


A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples


Abstract
:1. Introduction
2. Results and Discussion
2.1. Selection of the Surrogate Analyte for Quantification of siRNA
2.2. Optimization of LC-MS/MS Conditions
2.3. Optimization of Hybridization Extraction Conditions
2.3.1. Capture Probes
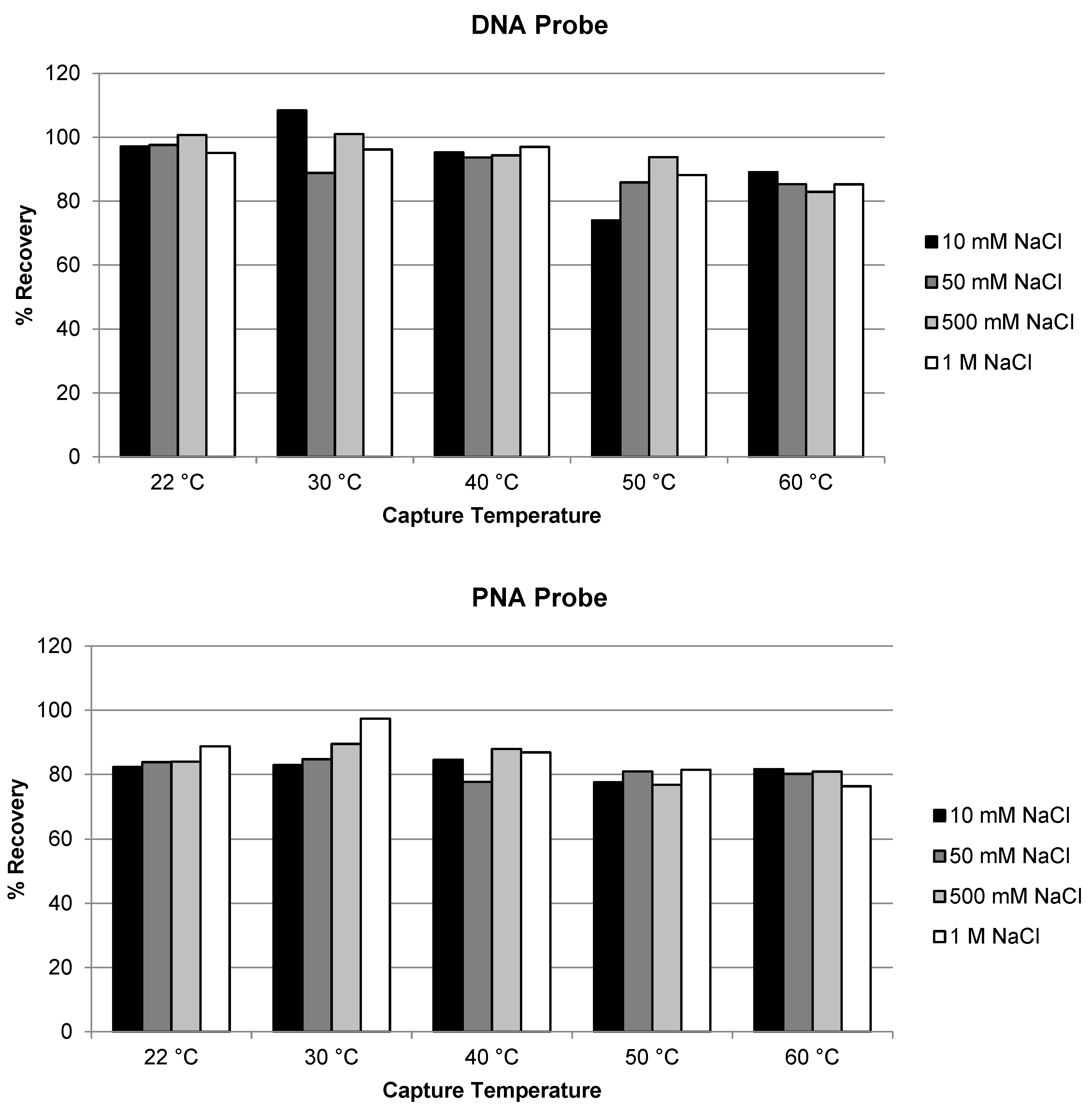
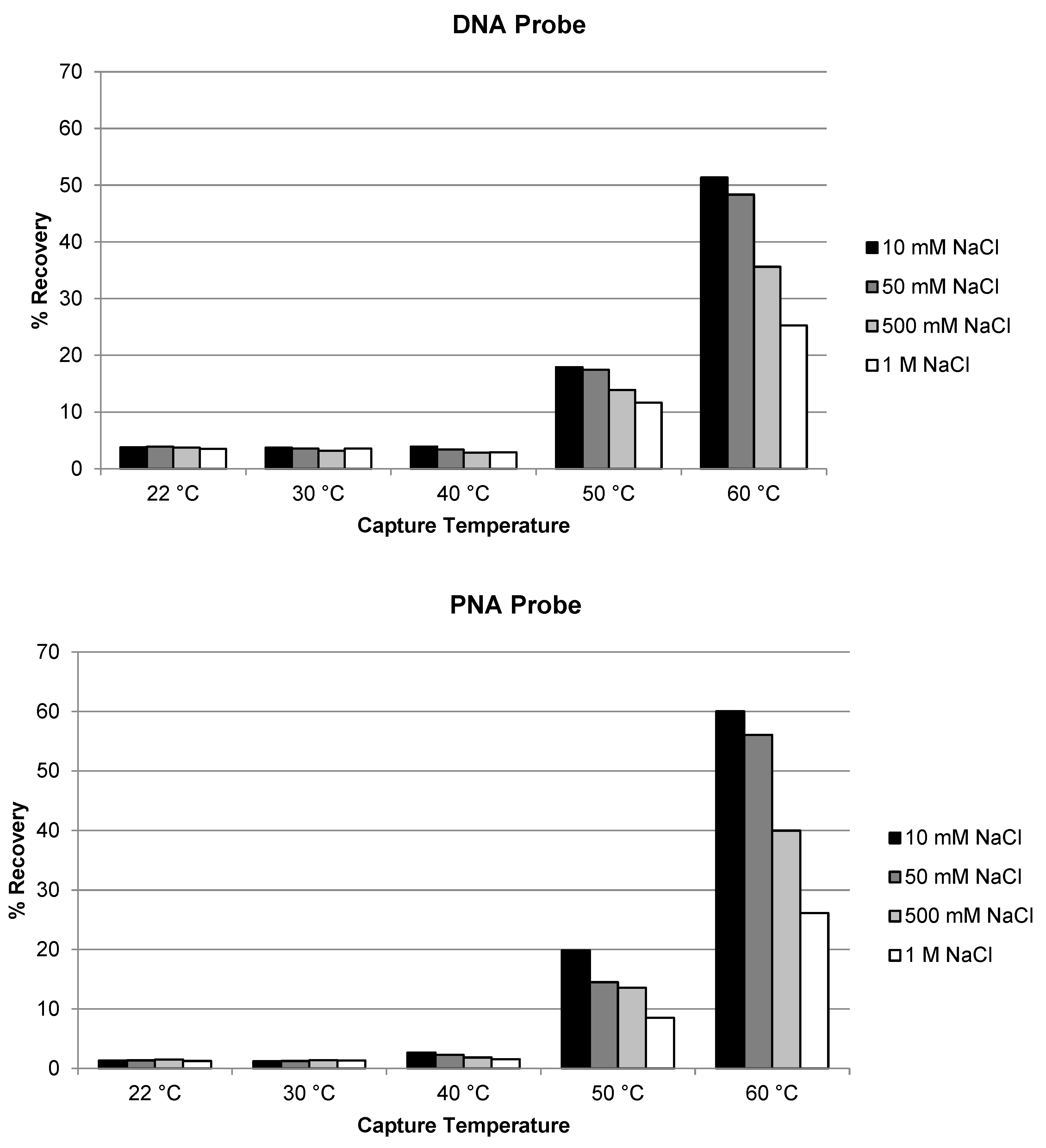
2.3.2. Temperature and Salt Concentration on the Extraction Recovery of Antisense Strand and siRNA Duplex
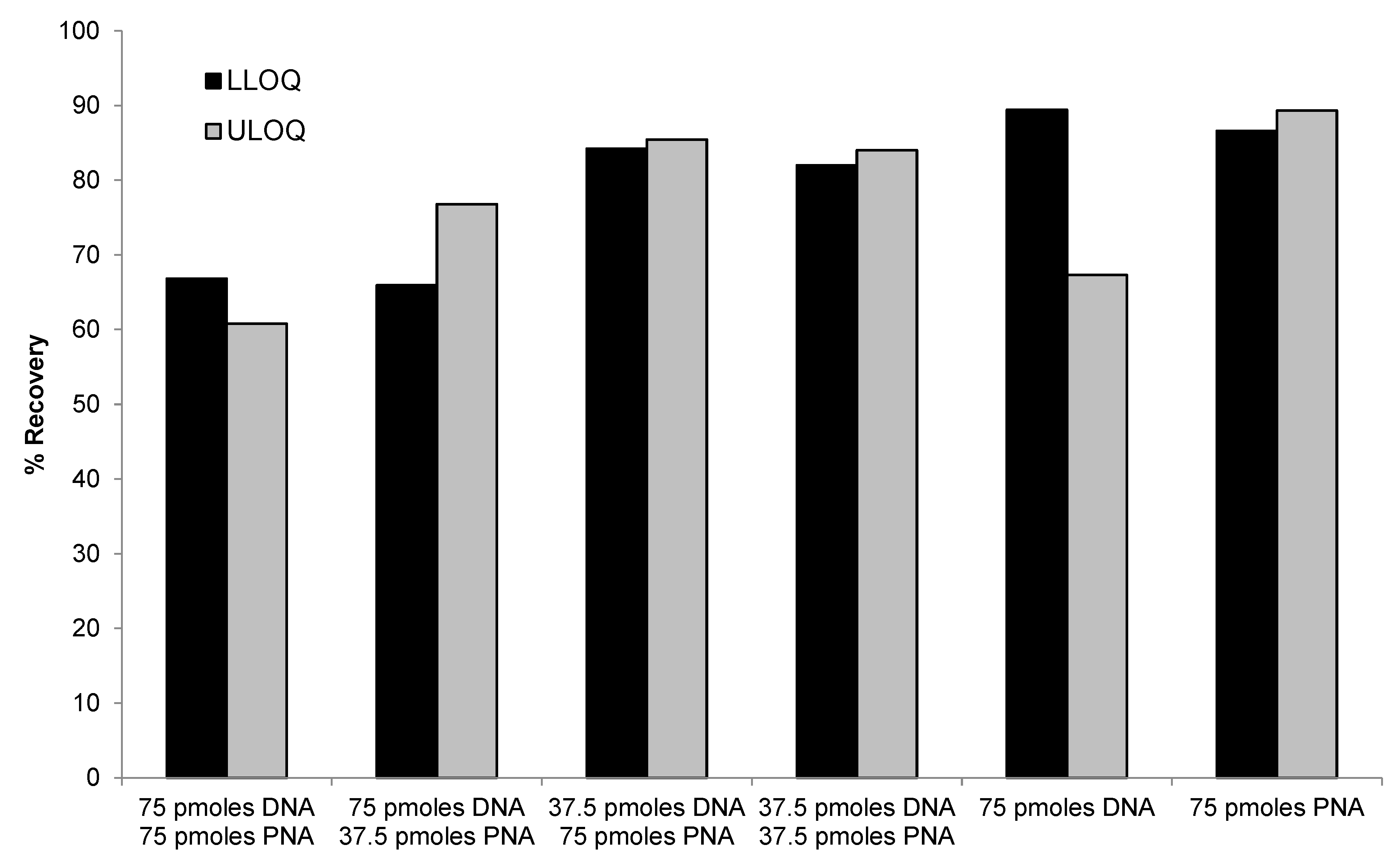
2.3.3. DNA Probe Concentration and Incubation Time on the Recovery
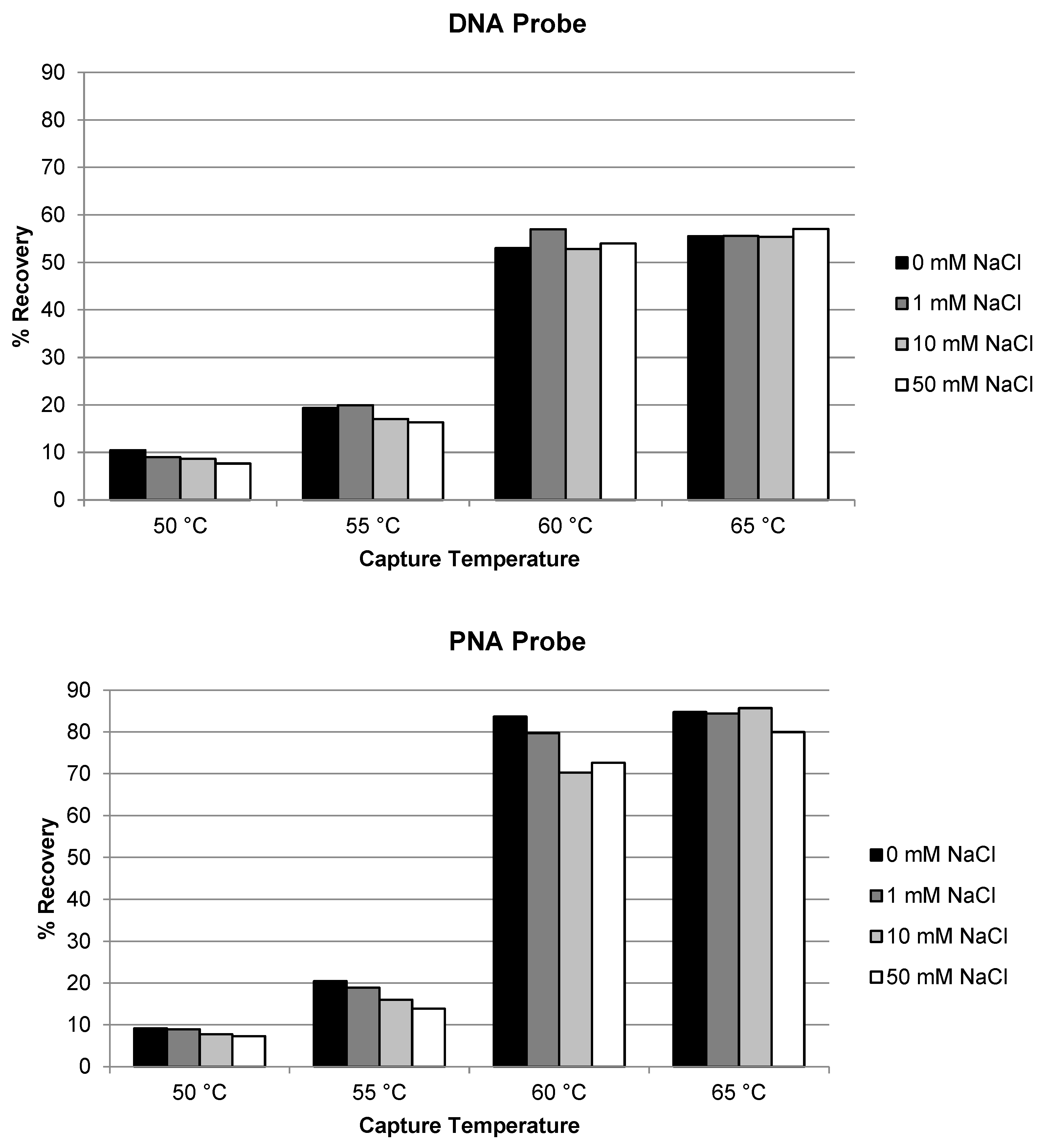
2.3.4. PNA Probe vs. DNA Probe
2.4. Equivalence of the Assay for the Analysis of siRNA-01 Duplex and Single-Stranded AS1
2.5. Qualification and Performance of the Hybridization LC-MS/MS Method
2.5.1. Accuracy, Precision, and Curve Linearity
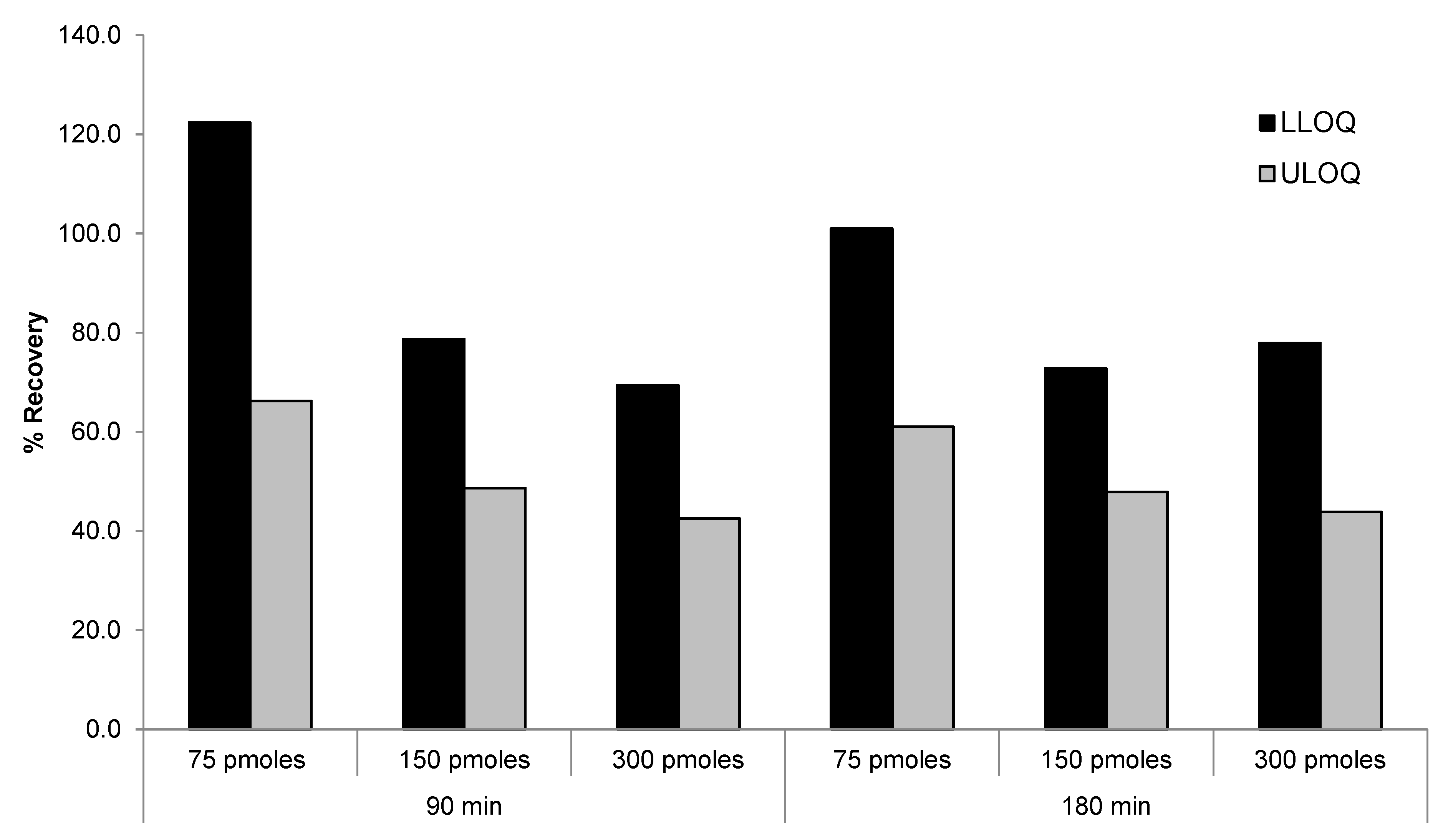
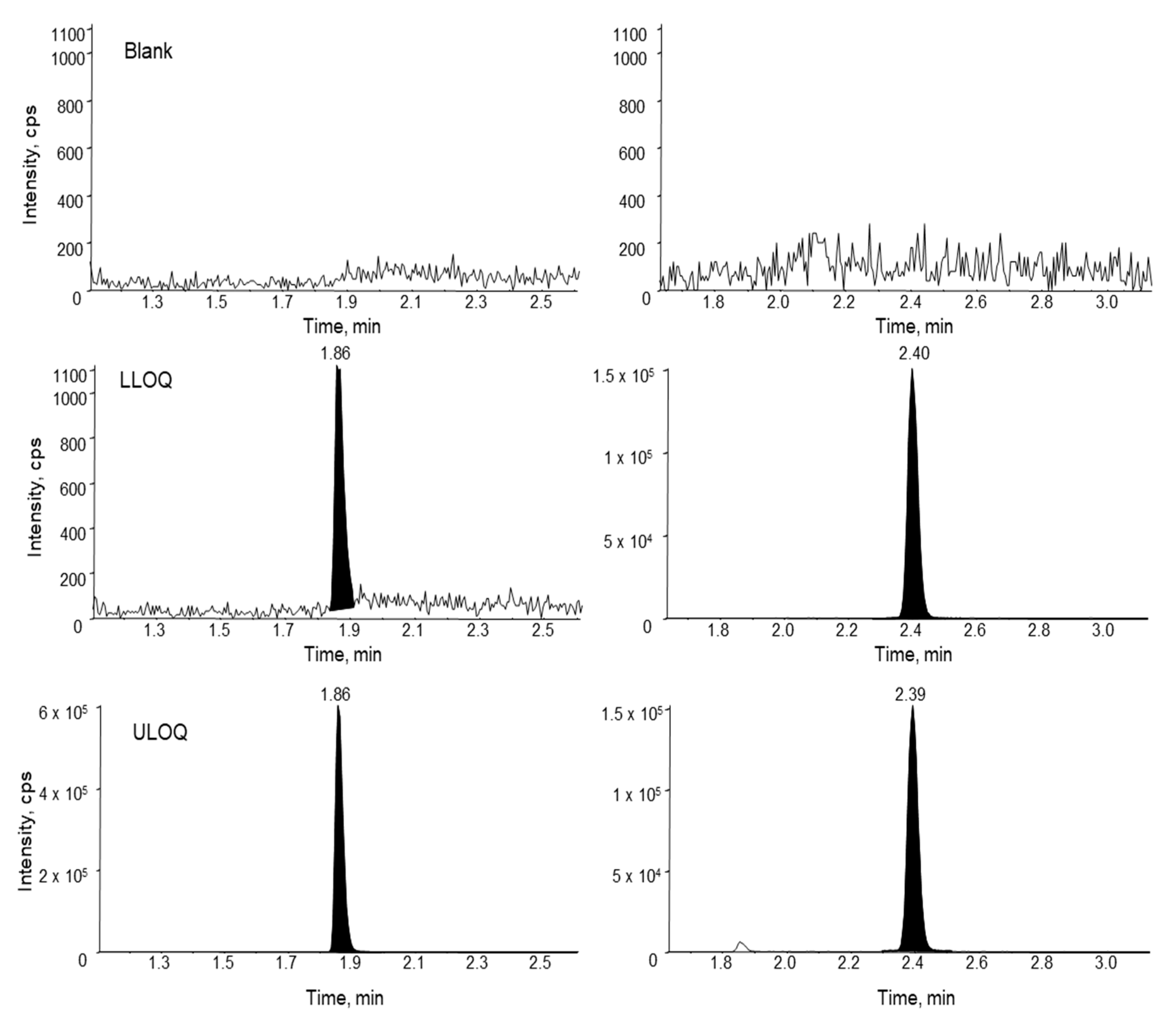
2.5.2. Specificity and Sensitivity
2.5.3. Extraction Recovery and Matrix Effect
2.5.4. Stability
2.5.5. Metabolite Interference
2.5.6. Dilution Integrity and Nonspecific Binding Test
2.6. CSF Assay
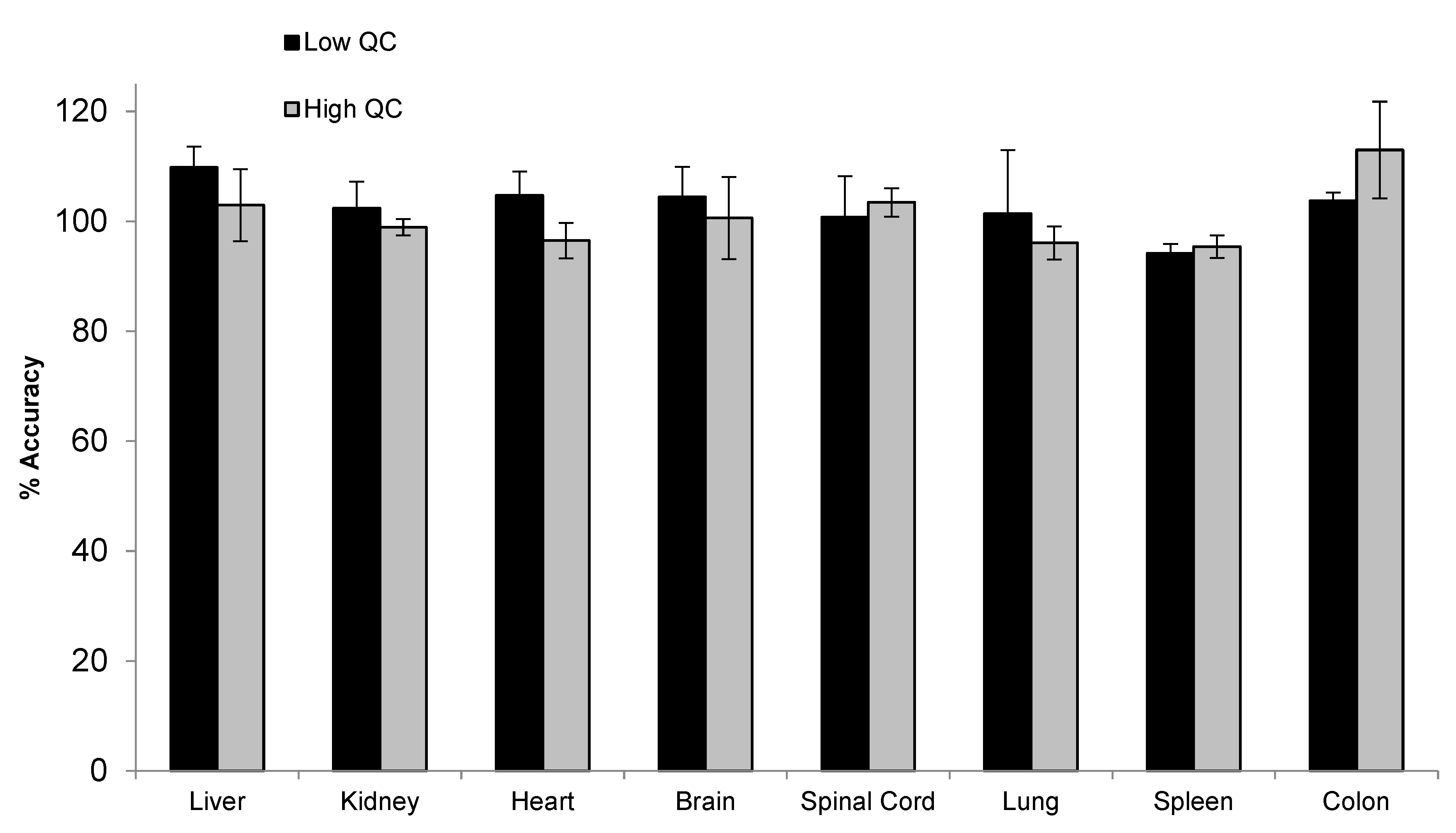
2.7. Tissue Assay
3. Materials and Methods
3.1. Chemicals, Reagents, Materials, and Instrumentation
3.2. Preparation of Calibration Standard and Quality Control Samples
3.3. Sample Preparation
3.4. UHPLC-MS/MS Conditions
