
Molecular Characteristics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis

 , ,
, ,
Abstract
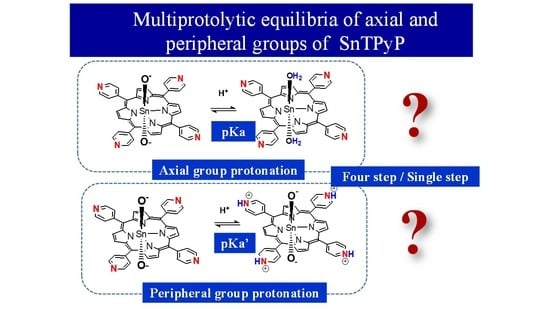
1. Introduction
2. Results and Discussion
2.1. Molecular Catalyst Sensitized System (MCSS)
2.2. Acid-Base Equilibria of Axial Ligands Water of SnTPyP in the Ground State
2.3. Acid-Base Equilibria in the Excited State
2.4. One-Electron Oxidation of SnTPyP
∆E([SnTPyP(OH)2]+• − [SnT(PyPH+)(OH)(O•)]+•) = −19.0 kcal/mol
∆E([SnT(PyH+)P(OH)2]2+• − [SnT(PyH2+)P(OH)(O•)]2+•) = −16.2 kcal/mol
∆E([SnT(PyH2+)P(OH)2]3+• − [SnT(PyH3+)P(OH)(O•)]3+•) = −13.3 kcal/mol
∆E([SnT(PyH3+)P(OH)2]4+• − [SnT(PyH4+)P(OH)(O•)]4+•) = −10.4 kcal/mol
2.5. Spin Density of the One-Electron Oxidized Species
3. Materials and Methods
3.1. Materials
3.2. Measurements
3.3. Density Functional Theory (DFT) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hammarström, L. Artificial Photosynthesis: A closing remarks. Faraday Discuss. 2017, 198, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Pray, L.A. Powering the World with Sunlight: A White Paper Describing the Discussions and Outcomes of the 1st Annual Chemical Sciences and Society Symposium (CS3), Kloster Seeon, Germany. 2009. Available online: https://www.gdch.de/fileadmin/downloads/Veranstaltungen/Sonderveranstaltungen/cs3wp.pdf (accessed on 10 January 2023).
- Olabi, G.; Abdelkareem, M. Renewable energy and climate change. Renew. Sustain. Energy Rev. 2022, 158, 112111. [Google Scholar] [CrossRef]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Gersten, S.W.; Samuels, G.; Meyer, T. Catalytic oxidation of water by an oxo-bridged Ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy) (CO)3 or Ru(bipy)32+-Co2+ combinations as homogeneous catalyst. J. Chem. Soc. Chem. Commun. 1983, 536–538. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Photochemical and electrochemical reduction of Carbon dioxide to carbon monoxide Mediated (2,2’ bipyridine) tricarbonyl chlororhenium (I) and related complexes as a homogeneous catalyst. Helv. Chim. Acta 1986, 69, 1990–2012. [Google Scholar] [CrossRef]
- Licht, S.; Wang, B.; Mukerji, S.; Soga, T.; Umeno, M.; Tributsch, H. Over 18% solar energy conversion to generation of Hydrogen fuel; theory and experiment for efficient solar water splitting. Int. J. Hydrogen Energy 2001, 26, 653–659. [Google Scholar] [CrossRef]
- Luo, J.; Im, J.H.; Mayer, M.; Schreier, M.; Nazeeruddin, M.; Park, N.-G.; Tilley, S.; Fan, H.; Grätzel, M. Water photolysis at 12.3% efficiency via perovskite photovoltaics and Earth-abundant catalyst. Science 2014, 345, 1593–1596. [Google Scholar] [CrossRef]
- Bonke, S.A.; Wiechen, M.; MacFarlane, D.; Spiccia, L. Renewable fuels from concentrated solar power: Towards practical artificial photosynthesis. Energy Environ. Sci. 2015, 8, 2791–2796. [Google Scholar] [CrossRef]
- ANakamura, K.; Ota, Y.; Koike, K.; Hidaka, Y.; Nishioka, K.; Sugiyama, M.; Fujii, K. A 24.4% solar to hydrogen energy conversion efficiency by combining concentrator photovoltaic modules and electrochemical cells. Appl. Phys. Express 2015, 8, 107101. [Google Scholar] [CrossRef]
- Jia, J.; Seitz, L.; Benck, J.; Huo, Y.; Chen, Y.; Ng, J.D.; Bilir, T.; Harris, J.; Jaramillo, T. Solar water splitting by photovoltaic electrolysis with a solar to hydrogen efficiency over 30%. Nat. Commun. 2016, 7, 13237. [Google Scholar] [CrossRef]
