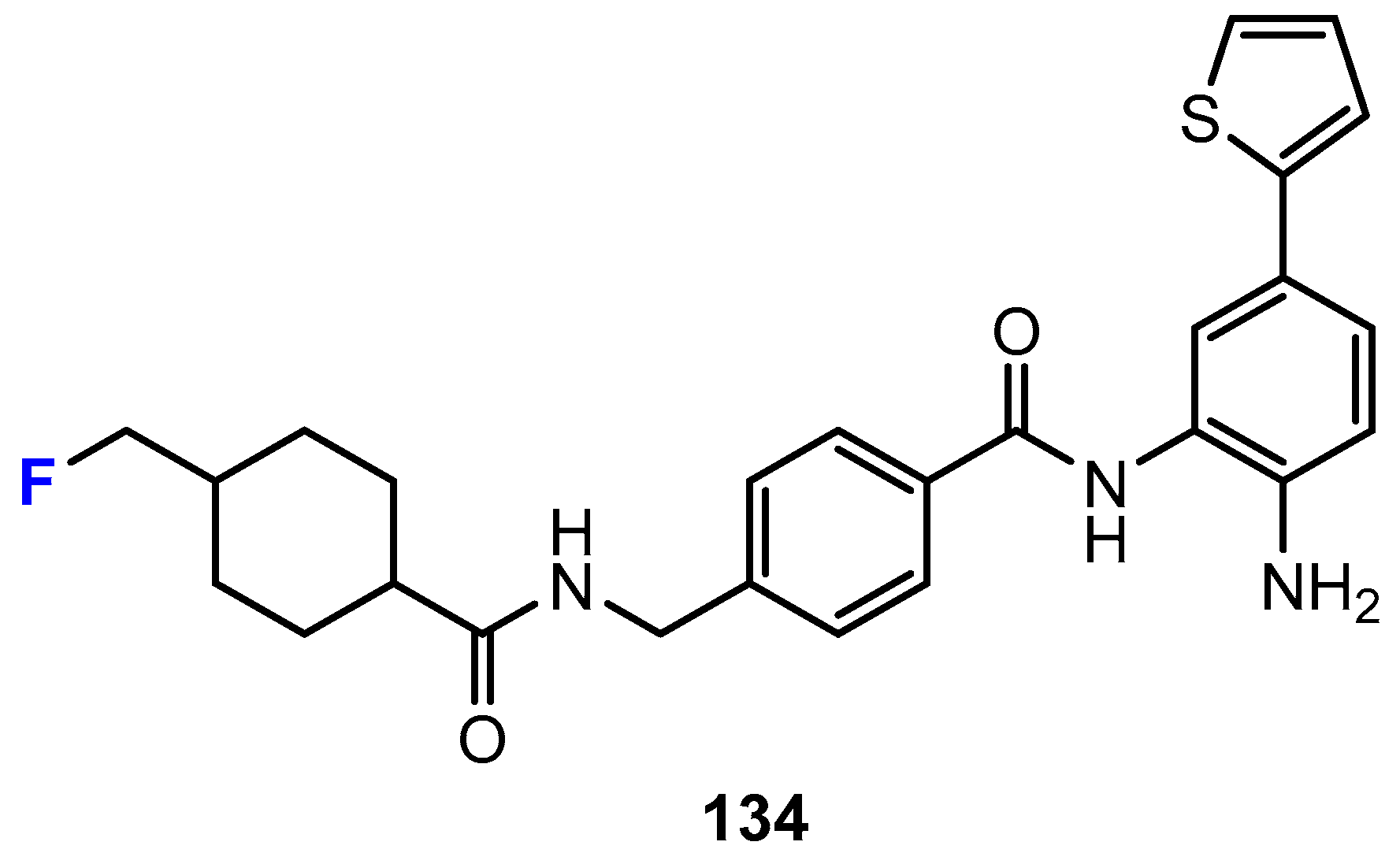
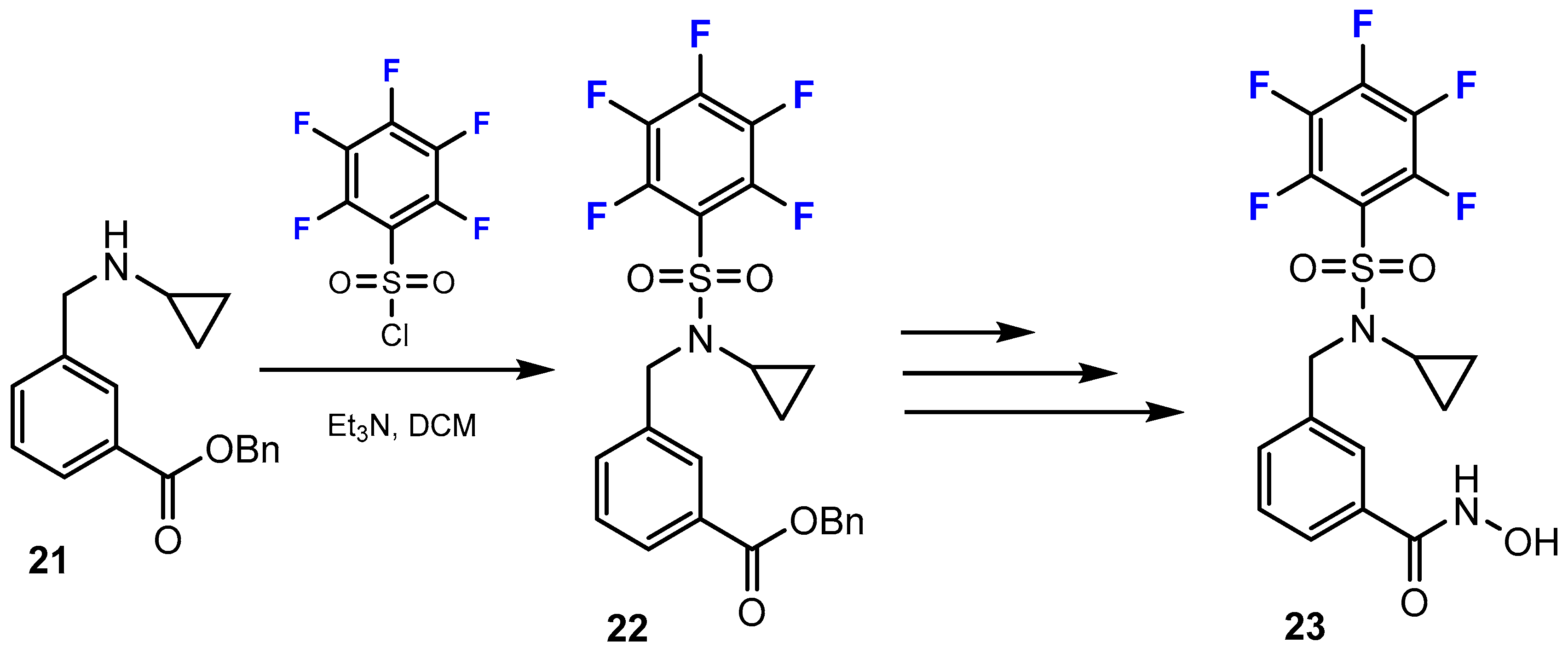
3.2. Selectivity
3.3. Labeling
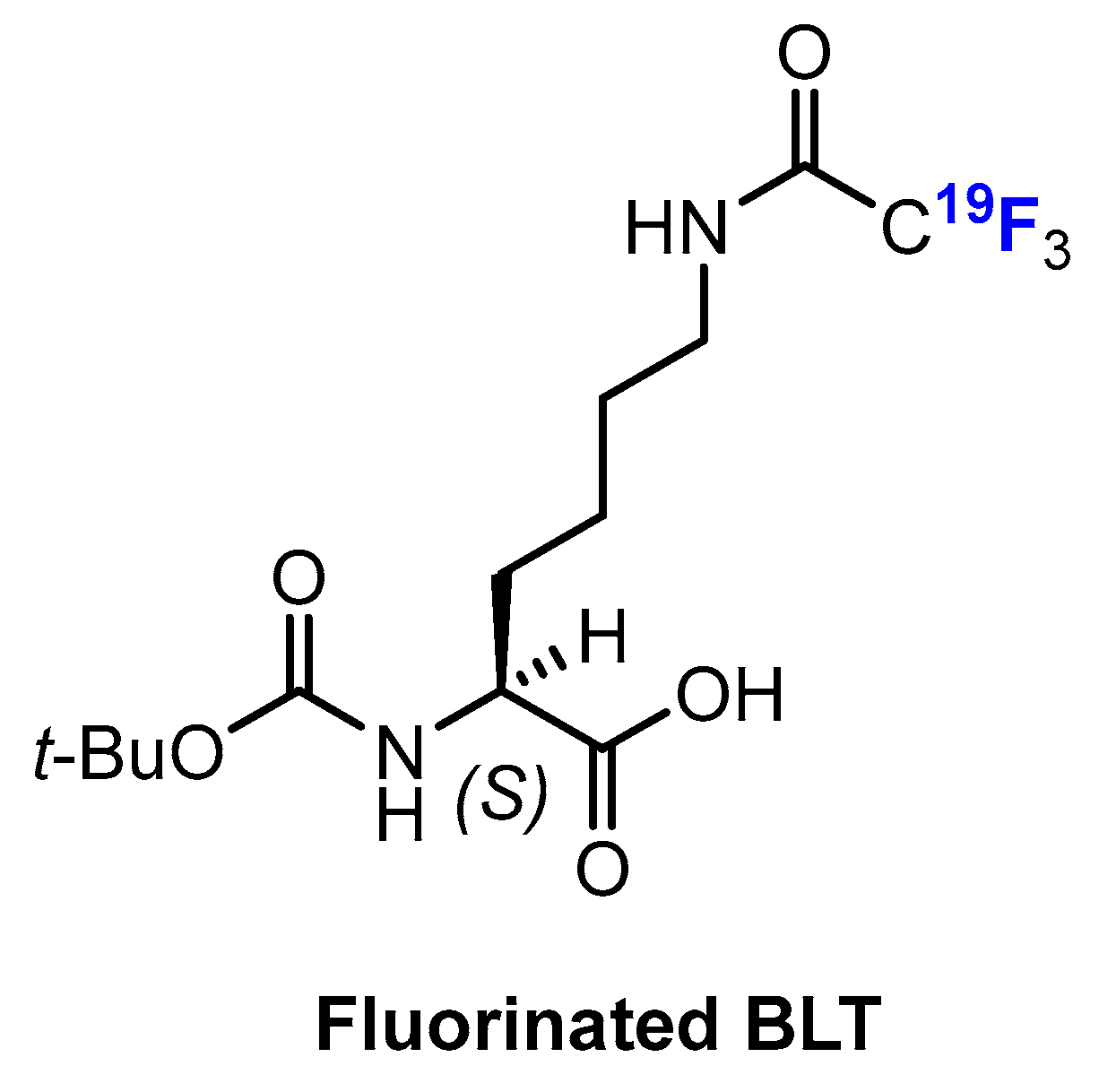
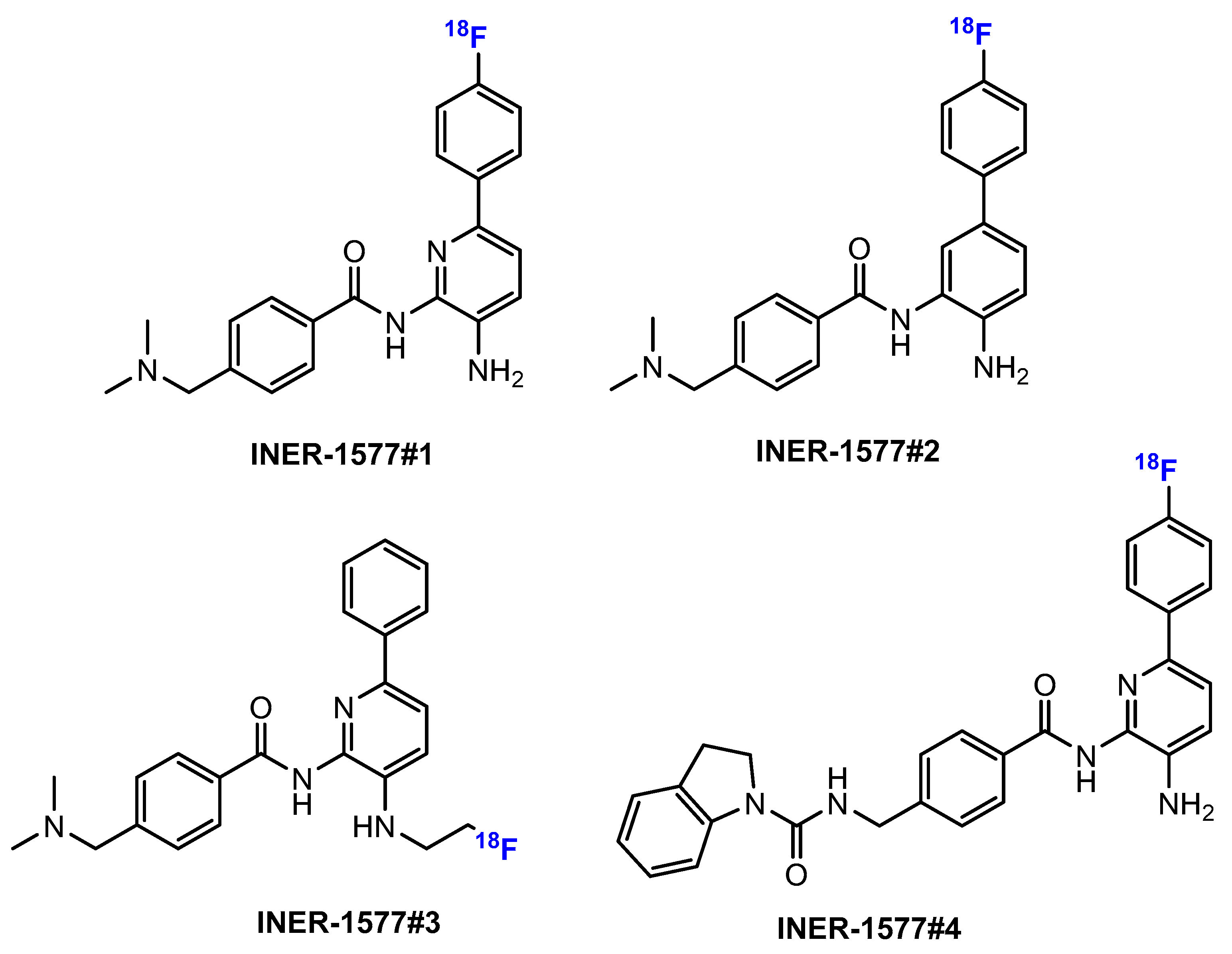
3.3.1. Labeling Using 19F
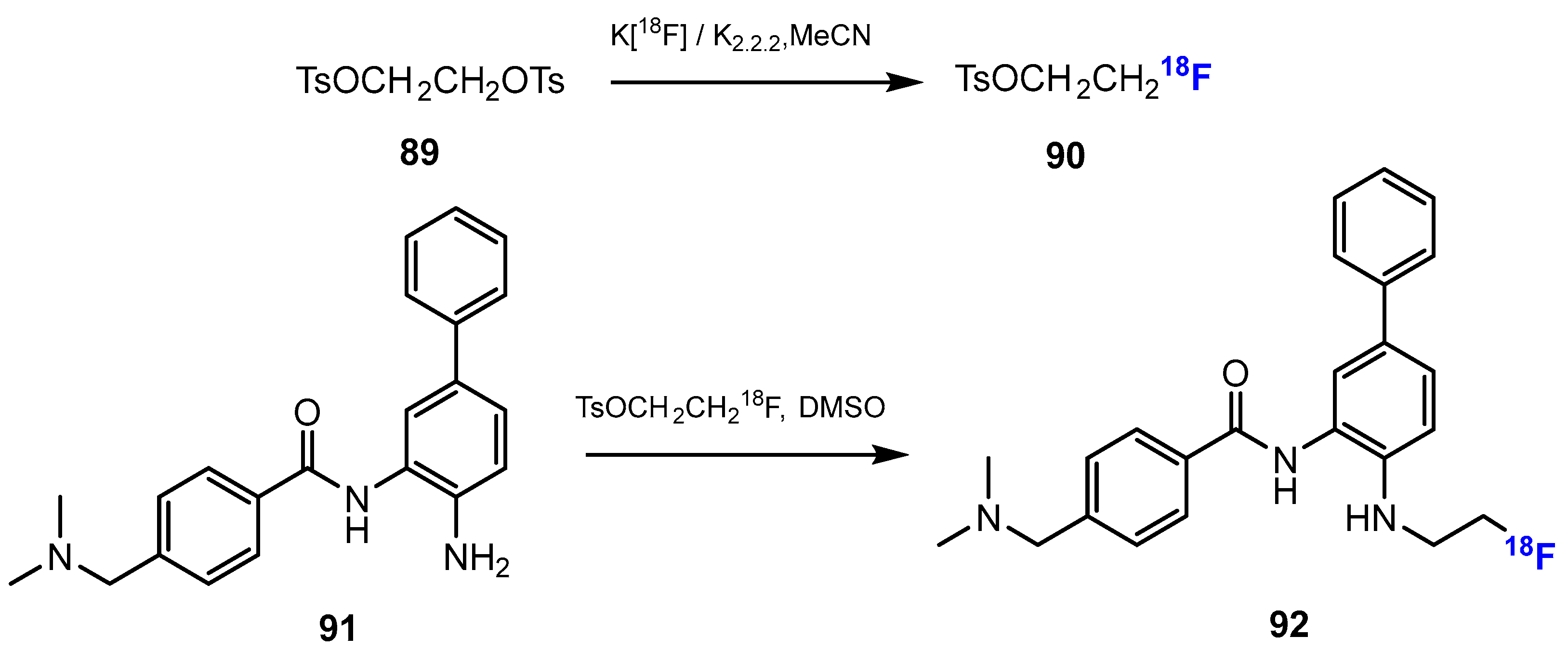
3.3.2. Labeling Using 18F
4. Issues of Fluorination
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tempany, C.M.C.; Jayender, J.; Kapur, T.; Bueno, R.; Golby, A.; Agar, N.; Jolesz, F.A. Multimodal imaging for improved diagnosis and treatment of cancers. Cancer 2015, 121, 817–827. [Google Scholar] [CrossRef]
- Bünger, S.; Laubert, T.; Roblick, U.J.; Habermann, J.K. Serum biomarkers for improved diagnostic of pancreatic cancer: A current overview. J. Cancer Res. Clin. Oncol. 2011, 137, 375–389. [Google Scholar] [CrossRef]
- Dalal, N.; Jalandra, R.; Sharma, M.; Prakash, H.; Makharia, G.K.; Solanki, P.R.; Singh, R.; Kumar, A. Omics technologies for improved diagnosis and treatment of colorectal cancer: Technical advancement and major perspectives. Biomed. Pharmacother. 2020, 131, 110648. [Google Scholar] [CrossRef]
- Steiner, N.; Schober, P.; Willenbacher, W.; Kircher, B.; Gunsilius, E.; Wolf, D.; Nachbaur, D. Autologous and Allogeneic Stem Cell Transplantation as Salvage Treatment Options for Relapsed/Refractory Multiple Myeloma: A Single-center Experience over 20 Years. Anticancer Res. 2022, 42, 5825–5832. [Google Scholar] [CrossRef]
- Steiner, N.; Göbel, G.; Mauser, L.; Mühlnikel, L.; Fischinger, M.; Künz, T.; Willenbacher, W.; Hetzenauer, G.; Rudzki, J.; Nussbaumer, W.; et al. Poor Mobilizers in Lymphoma but Not Myeloma Patients Had Significantly Poorer Progression-Free Survival after Autologous Stem Cell Transplantation: Results of a Large Retrospective, Single-Center Observational Study. Cancers 2023, 15, 608. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Baig, M.H.; Adil, M.; Khan, R.; Dhadi, S.; Ahmad, K.; Rabbani, G.; Bashir, T.; Imran, M.A.; Husain, F.M.; Lee, E.J.; et al. Enzyme targeting strategies for prevention and treatment of cancer: Implications for cancer therapy. Semin. Cancer Biol. 2019, 56, 1–11. [Google Scholar] [CrossRef]
- Li, G.; Tian, Y.; Zhu, W.-G. The Roles of Histone Deacetylases and Their Inhibitors in Cancer Therapy. Front. Cell Dev. Biol. 2020, 8, 576946. [Google Scholar] [CrossRef]
- Ruzic, D.; Djoković, N.; Srdić-Rajić, T.; Echeverria, C.; Nikolic, K.; Santibanez, J.F. Targeting Histone Deacetylases: Opportunities for Cancer Treatment and Chemoprevention. Pharmaceutics 2022, 14, 209. [Google Scholar] [CrossRef]
- Jin, Y.; Liu, T.; Luo, H.; Liu, Y.; Liu, D. Targeting Epigenetic Regulatory Enzymes for Cancer Therapeutics: Novel Small-Molecule Epidrug Development. Front. Oncol. 2022, 12, 848221. [Google Scholar] [CrossRef]