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Moumné, L.; Marie, A.-C.; Crouvezier, N. Oligonucleotide Therapeutics: From Discovery and Development to Patentability. Pharmaceutics 2022, 14, 260. [Google Scholar] [CrossRef]
- Weidolf, L.; Björkbom, A.; Dahlén, A.; Elebring, M.; Gennemark, P.; Hölttä, M.; Janzén, D.; Li, X.; Andersson, S. Distribution and biotransformation of therapeutic antisense oligonucleotides and conjugates. Drug Discov. Today 2021, 26, 2244–2258. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, G.A.; Oldfield, P.R. Bioanalysis of siRNA and oligonucleotide therapeutics in biological fluids and tissues. Bioanalysis 2009, 1, 595–609. [Google Scholar] [CrossRef]
- Norris, D.A.; Post, N.; Yu, R.Z.; Greenlee, S.; Wang, Y. Bioanalysis considerations on the pharmacokinetic evaluation of antisense therapeutics. Bioanalysis 2019, 11, 1909–1912. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ji, C. Advances in quantitative bioanalysis of oligonucleotide biomarkers and therapeutics. Bioanalysis 2015, 8, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Oligonucleotide bioanalysis: Sensitivity versus specificity. Bioanalysis 2011, 3, 1299–1303. [Google Scholar] [CrossRef]
- Shin, M.; Meda Krishnamurthy, P.; Devi, G.; Watts, J.K. Quantification of Antisense Oligonucleotides by Splint Ligation and Quantitative Polymerase Chain Reaction. Nucleic Acid Ther. 2021, 32, 66–73. [Google Scholar] [CrossRef]