- Kudo, A.; Miseki, Y. Heterogeneous photocatalyst materials for water splitting. Chem. Soc. Rev. 2009, 38, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Abe, R. Recent progress on photocatalytic and photoelectrochemical water splitting under visible light irradiation. J. Photochem. Photobiol. C Photochem. Rev. 2010, 11, 179–209. [Google Scholar] [CrossRef]
- Maeda, K. Photocatalytic water splitting using semiconductor particles: History and recent developments. Photochem. Photobiol. C Photochem. Rev. 2011, 12, 237–268. [Google Scholar] [CrossRef]
- Maeda, K. Z-scheme Water splitting using two different semiconductor Photocatalysts. ACS Catal. 2013, 3, 1486–1503. [Google Scholar] [CrossRef]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef]
- Lewis, N.S. Introduction-Solar Energy conversion. Chem. Rev. 2015, 115, 12631–12632. [Google Scholar] [CrossRef]
- Wang, Y.; Suzuki, H.; Xie, J.; Tomita, O.; Martin, D.; Higashi, M.; Kong, D.; Abe, R.; Tang, J. Mimicking Photosynthesis: Solar to renewable H2 fuel synthesis by Z scheme water splitting systems. Chem. Rev. 2018, 118, 5201–5241. [Google Scholar] [CrossRef]
- Wang, Q.; Domen, K. Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies. Chem. Rev. 2020, 120, 919–985. [Google Scholar] [CrossRef]
- Maeda, K.; Teramura, K.; Lu, D.; Takata, T.; Saito, N.; Inoue, Y.; Domen, K. Photocatalyst releasing hydrogen from water. Nature 2006, 440, 295. [Google Scholar] [CrossRef]
- Wang, Q.; Nakabayashi, M.; Hisatomi, T.; Sun, S.; Akiyama, S.; Wang, Z.; Pan, Z.; Xiao, X.; Watanabe, T.; Yamada, T.; et al. Oxysulphide photocatalyst for visible-light-driven overall water splitting. Nat. Mater. 2019, 18, 827–832. [Google Scholar] [CrossRef]
- Takata, T.; Jiang, J.; Sakata, Y.; Nakabayashi, M.; Shibata, N.; Nandal, V.; Seki, K.; Hisatomi, T.; Domen, K. Photocatalytic water splitting with a quantum efficiency of almost unity. Nature 2020, 581, 411–414. [Google Scholar] [CrossRef]
- Zhou, P.; Navid, I.; Ma, Y.; Xiao, Y.; Wang, P.; Ye, Z.; Zhou, B.; Sun, K.; Mi, Z. Solar-to-hydrogen efficiency of more than 9% in photocatalytic water splitting. Nature 2023, 613, 66–70. [Google Scholar] [CrossRef]
- House, R.L.; Iha, N.; Coppo, R.; Alibabaei, L.; Sherman, B.; Kang, P.; Brennaman, M.; Hoertz, P.; Meyer, T. Artificial photosynthesis: Where are we now? Where we can go? J. Photochem. Photobiol. C Photochem. Rev. 2015, 25, 32–45. [Google Scholar] [CrossRef]
- Ashford, D.L.; Lapides, A.; Vannucci, A.; Hanson, K.; Torelli, D.; Harrison, D.; Templeton, J.; Meyer, T. Water oxidation by an electropolymerized catalyst on derivatized mesoporous metal oxide electrodes. J. Am. Chem. Soc. 2014, 136, 6578–6581. [Google Scholar] [CrossRef]
- Gao, Y.; Ding, X.; Liu, J.; Wang, L.; Lu, Z.; Li, L.; Sun, L. Visible light driven water splitting in a molecular device with unprecedentedly high photocurrent density. J. Am. Chem. Soc. 2013, 135, 4219–4222. [Google Scholar] [CrossRef]
- Li, F.; Fan, K.; Xu, B.; Gabrielsson, E.; Daniel, Q.; Li, L.; Sun, L. Organic Dye- sensitized tandem electrochemical cell for light driven total water splitting. J. Am. Chem. Soc. 2015, 137, 9153–9159. [Google Scholar] [CrossRef]