- Behnisch-Cornwell, S.; Grathwol, C.W.; Schulig, L.; Voigt, A.; Baecker, D.; Link, A.; Bednarski, P.J. Correlation Analysis of Protein Expression of 10 HDAC/Sirtuin Isoenzymes with Sensitivities of 23 Anticancer Drugs in 17 Cancer Cell Lines and Potentiation of Drug Activity by Co-Treatment with HDAC Inhibitors. Cancers 2022, 14, 187. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Huy, L.D.; Ngoc, H.B.; Ngoc, T.T.M.; Dung, D.T.M.; Park, E.J.; Song, I.K.; Kang, J.S.; Kwon, J.-H.; et al. Novel 4-Oxoquinazoline-Based N-Hydroxypropenamides as Histone Deacetylase Inhibitors: Design, Synthesis, and Biological Evaluation. ACS Omega 2021, 6, 4907–4920. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Hai, P.-T.; Thuan, N.T.; Huong, L.-T.-T.; Park, E.J.; Young Ji, A.; Soon Kang, J.; Phuong Dung, P.T.; Han, S.-B.; et al. Quinazolin-4(3H)-one-Based Hydroxamic Acids: Design, Synthesis and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodivers. 2019, 16, e1800502. [Google Scholar] [CrossRef]
- Minh, V.N.; Thanh, T.N.; Lien, T.H.; Anh, T.P.D.; Cuong, D.H.; Nam, H.N.; Hai, T.P.; Minh-Ngoc, L.; Le-Thi-Thu, H.; Chinh, V.L.; et al. Design, Synthesis and Biological Evaluation of Novel N-hydroxyheptanamides Incorporating 6-hydroxy-2-methylquinazolin-4(3H)-ones as Histone Deacetylase Inhibitors and Cytotoxic Agents. Anti-Cancer Agents Med. Chem. 2019, 19, 1543–1557. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Choi, J.E.; Kang, J.S.; Dung, P.T.P.; Han, S.-B.; Nam, N.-H. Quinazoline-Based Hydroxamic Acids: Design, Synthesis, and Evaluation of Histone Deacetylase Inhibitory Effects and Cytotoxicity. Chem. Biodivers. 2018, 15, e1800027. [Google Scholar] [CrossRef]
- Hieu, D.T.; Anh, D.T.; Tuan, N.M.; Hai, P.-T.; Huong, L.-T.-T.; Kim, J.; Kang, J.S.; Vu, T.K.; Dung, P.T.P.; Han, S.-B.; et al. Design, synthesis and evaluation of novel N-hydroxybenzamides/N-hydroxypropenamides incorporating quinazolin-4(3H)-ones as histone deacetylase inhibitors and antitumor agents. Bioorganic Chem. 2018, 76, 258–267. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Dung, D.T.M.; Dung, P.T.P.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Tung, T.T.; et al. Design, synthesis and evaluation of novel indirubin-based N-hydroxybenzamides, N-hydroxypropenamides and N-hydroxyheptanamides as histone deacetylase inhibitors and antitumor agents. Bioorganic Med. Chem. Lett. 2020, 30, 127537. [Google Scholar] [CrossRef]
- Anh, D.T.; Hai, P.-T.; Huong, L.-T.-T.; Park, E.J.; Jun, H.W.; Kang, J.S.; Kwon, J.-H.; Dung, D.T.M.; Anh, V.T.; Hue, V.T.M.; et al. Exploration of certain 1,3-oxazole- and 1,3-thiazole-based hydroxamic acids as histone deacetylase inhibitors and antitumor agents. Bioorganic Chem. 2020, 101, 103988. [Google Scholar] [CrossRef]
- Jampilek, J. Heterocycles in Medicinal Chemistry. Molecules 2019, 24, 3839. [Google Scholar] [CrossRef]
- Gomtsyan, A. Heterocycles in drugs and drug discovery. Chem. Heterocycl. Compd. 2012, 48, 7–10. [Google Scholar] [CrossRef]
- Gupta, P.S. Roles of Fluorine in Drug Design and Drug Action. Lett. Drug Des. Discov. 2019, 16, 1089–1109. [Google Scholar] [CrossRef]
- Grygorenko, O.O.; Melnykov, K.P.; Holovach, S.; Demchuk, O. Fluorinated Cycloalkyl Building Blocks for Drug Discovery. ChemMedChem 2022, 17, e202200365. [Google Scholar] [CrossRef]
- Richardson, P. Applications of fluorine to the construction of bioisosteric elements for the purposes of novel drug discovery. Expert Opin. Drug Discov. 2021, 16, 1261–1286. [Google Scholar] [CrossRef]