- Wang, L.; Meng, M.; Reuschel, S. Regulated bioanalysis of oligonucleotide therapeutics and biomarkers: qPCR versus chromatographic assays. Bioanalysis 2013, 5, 2747–2751. [Google Scholar] [CrossRef]
- Lin, Z.J.; Li, W.; Dai, G. Application of LC–MS for quantitative analysis and metabolite identification of therapeutic oligonucleotides. J. Pharm. Biomed. Anal. 2007, 44, 330–341. [Google Scholar] [CrossRef]
- van Dongen, W.D.; Niessen, W.M.A. Bioanalytical LC–MS of therapeutic oligonucleotides. Bioanalysis 2011, 3, 541–564. [Google Scholar] [CrossRef]
- Basiri, B.; Bartlett, M.G. LC–MS of oligonucleotides: Applications in biomedical research. Bioanalysis 2014, 6, 1525–1542. [Google Scholar] [CrossRef]
- Sutton, J.M.; Kim, J.; El Zahar, N.M.; Bartlett, M.G. Bioanalysis and Biotransformation of Oligonucleotide Therapeutics by Liquid Chromatography-Mass Spectrometry. Mass Spectrom. Rev. 2021, 40, 334–358. [Google Scholar] [CrossRef] [PubMed]
- Dillen, L.; Sips, L.; Greway, T.; Verhaeghe, T. Quantitative analysis of imetelstat in plasma with LC–MS/MS using solid-phase or hybridization extraction. Bioanalysis 2017, 9, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Sips, L.; Ediage, E.N.; Ingelse, B.; Verhaeghe, T.; Dillen, L. LC–MS quantification of oligonucleotides in biological matrices with SPE or hybridization extraction. Bioanalysis 2019, 11, 1941–1954. [Google Scholar] [CrossRef]