- Sahara, G.; Kumagai, H.; Maeda, K.; Kaeffer, N.; Artero, V.; Higashi, M.; Abe, R.; Ishitani, O. Photoelectrochemical reduction of CO2 coupled to water oxidation using a photocathode with a Ru (II)-Re (I) complex photocatalyst and a CoOx/TaON photoanode. J. Am. Chem. Soc. 2016, 138, 14152–14158. [Google Scholar] [CrossRef]
- Kumagai, H.; Sahara, G.; Maeda, K.; Higashi, M.; Abe, R.; Ishitani, O. Hybrid photocathode consisting of a CuGaO2 p-type semiconductor and a Ru (II)–Re (I) supramolecular photocatalyst: Non-biased visible-light-driven CO2 reduction with water oxidation. Chem. Sci. 2017, 8, 4242–4249. [Google Scholar] [CrossRef]
- Kamata, R.; Kumagai, H.; Yamazaki, Y.; Higashi, M.; Abe, R.; Ishitani, O. Durable photoelectrochemical CO2 reduction with a water oxidation using a visible-light driven photocathode. J. Mater. Chem. A 2021, 9, 1517–1529. [Google Scholar] [CrossRef]
- Kuttassery, F.; Kumagai, H.; Kamata, R.; Ebato, Y.; Higashi, M.; Suzuki, H.; Abe, R.; Ishitani, O. Supramolecular photocatalyst fixed on the inside of the polypyrrole layer in the dye sensitized molecular photocathode: Application to photocatalytic CO2 reduction coupled with water oxidation. Chem. Sci. 2021, 12, 13216–13232. [Google Scholar] [CrossRef] [PubMed]
- Kou, Y.; Nakatani, S.; Sunagawa, G.; Tachikawa, Y.; Masui, D.; Shimada, T.; Takagi, S.; Tryk, D.; Nabetani, Y.; Tachibana, H.; et al. Visible light induced reduction of carbon dioxide sensitized by a porphyrin–rhenium dyad metal complex on p-type semiconducting NiO as reduction terminal end of an artificial photosynthetic system. J. Catal. 2014, 310, 57–66. [Google Scholar] [CrossRef]
- Kou, Y.; Nabetani, Y.; Masui, D.; Shimada, T.; Takagi, S.; Tachibana, H.; Inoue, H. Direct detection of Key reaction intermediates in photochemical CO2 reduction sensitized by a Rhenium bipyridine complex. J. Am. Chem. Soc. 2014, 136, 6021–6030. [Google Scholar] [CrossRef]
- Nakazato, R.; Kou, Y.; Yamamoto, D.; Shimada, T.; Ishida, T.; Takagi, S.; Munakata, H.; Kanamura, K.; Tachibana, H.; Inoue, H. Effect of Li ions doping into p-type semiconductor NiO as a hole injection/transfer medium in the CO2 reduction sensitized/catalyzed by Zn porphyrin/Re complex upon visible light irradiation. Res. Chem. Intermed. 2021, 47, 269–285. [Google Scholar] [CrossRef]
- Kou, Y.; Nabetani, Y.; Nakazato, R.; Pratheesh, N.; Sato, T.; Nozawa, S.; Adachi, S.; Tachibana, H.; Inoue, H. Mechanism of photoreduction of Carbon dioxide catalysed by the benchmarking rhenium dimethyl bipyridine complexes; operando measurement by XAFS and FT-IR. J. Catal. 2022, 405, 508–519. [Google Scholar] [CrossRef]
- Okamura, M.; Kondo, M.; Kuga, R.; Kurashige, Y.; Yanai, T.; Hayami, S.; Praneeth, V.; Yoshida, M.; Yoneda, K.; Kawata, S.; et al. A pentanuclear iron catalyst designed for water oxidation. Nature 2016, 530, 465–468. [Google Scholar] [CrossRef]
- Rao, H.; Schmidt, L.; Bonin, J.; Robert, M. Visible-light driven methane formation from CO2 with molecular iron catalyst. Nature 2017, 548, 74–77. [Google Scholar] [CrossRef]
- Li, D.; Kassymova, M.; Cai, X.; Zang, S.-Q.; Jiang, H.-L. Photocatalytic CO2 reduction over metal-organic framework-based materials. Coord. Chem. Rev. 2020, 412, 213262. [Google Scholar] [CrossRef]
- Kuramochi, Y.; Satake, A. Porphyrins Acting as Photosensitizers in the Photocatalytic CO2 Reduction Reaction. Catalysts 2023, 13, 282. [Google Scholar] [CrossRef]