- Hevey, R. The Role of Fluorine in Glycomimetic Drug Design. Chem. Eur. J. 2021, 27, 2240–2253. [Google Scholar] [CrossRef]
- Páez-Franco, J.C.; Zermeño-Ortega, M.R.; de la O.-Contreras, C.M.; Canseco-González, D.; Parra-Unda, J.R.; Avila-Sorrosa, A.; Enríquez, R.G.; Germán-Acacio, J.M.; Morales-Morales, D. Relevance of Fluorinated Ligands to the Design of Metallodrugs for Their Potential Use in Cancer Treatment. Pharmaceutics 2022, 14, 402. [Google Scholar] [CrossRef]
- Pal, S.; Chandra, G.; Patel, S.; Singh, S. Fluorinated Nucleosides: Synthesis, Modulation in Conformation and Therapeutic Application. Chem. Rec. 2022, 22, e202100335. [Google Scholar] [CrossRef]
- Baecker, D.; Obermoser, V.; Kirchner, E.A.; Hupfauf, A.; Kircher, B.; Gust, R. Fluorination as tool to improve bioanalytical sensitivity and COX-2-selective antitumor activity of cobalt alkyne complexes. Dalton Trans. 2019, 48, 15856–15868. [Google Scholar] [CrossRef]
- Sagasser, J.; Ma, B.N.; Baecker, D.; Salcher, S.; Hermann, M.; Lamprecht, J.; Angerer, S.; Obexer, P.; Kircher, B.; Gust, R. A New Approach in Cancer Treatment: Discovery of Chlorido[N,N′-disalicylidene-1,2-phenylenediamine]iron(III) Complexes as Ferroptosis Inducers. J. Med. Chem. 2019, 62, 8053–8061. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef]
- Gong, W.; Wu, R.; Zhang, Y. Thiol versus hydroxamate as zinc binding group in HDAC inhibition: An ab initio QM/MM molecular dynamics study. J. Comput. Chem. 2015, 36, 2228–2235. [Google Scholar] [CrossRef]
- Bondarev, A.D.; Attwood, M.M.; Jonsson, J.; Chubarev, V.N.; Tarasov, V.V.; Schiöth, H.B. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br. J. Clin. Pharmacol. 2021, 87, 4577–4597. [Google Scholar] [CrossRef]
- Aboukhatwa, S.M.; Hanigan, T.W.; Taha, T.Y.; Neerasa, J.; Ranjan, R.; El-Bastawissy, E.E.; Elkersh, M.A.; El-Moselhy, T.F.; Frasor, J.; Mahmud, N.; et al. Structurally Diverse Histone Deacetylase Photoreactive Probes: Design, Synthesis, and Photolabeling Studies in Live Cells and Tissue. ChemMedChem 2019, 14, 1096–1107. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Li, J.; Hou, J.; Qu, Y.; Yu, C.; He, F.; Xu, W.; Wu, J. Design, synthesis, and preliminary bioactivity evaluation of N1-hydroxyterephthalamide derivatives with indole cap as novel histone deacetylase inhibitors. Chem. Biol. Drug Des. 2017, 89, 38–46. [Google Scholar] [CrossRef]
- Goehringer, N.; Peng, Y.; Nitzsche, B.; Biermann, H.; Pradhan, R.; Schobert, R.; Herling, M.; Höpfner, M.; Biersack, B. Improved Anticancer Activities of a New Pentafluorothio-Substituted Vorinostat-Type Histone Deacetylase Inhibitor. Pharmaceuticals 2021, 14, 1319. [Google Scholar] [CrossRef]
- Meyners, C.; Wolff, B.; Kleinschek, A.; Krämer, A.; Meyer-Almes, F.-J. Perfluorinated hydroxamic acids are potent and selective inhibitors of HDAC-like enzymes from Pseudomonas aeruginosa. Bioorganic Med. Chem. Lett. 2017, 27, 1508–1512. [Google Scholar] [CrossRef]
- Toutah, K.; Nawar, N.; Timonen, S.; Sorger, H.; Raouf, Y.S.; Bukhari, S.; von Jan, J.; Ianevski, A.; Gawel, J.M.; Olaoye, O.O.; et al. Development of HDAC Inhibitors Exhibiting Therapeutic Potential in T-Cell Prolymphocytic Leukemia. J. Med. Chem. 2021, 64, 8486–8509. [Google Scholar] [CrossRef]