- Basiri, B.; Sutton, J.M.; Hooshfar, S.; Byrnes, C.C.; Murph, M.M.; Bartlett, M.G. Direct identification of microribonucleic acid miR-451 from plasma using liquid chromatography mass spectrometry. J. Chromatogr. A 2019, 1584, 97–105. [Google Scholar] [CrossRef]
- Li, P.; Gong, Y.; Kim, J.; Liu, X.; Gilbert, J.; Kerns, H.M.; Groth, R.; Rooney, M. Hybridization Liquid Chromatography–Tandem Mass Spectrometry: An Alternative Bioanalytical Method for Antisense Oligonucleotide Quantitation in Plasma and Tissue Samples. Anal. Chem. 2020, 92, 10548–10559. [Google Scholar] [CrossRef]
- Guimaraes, G.; Yuan, L.; Li, P. Antisense Oligonucleotide In Vitro Protein Binding Determination in Plasma, Brain, and Cerebral Spinal Fluid Using Hybridization LC-MS/MS. Drug Metab. Dispos. 2022, 50, 268–276. [Google Scholar] [CrossRef]
- Studzińska, S.; Skoczylas, M.; Bocian, S.; Dembska, A.; Buszewski, B. Attachment of hybridizable oligonucleotides to a silica support and its application for selective extraction of unmodified and antisense oligonucleotides from serum samples. RSC Adv. 2020, 10, 16221–16230. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Yuan, L. Microflow LC–MS/MS to improve sensitivity for antisense oligonucleotides bioanalysis: Critical role of sample cleanness. Bioanalysis 2022, 14, 1365–1376. [Google Scholar] [CrossRef]
- Li, P.; Dupuis, J.-F.; Vrionis, V.; Mekhssian, K.; Magee, T.; Yuan, L. Validation and application of hybridization liquid chromatography-tandem mass spectrometry methods for quantitative bioanalysis of antisense oligonucleotides. Bioanalysis 2022, 14, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef] [PubMed]