- Sakurai, H.; Masukawa, H.; Kitashima, M.; Inoue, K. Photobiological hydrogen production: Bioenergetics and challenges for its practical application. J. Photochem. Photobiol. C Photochem. Rev. 2013, 17, 1–25. [Google Scholar] [CrossRef]
- Masukawa, H.; Sakurai, H.; Hausinger, R.; Inoue, K. Increased heterocyst frequency by patN disruption in Anabaena leads to enhanced photobiological hydrogen production at high light intensity and high cell density. Appl. Microbiol. Biotechnol. 2017, 101, 2177–2188. [Google Scholar] [CrossRef]
- Liu, C.; Colón, B.; Ziesack, M.; Silver, P.A.; Nocera, D.G. Water splitting- biosynthetic system with CO2 reduction efficiencies exceeding photosynthesis. Science 2016, 352, 1210–1213. [Google Scholar] [CrossRef]
- Kuttassery, F.; Mathew, S.; Remello, S.; Thomas, A.; Sano, K.; Ohsaki, Y.; Nabetani, Y.; Tachibana, H.; Inoue, H. Alternative route to bypass the bottle-neck of water oxidation: Two- electron oxidation of water catalyzed by earth-abundant metalloporphyrins. Coord. Chem. Rev. 2018, 377, 64–72. [Google Scholar] [CrossRef]
- Inoue, H.; Shimada, T.; Kou, Y.; Nabetani, Y.; Masui, D.; Takagi, S.; Tachibana, H. The water oxidation bottleneck in artificial photosynthesis: How we can get through it? An alternative route involving two-electron process. ChemSusChem 2011, 4, 173–179. [Google Scholar] [CrossRef]
- Inoue, H.; Sumitani, M.; Sekita, A.; Hida, M. Photochemical epoxidation of alkenes by visible light in a redox system involving tetraphenylporphyrinatoantimony (V) and water. J. Chem. Soc. Chem. Commun. 1987, 1681–1682. [Google Scholar] [CrossRef]
- Inoue, H.; Okamoto, T.; Kameo, Y.; Sumitani, M.; Fujiwara, A.; Ishibashi, D.; Hida, M. Photochemical epoxidation of cyclohexene sensitized by tetraphenylporphyrinatoantimony (V) in the presence of water acting both as an electron and oxygen donor. J. Chem. Soc. Perkin Trans. 1 1994, 105–111. [Google Scholar] [CrossRef]
- Takagi, S.; Suzuki, M.; Shiragami, T.; Inoue, H. Photochemical P-450 oxygenation of cyclohexene with water sensitized by dihydroxy coordinated (tetraphenyl porphyrinato) antimony (V) hexafluorophosphate. J. Am. Chem. Soc. 1997, 119, 8712–8713. [Google Scholar] [CrossRef]
- Funyu, S.; Isobe, T.; Takagi, S.; Tryk, D.; Inoue, H. Highly efficient and selective epoxidation of alkenes by photochemical oxygenation sensitized by Ruthenium (II) porphyrins with water as both electron and oxygen donor. J. Am. Chem. Soc. 2003, 125, 5734–5740. [Google Scholar] [CrossRef]
- Inoue, H.; Funyu, S.; Shimada, Y.; Takagi, S. Artificial photosynthesis via two-electron conversion: Photochemical oxygenation sensitized by ruthenium porphyrins with water as both electron and oxygen atom donor. Pure Appl. Chem. 2005, 77, 1019–1033. [Google Scholar] [CrossRef]
- Funyu, S.; Kinai, M.; Masui, D.; Takagi, S.; Shimada, T.; Tachibana, H.; Inoue, H. Key reaction intermediates of the photochemical oxygenation of alkene sensitized by RuII porphyrin with water by visible light. Photochem. Photobiol. Sci. 2010, 9, 931–936. [Google Scholar] [CrossRef]
- Shimada, T.; Kumagai, A.; Funyu, S.; Takagi, S.; Masui, D.; Nabetani, Y.; Tachibana, H.; Tryk, D.; Inoue, H. How is the water molecule activated on the metalloporphyrins? Oxygenation of substrate induced through one-photon/two-electron conversion in artificial photosynthesis. Faraday Discuss. 2012, 155, 145–163. [Google Scholar] [CrossRef]