- Walton, J.W.; Cross, J.M.; Riedel, T.; Dyson, P.J. Perfluorinated HDAC inhibitors as selective anticancer agents. Org. Biomol. Chem. 2017, 15, 9186–9190. [Google Scholar] [CrossRef]
- Vu, K.T.; Thanh, T.N.; Minh, V.N.; Linh, H.N.; Thao, T.P.N.; Nguyen, T.B.T.; Hien, T.D.; Chinh, V.L.; Duc, H.T.; Anh, D.L.; et al. Novel Conjugated Quinazolinone-Based Hydroxamic Acids: Design, Synthesis and Biological Evaluation. Med. Chem. 2021, 17, 732–749. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. Eur. J. Med. Chem. 2022, 229, 114058. [Google Scholar] [CrossRef]
- Erdeljac, N.; Bussmann, K.; Schöler, A.; Hansen, F.K.; Gilmour, R. Fluorinated Analogues of the Histone Deacetylase Inhibitor Vorinostat (Zolinza): Validation of a Chiral Hybrid Bioisostere, BITE. ACS Med. Chem. Lett. 2019, 10, 1336–1340. [Google Scholar] [CrossRef]
- Ariawan, A.D.; Mansour, F.; Richardson, N.; Bhadbhade, M.; Ho, J.; Hunter, L. The Effect of Vicinal Difluorination on the Conformation and Potency of Histone Deacetylase Inhibitors. Molecules 2021, 26, 3974. [Google Scholar] [CrossRef]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.-N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef]
- Hendricks, J.A.; Keliher, E.J.; Marinelli, B.; Reiner, T.; Weissleder, R.; Mazitschek, R. In Vivo PET Imaging of Histone Deacetylases by 18F-Suberoylanilide Hydroxamic Acid (18F-SAHA). J. Med. Chem. 2011, 54, 5576–5582. [Google Scholar] [CrossRef]
- Strebl, M.G.; Wang, C.; Schroeder, F.A.; Placzek, M.S.; Wey, H.-Y.; Van de Bittner, G.C.; Neelamegam, R.; Hooker, J.M. Development of a Fluorinated Class-I HDAC Radiotracer Reveals Key Chemical Determinants of Brain Penetrance. ACS Chem. Neurosci. 2016, 7, 528–533. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic Diphenylamide 106 Is a Slow, Tight-binding Inhibitor of Class I Histone Deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef]
- Jayathilaka, N.; Han, A.; Gaffney, K.J.; Dey, R.; Jarusiewicz, J.A.; Noridomi, K.; Philips, M.A.; Lei, X.; He, J.; Ye, J.; et al. Inhibition of the function of class IIa HDACs by blocking their interaction with MEF2. Nucleic Acids Res. 2012, 40, 5378–5388. [Google Scholar] [CrossRef]
- Bonomi, R.; Mukhopadhyay, U.; Shavrin, A.; Yeh, H.-H.; Majhi, A.; Dewage, S.W.; Najjar, A.; Lu, X.; Cisneros, G.A.; Tong, W.P.; et al. Novel Histone Deacetylase Class IIa Selective Substrate Radiotracers for PET Imaging of Epigenetic Regulation in the Brain. PLoS ONE 2015, 10, e0133512. [Google Scholar] [CrossRef]
- La, M.T.; Jeong, B.-H.; Kim, H.-K. Design and Synthesis of Novel N-(2-aminophenyl)benzamide Derivatives as Histone Deacetylase Inhibitors and Their Antitumor Activity Study. Bull. Korean Chem. Soc. 2021, 42, 740–743. [Google Scholar] [CrossRef]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.-H.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bülbül, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2022, 23, 369. [Google Scholar] [CrossRef]
- Schäker-Hübner, L.; Haschemi, R.; Büch, T.; Kraft, F.B.; Brumme, B.; Schöler, A.; Jenke, R.; Meiler, J.; Aigner, A.; Bendas, G.; et al. Balancing Histone Deacetylase (HDAC) Inhibition and Drug-likeness: Biological and Physicochemical Evaluation of Class I Selective HDAC Inhibitors. ChemMedChem 2022, 17, e202100755. [Google Scholar] [CrossRef]