- McDougall, R.; Ramsden, D.; Agarwal, S.; Agarwal, S.; Aluri, K.; Arciprete, M.; Brown, C.R.; Castellanos-Rizaldos, E.; Charisse, K.; Chong, S.; et al. The Nonclinical Disposition and PK/PD Properties of GalNAc-conjugated siRNA Are Highly Predictable and Build Confidence in Translation to Man. Drug Metab. Dispos. 2022, 50, 781–797. [Google Scholar] [CrossRef]
- Apffel, A.; Chakel, J.A.; Fischer, S.; Lichtenwalter, K.; Hancock, W.S. Analysis of Oligonucleotides by HPLC−Electrospray Ionization Mass Spectrometry. Anal. Chem. 1997, 69, 1320–1325. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PNA Probe 75 pmols/Sample | LLOQ QC 2.00 ng/mL | Low QC 6.00 ng/mL | Mid QC 500 ng/mL | High QC 750 ng/mL |
|---|---|---|---|---|
| Measured Conc. | 2.14 | 5.78 | 502.18 | 758.45 |
| 2.03 | 6.32 | 417.41 | 726.41 | |
| 1.79 | 6.21 | 446.34 | 726.46 | |
| 1.95 | 6.00 | 472.78 | 788.82 | |
| Mean | 1.98 | 6.08 | 459.68 | 750.03 |
| S.D. | 0.15 | 0.24 | 36.25 | 29.94 |
| N | 4 | 4 | 4 | 4 |
| % CV | 7.5 | 4.0 | 7.9 | 4.0 |
| % Nominal | 98.7 | 101.3 | 91.9 | 100.0 |
| PNA and DNA Probes 37.5 pmols Each/Sample | LLOQ QC 2.00 ng/mL | Low QC 6.00 ng/mL | Mid QC 500 ng/mL | High QC 750 ng/mL |
| Measured Conc. | 1.98 | 6.25 | 526.62 | 751.12 |
| 2.09 | 6.07 | 457.77 | 745.37 | |
| 2.24 | 6.04 | 533.94 | 752.48 | |