- Kurimoto, K.; Yamazaki, T.; Suzuri, Y.; Nabetani, Y.; Onuki, S.; Takagi, S.; Shimada, T.; Tachibana, H.; Inoue, H. Hydrogen evolution coupled with photochemical oxygenation of cyclohexene with water sensitized by Tin (IV) porphyrins by visible light. Photochem. Photobiol. Sci. 2014, 13, 154–156. [Google Scholar] [CrossRef]
- Mathew, S.; Kuttassery, F.; Gomi, Y.; Yamamoto, D.; Kiyooka, R.; Onuki, S.; Nabetani, Y.; Tachibana, H.; Inoue, H. Photochemical oxygenation of cyclohexene with water sensitized by aluminum (III) porphyrins with visible light. J. Photochem. Photobiol. A Chem. 2015, 313, 137–142. [Google Scholar] [CrossRef]
- Remello, S.N.; Hirano, T.; Kuttassery, F.; Nabetani, Y.; Yamamoto, D.; Onuki, S.; Tachibana, H.; Inoue, H. Visible light induced oxygenation of alkenes with water sensitized by silicon porphyrins with the second most earth-abundant metal. J. Photochem. Photobiol. A Chem. 2015, 313, 176–183. [Google Scholar] [CrossRef]
- Kuttassery, F.; Mathew, S.; Sagawa, S.; Remello, S.; Thomas, A.; Yamamoto, D.; Onuki, S.; Nabetani, Y.; Tachibana, H.; Inoue, H. One electron initiated two-electron oxidation of water by aluminum porphyrins with earth’s most abundant metal. ChemSusChem 2017, 10, 1909–1915. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Kuttassery, F.; Mathew, S.; Remello, S.; Ohsaki, Y.; Yamamoto, D.; Nabetani, Y.; Tachibana, H.; Inoue, H. Protolytic behavior axially coordinated hydroxy group of Tin (IV) porphyrins as promising molecular catalysts for water oxidation. J. Photochem. Photobiol. A Chem. 2018, 358, 402–410. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Thomas, A.; Kuttassery, F.; Mathew, S.; Remello, S.; Shimada, T.; Ishida, T.; Takagi, S.; Tachibana, H.; Inoue, H. Two electron oxidation of water to form hydrogen peroxide initiated by one-electron oxidation of Tin (IV)-porphyrins. J. Photochem. Photobiol. A Chem. 2020, 401, 112732. [Google Scholar] [CrossRef]
- Sebastian, A.; Remello, S.; Kuttassery, F.; Mathew, S.; Ohsaki, Y.; Tachibana, H.; Inoue, H. Protolytic behavior of water soluble Zinc (II) porphyrin and the electrocatalytic two-electron oxidation of water to form hydrogen peroxide. J. Photochem. Photobiol. A Chem. 2020, 400, 112619. [Google Scholar] [CrossRef]
- Shiragami, T.; Nakamura, H.; Matsumoto, J.; Yasuda, M.; Suzuri, Y.; Tachibana, H.; Inoue, H. Two electron oxidation of water to form hydrogen peroxide sensitized by di (hydroxo) Porphyrin GeIV complex under visible-light irradiation. J. Photochem. Photobiol. A Chem. 2015, 313, 131–136. [Google Scholar] [CrossRef]
- Kuttassery, F.; Sagawa, S.; Mathew, S.; Nabetani, Y.; Iwase, A.; Kudo, A.; Tachibana, H.; Inoue, H. Water splitting on Aluminum porphyrins to form hydrogen and hydrogen peroxide by one-photon of visible light. ACS Appl. Energy Mater. 2019, 2, 8045–8051. [Google Scholar] [CrossRef]
- Mathew, S.; Kuttassery, F.; Remello, S.; Thomas, A.; Yamamoto, D.; Onuki, S.; Nabetani, Y.; Tachibana, H.; Inoue, H. Two electron oxidation of water through one photon excitation of aluminum porphyrins: Molecular mechanism and key intermediates. ChemPhotoChem 2018, 2, 240–248. [Google Scholar] [CrossRef]
- Kuttassery, F.; Mathew, S.; Tachibana, H.; Inoue, H. How one photon can induce water splitting into hydrogen peroxide and hydrogen by aluminum porphyrins. Rationale of thermodynamics. Sustain. Energy Fuels 2020, 4, 1945–1953. [Google Scholar] [CrossRef]