- Li, M.H.; Shiue, C.Y.; Chang, H.-C.; Chu, H.H. Synthesis of [18F]benzamide ([18F]INER-1577) as Histone Deacetylase (HDACs) Imaging Agent. J. Nucl. Med. 2016, 57, 2664. [Google Scholar]
- Li, M.-H.; Chang, H.-C.; Feng, C.-F.; Yu, H.-W.; Shiue, C.-Y. Synthesis and Evaluation of <sup>18</sup>F-INER-1577-3 as a Central Nervous System (CNS) Histone Deacetylase Imaging Agent. Curr. Med. Imaging 2020, 16, 978–990. [Google Scholar] [CrossRef]
- Tavares, M.T.; Kozikowski, A.P.; Shen, S. Mercaptoacetamide: A promising zinc-binding group for the discovery of selective histone deacetylase 6 inhibitors. Eur. J. Med. Chem. 2021, 209, 112887. [Google Scholar] [CrossRef]
- Chuman, Y.; Ueyama, M.; Sano, S.; Wu, F.; Kiyota, Y.; Higashi, T.; Osada, S.; Sakaguchi, K. Effects of E/Z Configuration of Fluoroalkene-containing HDAC Inhibitors on Selectivity for HDAC Isoforms. Chem. Lett. 2013, 42, 833–835. [Google Scholar] [CrossRef]
- Wen, J.; Niu, Q.; Liu, J.; Bao, Y.; Yang, J.; Luan, S.; Fan, Y.; Liu, D.; Zhao, L. Novel thiol-based histone deacetylase inhibitors bearing 3-phenyl-1H-pyrazole-5-carboxamide scaffold as surface recognition motif: Design, synthesis and SAR study. Bioorganic Med. Chem. Lett. 2016, 26, 375–379. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Lu, Q.; Yang, Y.-T.; Chen, C.-S.; Davis, M.; Byrd, J.C.; Etherton, M.R.; Umar, A.; Chen, C.-S. Zn2+-Chelating Motif-Tethered Short-Chain Fatty Acids as a Novel Class of Histone Deacetylase Inhibitors. J. Med. Chem. 2004, 47, 467–474. [Google Scholar] [CrossRef]
- Lübke, M.; Jung, M.; Haufe, G. New histone deacetylase inhibitors based on 4-fluoro-2-amino acid esters: Synthesis and activity. J. Fluor. Chem. 2013, 152, 144–156. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Gao, D.; Yu, X.; Wang, J.; Lei, X. A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling. Eur. J. Med. Chem. 2019, 182, 111672. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Gao, D.; Yu, X.; Wang, J.; Lei, X. Corrigendum to “A fluorine scan on the Zn2+-binding thiolate side chain of HDAC inhibitor largazole: Synthesis, biological evaluation, and molecular modeling”. Eur. J. Med. Chem. 2019, 182, 111672, Erratum in Eur. J. Med. Chem. 2020, 198, 112340. [Google Scholar] [CrossRef]
- Hong, J.; Luesch, H. Largazole: From discovery to broad-spectrum therapy. Nat. Prod. Rep. 2012, 29, 449–456. [Google Scholar] [CrossRef]
- Schwaid, A.G.; Cornella-Taracido, I. Causes and Significance of Increased Compound Potency in Cellular or Physiological Contexts. J. Med. Chem. 2018, 61, 1767–1773. [Google Scholar] [CrossRef]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef]
- Dimova, D.; Stumpfe, D.; Bajorath, J. Specific chemical changes leading to consistent potency increases in structurally diverse active compounds. MedChemComm 2014, 5, 742–749. [Google Scholar] [CrossRef]