| 2.01 | 6.42 | 456.61 | 742.90 | |
| Mean | 2.08 | 6.19 | 493.73 | 747.97 |
| S.D. | 0.12 | 0.18 | 42.31 | 4.57 |
| N | 4 | 4 | 4 | 4 |
| % CV | 5.6 | 2.8 | 8.6 | 0.6 |
| % Nominal | 103.9 | 103.2 | 98.7 | 99.7 |
| siRNA-01 Low QC 6.00 ng/mL | AS1 Low QC 6.00 ng/mL | siRNA-01 + AS1 Low QC 3.00 + 3.00 ng/mL | siRNA-01 High QC 750 ng/mL | AS1 High QC 750 ng/mL | siRNA-01 + AS1 High QC 375 + 375 ng/mL | |
|---|---|---|---|---|---|---|
| 6.46 | 6.27 | 5.80 | 696.41 | 752.41 | 733.57 | |
| 6.55 | 5.99 | 5.99 | 677.39 | 813.93 | 705.49 | |
| 6.92 | 6.50 | 5.10 | 782.65 | 761.66 | 680.36 | |
| 5.62 | 5.53 | 5.41 | 799.26 | 759.01 | 822.64 | |
| Mean | 6.39 | 6.07 | 5.58 | 738.93 | 771.75 | 735.52 |
| S.D. | 0.55 | 0.42 | 0.40 | 60.95 | 28.39 | 62.02 |
| n | 4 | 4 | 4 | 4 | 4 | 4 |
| % C.V. | 8.6 | 6.9 | 7.2 | 8.2 | 3.7 | 8.4 |
| % Nominal | 106.5 | 101.2 | 92.9 | 98.5 | 102.9 | 98.1 |
| Run 1 | LLOQ QC 2.00 ng/mL | Low QC 6.00 ng/mL | Mid QC 500 ng/mL | High QC 750 ng/mL |
|---|---|---|---|---|
| Measured Conc. | 1.98 | 5.87 | 527.11 | 681.34 |
| 1.94 | 6.50 | 441.75 | 674.75 | |
| 2.05 | 5.91 | 464.04 | 756.63 | |
| 2.29 | 5.56 | 469.18 | 753.45 | |
| Mean | 2.06 | 5.96 | 475.52 | 716.54 |
| S.D. | 0.16 | 0.40 | 36.40 | 44.55 |
| n | 4 | 4 | 4 | 4 |
| % CV | 7.7 | 6.6 | 7.7 | 6.2 |
| % Nominal | 103.1 | 99.3 | 95.1 | 95.5 |
| Run 2 | LLOQ QC 2.00 ng/mL | Low QC 6.00 ng/mL | Mid QC 500 ng/mL | High QC 750 ng/mL |
| Measured Conc. | 1.78 | 5.83 | 493.31 | 746.88 |