- Mathew, S.; Sebastian, A.; Kuttassery, F.; Yamauchi, Y.; Isobe, T.; Hatanaka, T.; Funahashi, Y.; Tachibana, H.; Inoue, H. Supramolecular Aluminum-based molecular catalyst for water oxidation into H2O2 in saline water. Sustain. Energy Fuels 2022, 6, 4256–4264. [Google Scholar] [CrossRef]
- Thomas, A.; Kuttassery, F.; Remello, S.; Mathew, S.; Yamamoto, D.; Onuki, S.; Nabetani, Y.; Tachibana, H.; Inoue, H. Facile synthesis of water soluble cationic Tin (IV) porphyrins and water insoluble Tin (IV) porphyrins in water at ambient temperature. Bull. Chem. Soc. Jpn. 2016, 89, 902–904. [Google Scholar] [CrossRef]
- Ohsaki, Y.; Thomas, A.; Kuttassery, F.; Mathew, S.; Remello, S.; Nabetani, Y.; Shimada, T.; Takagi, S.; Tachibana, H.; Inoue, H. How does the Tin (IV) insertion to porphyrin proceed in water at ambient temperature? Reinvestigation by time dependent 1H NMR and detection of intermediates. Inorg. Chim. Acta 2018, 482, 914–924. [Google Scholar] [CrossRef]
- Reed, A.; Curtiss, L.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.; Weinhold, F. Natural bond orbital methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Fujishima, A.; Einaga, Y.; Rao, T.; Tryk, D. Diamond Electrochemistry; Elsevier: Tokyo, Japan, 2005. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SnTPyP | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pKa | 0.7 | 1.4 | 2.4 | 3.0 | 3.9 | 4.3 | 5.0 | 5.9 | ||||||||||
| Fluorescence lifetime/ns | 0.88 | 0.88 | 0.93 | 0.95 | 0.92 | 0.92 | 0.92 | 0.93 | 1.57 | |||||||||
| Oxidation potential /Volt vs. SHE | - | 1.79 | Eox = −0.056 pH + 1.82 a | 1.58 | 1.55 | 1.54 | ||||||||||||
| Spin density b | 0.001 0.001 | 0.002 0.007 | 0.011 0.011 | 0.012 0.012 | 0.011 0.011 | 0.010 0.010 | 0.010 0.010 | 0.993 0.002 | 0.988 0.001 | |||||||||
| ∆E(TiO2) c /eV (pH) | - | −0.11 (1.0) | −0.13 (2.0) | −0.13 (2.5) | −0.13 (3.5) | −0.13 (4.0) | −0.11 (4.5) | −0.09 (5.5) | −0.01 (7.0) | |||||||||
| ∆E(SnO2) d /eV (pH) | - | −0.30 (1.0) | −0.32 (2.0) | −0.32 (2.5) | −0.32 (3.5) | −0.32 (4.0) | −0.30 (4.5) | −0.28 (5.5) | −0.20 (7.0) | |||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, A.; Ohsaki, Y.; Nakazato, R.; Kuttassery, F.; Mathew, S.; Remello, S.N.; Tachibana, H.; Inoue, H. Molecular Characteristics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis. Molecules 2023, 28, 1882. https://doi.org/10.3390/molecules28041882
Thomas A, Ohsaki Y, Nakazato R, Kuttassery F, Mathew S, Remello SN, Tachibana H, Inoue H. Molecular Characteristics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis. Molecules. 2023; 28(4):1882. https://doi.org/10.3390/molecules28041882
Chicago/Turabian StyleThomas, Arun, Yutaka Ohsaki, Ryosuke Nakazato, Fazalurahman Kuttassery, Siby Mathew, Sebastian Nybin Remello, Hiroshi Tachibana, and Haruo Inoue. 2023. "Molecular Characteristics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis" Molecules 28, no. 4: 1882. https://doi.org/10.3390/molecules28041882
APA StyleThomas, A., Ohsaki, Y., Nakazato, R., Kuttassery, F., Mathew, S., Remello, S. N., Tachibana, H., & Inoue, H. (2023). Molecular Characteristics of Water-Insoluble Tin-Porphyrins for Designing the One-Photon-Induced Two-Electron Oxidation of Water in Artificial Photosynthesis. Molecules, 28(4), 1882. https://doi.org/10.3390/molecules28041882