- Erdeljac, N.; Kehr, G.; Ahlqvist, M.; Knerr, L.; Gilmour, R. Exploring physicochemical space via a bioisostere of the trifluoromethyl and ethyl groups (BITE): Attenuating lipophilicity in fluorinated analogues of Gilenya® for multiple sclerosis. Chem. Commun. 2018, 54, 12002–12005. [Google Scholar] [CrossRef]
- Altomonte, S.; Zanda, M. Synthetic chemistry and biological activity of pentafluorosulphanyl (SF5) organic molecules. J. Fluor. Chem. 2012, 143, 57–93. [Google Scholar] [CrossRef]
- Salmi-Smail, C.; Fabre, A.; Dequiedt, F.; Restouin, A.; Castellano, R.; Garbit, S.; Roche, P.; Morelli, X.; Brunel, J.M.; Collette, Y. Modified Cap Group Suberoylanilide Hydroxamic Acid Histone Deacetylase Inhibitor Derivatives Reveal Improved Selective Antileukemic Activity. J. Med. Chem. 2010, 53, 3038–3047. [Google Scholar] [CrossRef]
- Luckhurst, C.A.; Breccia, P.; Stott, A.J.; Aziz, O.; Birch, H.L.; Bürli, R.W.; Hughes, S.J.; Jarvis, R.E.; Lamers, M.; Leonard, P.M.; et al. Potent, Selective, and CNS-Penetrant Tetrasubstituted Cyclopropane Class IIa Histone Deacetylase (HDAC) Inhibitors. ACS Med. Chem. Lett. 2016, 7, 34–39. [Google Scholar] [CrossRef]
- Yao, D.; Li, C.; Jiang, J.; Huang, J.; Wang, J.; He, Z.; Zhang, J. Design, synthesis and biological evaluation of novel HDAC inhibitors with improved pharmacokinetic profile in breast cancer. Eur. J. Med. Chem. 2020, 205, 112648. [Google Scholar] [CrossRef]
- Gawel, J.M.; Shouksmith, A.E.; Raouf, Y.S.; Nawar, N.; Toutah, K.; Bukhari, S.; Manaswiyoungkul, P.; Olaoye, O.O.; Israelian, J.; Radu, T.B.; et al. PTG-0861: A novel HDAC6-selective inhibitor as a therapeutic strategy in acute myeloid leukaemia. Eur. J. Med. Chem. 2020, 201, 112411. [Google Scholar] [CrossRef]
- Kim, S.L.; La, M.T.; Shin, M.W.; Kim, S.W.; Kim, H.K. A novel HDAC1 inhibitor, CBUD-1001, exerts anticancer effects by modulating the apoptosis and EMT of colorectal cancer cells. Int. J. Oncol. 2020, 57, 1027–1038. [Google Scholar] [CrossRef]
- Fischer, T.; Gazzola, S.; Riedl, R. Approaching Target Selectivity by De Novo Drug Design. Expert Opin. Drug Discov. 2019, 14, 791–803. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-generation of selective histone deacetylase inhibitors. RSC Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Shetty, M.G.; Pai, P.; Deaver, R.E.; Satyamoorthy, K.; Babitha, K.S. Histone deacetylase 2 selective inhibitors: A versatile therapeutic strategy as next generation drug target in cancer therapy. Pharmacol. Res. 2021, 170, 105695. [Google Scholar] [CrossRef]
- Suzuki-Karasaki, Y.; Suzuki-Karasaki, M.; Uchida, M.; Ochiai, T. Depolarization Controls TRAIL-Sensitization and Tumor-Selective Killing of Cancer Cells: Crosstalk with ROS. Front. Oncol. 2014, 4, 128. [Google Scholar] [CrossRef]
- Gryder, B.E.; Wu, L.; Woldemichael, G.M.; Pomella, S.; Quinn, T.R.; Park, P.M.C.; Cleveland, A.; Stanton, B.Z.; Song, Y.; Rota, R.; et al. Chemical genomics reveals histone deacetylases are required for core regulatory transcription. Nat. Commun. 2019, 10, 3004. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Baecker, D.; Sagasser, J.; Karaman, S.; Hörmann, A.A.; Gust, R. Development of methylated cobalt–alkyne complexes with selective cytotoxicity against COX-positive cancer cell lines. Arch. Pharm. 2022, 355, 2100408. [Google Scholar] [CrossRef]