| 1.82 | 5.06 | 460.57 | 754.49 | |
| 1.72 | 5.12 | 471.69 | 721.32 | |
| 1.75 | 6.09 | 492.21 | 702.19 | |
| Mean | 1.77 | 5.53 | 479.45 | 731.22 |
| S.D. | 0.04 | 0.51 | 16.04 | 24.00 |
| n | 4 | 4 | 4 | 4 |
| % CV | 2.4 | 9.3 | 3.3 | 3.3 |
| % Nominal | 88.3 | 92.1 | 95.9 | 97.5 |
| Low QC (6.00 ng/mL) | High QC (750 ng/mL) | |||||
|---|---|---|---|---|---|---|
| Analyte Matrix Factor | IS Matrix Factor | IS-Normalized Matrix Factor | Analyte Matrix Factor | IS Matrix Factor | IS-Normalized Matrix Factor | |
| Lot 1 | 0.92 | 0.97 | 0.95 | 1.00 | 1.01 | 0.99 |
| 0.95 | 1.02 | 0.93 | 0.98 | 0.98 | 1.00 | |
| 1.01 | 1.01 | 1.00 | 0.94 | 0.95 | 0.99 | |
| Lot 2 | 0.92 | 1.00 | 0.92 | 1.04 | 1.02 | 1.02 |
| 0.97 | 1.00 | 0.97 | 0.95 | 0.92 | 1.03 | |
| 0.99 | 0.96 | 1.03 | 1.08 | 1.00 | 1.08 | |
| Lot 3 | 1.02 | 1.01 | 1.01 | 0.99 | 0.98 | 1.01 |
| 0.98 | 1.03 | 0.95 | 0.98 | 1.00 | 0.98 | |
| 0.94 | 1.01 | 0.93 | 1.04 | 0.96 | 1.08 | |
| Mean | 0.97 | 1.02 | ||||
| S.D. | 0.040 | 0.040 | ||||
| n | 9 | 9 | ||||
| % C.V. | 4.1 | 3.9 | ||||
| Name | MW (kDa) | Sequence Length | Chemistry |
|---|---|---|---|
| AS1 | 6.9 | 21 | Mixed backbone with 2′-OMe and 2′-F modification |
| ASO-002 | 7.9 | 20 | Uniform MOE with PS backbone |
| Time (min) | Module | Events | Parameters |
|---|---|---|---|
| 0 | Pumps | Pump B Conc. | 0 |
| 0 | Pumps | A/B Total Flow | 0.400 |
| 0 | Pumps | Pump C Flow | 0.100 |
| 0.3 | Pumps | Pump B Conc. | 0 |