- Chen, Y.; Lopez-Sanchez, M.; Savoy, D.N.; Billadeau, D.D.; Dow, G.S.; Kozikowski, A.P. A Series of Potent and Selective, Triazolylphenyl-Based Histone Deacetylases Inhibitors with Activity against Pancreatic Cancer Cells and Plasmodium falciparum. J. Med. Chem. 2008, 51, 3437–3448. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, X. Recent advances in small molecular modulators targeting histone deacetylase 6. Future Drug Discov. 2020, 2, FDD53. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef]
- Sandrone, G.; Cukier, C.D.; Zrubek, K.; Marchini, M.; Vergani, B.; Caprini, G.; Fossati, G.; Steinkühler, C.; Stevenazzi, A. Role of Fluorination in the Histone Deacetylase 6 (HDAC6) Selectivity of Benzohydroxamate-Based Inhibitors. ACS Med. Chem. Lett. 2021, 12, 1810–1817. [Google Scholar] [CrossRef]
- Reßing, N.; Schliehe-Diecks, J.; Watson, P.R.; Sönnichsen, M.; Cragin, A.D.; Schöler, A.; Yang, J.; Schäker-Hübner, L.; Borkhardt, A.; Christianson, D.W.; et al. Development of Fluorinated Peptoid-Based Histone Deacetylase (HDAC) Inhibitors for Therapy-Resistant Acute Leukemia. J. Med. Chem. 2022, 65, 15457–15472. [Google Scholar] [CrossRef]
- Keuler, T.; König, B.; Bückreiß, N.; Kraft, F.B.; König, P.; Schäker-Hübner, L.; Steinebach, C.; Bendas, G.; Gütschow, M.; Hansen, F.K. Development of the first non-hydroxamate selective HDAC6 degraders. Chem. Commun. 2022, 58, 11087–11090. [Google Scholar] [CrossRef]
- Sankaranarayanapillai, M.; Tong, W.P.; Maxwell, D.S.; Pal, A.; Pang, J.; Bornmann, W.G.; Gelovani, J.G.; Ronen, S.M. Detection of histone deacetylase inhibition by noninvasive magnetic resonance spectroscopy. Mol. Cancer Ther. 2006, 5, 1325–1334. [Google Scholar] [CrossRef]
- Sankaranarayanapillai, M.; Tong, W.P.; Yuan, Q.; Bankson, J.A.; Dafni, H.; Bornmann, W.G.; Soghomonyan, S.; Pal, A.; Ramirez, M.S.; Webb, D.; et al. Monitoring Histone Deacetylase Inhibition In Vivo: Noninvasive Magnetic Resonance Spectroscopy Method. Mol. Imaging 2008, 7, 7290.2008.0011. [Google Scholar] [CrossRef]
- Jacobson, O.; Kiesewetter, D.O.; Chen, X. Fluorine-18 Radiochemistry, Labeling Strategies and Synthetic Routes. Bioconjugate Chem. 2015, 26, 1–18. [Google Scholar] [CrossRef]
- Ahamed, M.; Vermeulen, K.; Schnekenburger, M.; Moltzau, R.L.; Levy, O.F.; Marton, J.; Froeyen, M.; Olberg, E.D.; Diederich, M.; Bormans, G. Synthesis, Enzyme Assays and Molecular Docking Studies of Fluorinated Bioisosteres of Santacruzamate A as Potential HDAC Tracers. Lett. Drug Des. Discov. 2017, 14, 787–797. [Google Scholar] [CrossRef]
- Li, M.-H.; Shiue, C.-Y.; Chang, H.-C.; Feng, C.-F.; Chang, C.-H. Synthesis and evaluation of F-18-INER-1577 as HDACi Imaging Agent in Mice. J. Nucl. Med. 2017, 58, 1101. [Google Scholar]
- Daśko, M.; de Pascual-Teresa, B.; Ortín, I.; Ramos, A. HDAC Inhibitors: Innovative Strategies for Their Design and Applications. Molecules 2022, 27, 715. [Google Scholar] [CrossRef]
- Chen, W.; Hsiao, Y.; Li, M.; Tsai, M.; Feng, C.; Chang, H.; Yu, H.; Shiue, C. Determination of Fragmentation Schemes and Metabolites of Fluorinated Histone Deacetylase Inhibitors for Use as Positron Emission Tomography Imaging Agents Using HPLC-MS/MS. Int. J. Anal. Mass Spectrom. Chromatogr. 2018, 6, 1–19. [Google Scholar] [CrossRef]
- Halder, R.; Ritter, T. 18F-Fluorination: Challenge and Opportunity for Organic Chemists. J. Org. Chem. 2021, 86, 13873–13884. [Google Scholar] [CrossRef]
- Xing, L.; Blakemore, D.C.; Narayanan, A.; Unwalla, R.; Lovering, F.; Denny, R.A.; Zhou, H.; Bunnage, M.E. Fluorine in Drug Design: A Case Study with Fluoroanisoles. ChemMedChem 2015, 10, 715–726. [Google Scholar] [CrossRef]
- Venturini, F.; Navarrini, W.; Famulari, A.; Sansotera, M.; Dardani, P.; Tortelli, V. Direct trifluoro-methoxylation of aromatics with perfluoro-methyl-hypofluorite. J. Fluor. Chem. 2012, 140, 43–48. [Google Scholar] [CrossRef]
- Landelle, G.; Panossian, A.; Leroux, F.R. Trifluoromethyl ethers and -thioethers as tools for medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef]
- Caron, S. Where Does the Fluorine Come From? A Review on the Challenges Associated with the Synthesis of Organofluorine Compounds. Org. Process Res. Dev. 2020, 24, 470–480. [Google Scholar] [CrossRef]
- Kirk, K.L. Fluorination in Medicinal Chemistry: Methods, Strategies, and Recent Developments. Org. Process Res. Dev. 2008, 12, 305–321. [Google Scholar] [CrossRef]
- Ricaud, P.; Lefèvre, F. Chapter 1: Fluorine in the Atmosphere. In Advances in Fluorine Science; Tressaud, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 1–32. [Google Scholar]
- Pan, Y. The Dark Side of Fluorine. ACS Med. Chem. Lett. 2019, 10, 1016–1019. [Google Scholar] [CrossRef]
- Wermers, R.A.; Cooper, K.; Razonable, R.R.; Deziel, P.J.; Whitford, G.M.; Kremers, W.K.; Moyer, T.P. Fluoride Excess and Periostitis in Transplant Patients Receiving Long-Term Voriconazole Therapy. Clin. Infect. Dis. 2011, 52, 604–611. [Google Scholar] [CrossRef]
- Goncharov, N.V.; Jenkins, R.O.; Radilov, A.S. Toxicology of fluoroacetate: A review, with possible directions for therapy research. J. Appl. Toxicol. 2006, 26, 148–161. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Alauddin, M.M. Positron emission tomography (PET) imaging with (18)F-based radiotracers. Am. J. Nucl. Med. Mol. Imaging 2012, 2, 55–76. [Google Scholar]









































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
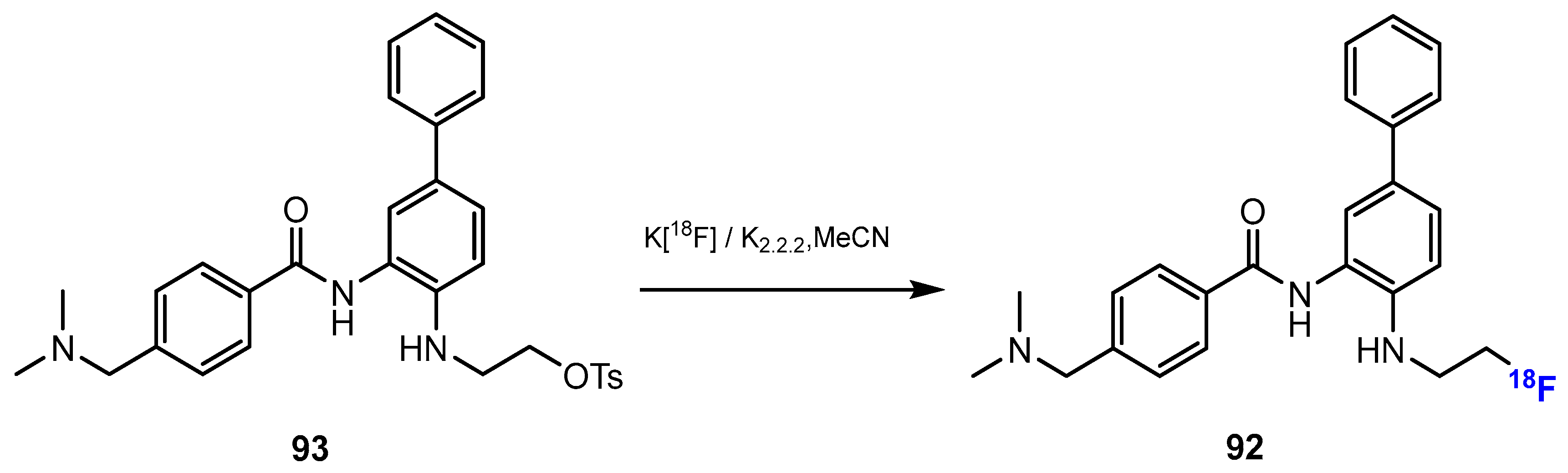{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tien Anh, D.; Hai Nam, N.; Kircher, B.; Baecker, D. The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors. Molecules 2023, 28, 1973. https://doi.org/10.3390/molecules28041973
Tien Anh D, Hai Nam N, Kircher B, Baecker D. The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors. Molecules. 2023; 28(4):1973. https://doi.org/10.3390/molecules28041973
Chicago/Turabian StyleTien Anh, Duong, Nguyen Hai Nam, Brigitte Kircher, and Daniel Baecker. 2023. "The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors" Molecules 28, no. 4: 1973. https://doi.org/10.3390/molecules28041973
APA StyleTien Anh, D., Hai Nam, N., Kircher, B., & Baecker, D. (2023). The Impact of Fluorination on the Design of Histone Deacetylase Inhibitors. Molecules, 28(4), 1973. https://doi.org/10.3390/molecules28041973