| 3.0 | Pumps | Pump B Conc. | 25 |
| 3.0 | Pumps | A/B Total Flow | 0.400 |
| 3.0 | Pumps | Pump C Flow | 0.100 |
| 3.1 | Pumps | Pump C Flow | 0 |
| 3.2 | Pumps | Pump B Conc. | 100 |
| 3.2 | Pumps | A/B Total Flow | 0.600 |
| 3.6 | Pumps | Pump B Conc. | 100 |
| 3.7 | Pumps | Pump B Conc. | 25 |
| 4.1 | Pumps | Pump B Conc. | 25 |
| 4.4 | Pumps | Pump B Conc. | 100 |
| 4.9 | Pumps | A/B Total Flow | 0.600 |
| 4.9 | Pumps | Pump B Conc. | 100 |
| 5.0 | Pumps | Pump C Flow | 0 |
| 5.1 | Pumps | Pump B Conc. | 0 |
| 5.1 | Pumps | A/B Total Flow | 0.400 |
| 5.1 | Pumps | Pump C Flow | 0.100 |
| 7.0 | Controller | Stop |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Dupuis, J.-F.; Mekhssian, K. A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples. Molecules 2023, 28, 1618. https://doi.org/10.3390/molecules28041618
Yuan L, Dupuis J-F, Mekhssian K. A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples. Molecules. 2023; 28(4):1618. https://doi.org/10.3390/molecules28041618
Chicago/Turabian StyleYuan, Long, Jean-François Dupuis, and Kevork Mekhssian. 2023. "A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples" Molecules 28, no. 4: 1618. https://doi.org/10.3390/molecules28041618
APA StyleYuan, L., Dupuis, J.-F., & Mekhssian, K. (2023). A Novel Hybridization LC-MS/MS Methodology for Quantification of siRNA in Plasma, CSF and Tissue Samples. Molecules, 28(4), 1618. https://doi.org/10.3390/molecules28041618