

Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target

Abstract
:1. Introduction
1.1. Epidemiology of OA
1.2. Correlation between OA and NO
2. NO and NO Synthase (NOS)
3. Physiological Effects of NO on Cartilage
3.1. NO and Osteoclasts
3.2. NO and Osteoblasts
3.3. NO and Chondrocytes
4. Pathological Effects of NO in OA
4.1. Characteristics of OA Chondrocytes
4.2. NO and Increased Matrix Degradation
4.3. NO and Apoptosis
4.4. NO and Inflammatory Mediators
4.5. NO and ROS/Reactive Nitrogen Oxide Species (RNOS)
4.6. NO and Pain in the OA Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathological Effects of NO in OA | Related Mediators or Signaling Pathways | Related Mechanisms | Reference |
|---|---|---|---|
| Increases cartilage matrix degradation | Aggrecanases and collagenases | NO mediates upregulation of aggrecanases and collagenases | Wu et al., 2019 [63] Brown et al., 2020 [65] |
| Enhances chondrocyte apoptosis | Caspase-3 and -9, tyrosine kinases, Bax, Cyt C, and ROS | NO induces ROS generation and co-activates capase-3 to enhance chondrocyte apoptosis through a mitochondria-dependent mechanism | Poderoso et al., 2019 [31] He et al., 2020 [75] |
| Inhibits chondrocyte autophagy | ERK, Akt and mTOR signaling pathways | NO reduces autophagic activity, autophagic flux, and expression of several autophagy-related genes | Akaraphutiporn et al., 2020 [59] |
| Induces synthesis and release of inflammatory mediators | Inflammatory mediators and NF-κB signaling | NO interacts with inflammatory mediators which lead to constant inflammatory molecules release and NF-κB activation | Chen et al., 2008 [84] Moon et al., 2018 [64] |
| Promotes inflammation and cell death | ROS and RNOS | NO combines with O2− to generate peroxynitrite which can cause DNA breaks and chondrocyte senescence, leading to cell necrosis | Copp et al., 2021 [85] |
| Cause central sensitization of the pain pathway | Inflammatory mediators and cGMP-dependent hyperalgesia | NO Promotes inflammation and structural changes of joint and play a role as neurotransmitter | Venkanna et al., 2014 [90] Aley et al., 1998 [91] |
5. NO in OA Treatment
5.1. iNOS Inhibitors
5.1.1. N-Monomethyl-L-Arginine (L-NMMA)
5.1.2. N-Iminoethyl-L-Lysine (L-NIL)
5.1.3. NG-nitro-L-Arginine Methyl Ester (L-NAME)
5.1.4. 1400W
5.1.5. Cindunistat
5.1.6. Aminoguanidine (AG)
5.2. Pharmaceuticals Inhibiting iNOS Expression
5.2.1. Glucocorticoids (GCs)
5.2.2. Hyaluronic Acid (HA)
5.2.3. CoenzymeQ10 (CoQ10)
5.2.4. Sargassum serratifolium
5.2.5. Wogonin
5.2.6. Myricitrin (Myr)
5.3. Applications of Materials Science
5.4. Physical Methods
6. Discussion and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, M.; Rey, K.; Besler, K.; Wang, C.; Choy, J. Immunobiology of Nitric Oxide and Regulation of Inducible Nitric Oxide Synthase. Results Probl. Cell Differ. 2017, 62, 181–207. [Google Scholar]
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis: Literature update. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar]
- O’Neill, T.W.; McCabe, P.S.; McBeth, J. Update on the epidemiology, risk factors and disease outcomes of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 2018, 32, 312–326. [Google Scholar]
- Beard, J.R.; Officer, A.; de Carvalho, I.A.; Sadana, R.; Pot, A.M.; Michel, J.P.; Lloyd-Sherlock, P.; Epping-Jordan, J.E.; Peeters, G.; Mahanani, W.R.; et al. The World report on ageing and health: A policy framework for healthy ageing. Lancet 2016, 387, 2145–2154. [Google Scholar]
- Rahmati, M.; Nalesso, G.; Mobasheri, A.; Mozafari, M. Aging and osteoarthritis: Central role of the extracellular matrix. Ageing Res Rev 2017, 40, 20–30. [Google Scholar]
- Vincent, T.L. Mechanoflammation in osteoarthritis pathogenesis. Semin Arthritis Rheum 2019, 49 (Suppl. S3), S36–S38. [Google Scholar]
- Abramoff, B.; Caldera, F.E. Osteoarthritis: Pathology, Diagnosis, and Treatment Options. Med. Clin. N. Am. 2020, 104, 293–311. [Google Scholar]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2016, 12, 580–592. [Google Scholar]
- Ersoy, Y.; Ozerol, E.; Baysal, O.; Temel, I.; MacWalter, R.S.; Meral, U.; Altay, Z.E. Serum nitrate and nitrite levels in patients with rheumatoid arthritis, ankylosing spondylitis, and osteoarthritis. Ann. Rheum. Dis. 2002, 61, 76–78. [Google Scholar]
- Subczynski, W.K.; Lomnicka, M.; Hyde, J.S. Permeability of nitric oxide through lipid bilayer membranes. Free Radic. Res. 1996, 24, 343–349. [Google Scholar]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar]
- Koppenol, W.H.; Traynham, J.G. Say NO to nitric oxide: Nomenclature for nitrogen- and oxygen-containing compounds. Methods Enzymol 1996, 268, 3–7. [Google Scholar]
- Davis, K.L.; Martin, E.; Turko, I.V.; Murad, F. Novel effects of nitric oxide. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 203–236. [Google Scholar]
- Korhonen, R.; Lahti, A.; Kankaanranta, H.; Moilanen, E. Nitric oxide production and signaling in inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 471–479. [Google Scholar]
- Kim, J.M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar]
- Shirazi, S.; Ravindran, S.; Cooper, L.F. Topography-mediated immunomodulation in osseointegration; Ally or Enemy. Biomaterials 2022, 291, 121903. [Google Scholar]
- Morimoto, A.; Kikuta, J.; Nishikawa, K.; Sudo, T.; Uenaka, M.; Furuya, M.; Hasegawa, T.; Hashimoto, K.; Tsukazaki, H.; Seno, S.; et al. SLPI is a critical mediator that controls PTH-induced bone formation. Nat. Commun. 2021, 12, 2136. [Google Scholar]
- Srivastava, R.K.; Sapra, L.; Mishra, P.K. Osteometabolism: Metabolic Alterations in Bone Pathologies. Cells 2022, 11, 3943. [Google Scholar]
- Siddiqui, J.A.; Partridge, N.C. Physiological Bone Remodeling: Systemic Regulation and Growth Factor Involvement. Physiology 2016, 31, 233–245. [Google Scholar]
- Kalyanaraman, H.; Ramdani, G.; Joshua, J.; Schall, N.; Boss, G.R.; Cory, E.; Sah, R.L.; Casteel, D.E.; Pilz, R.B. A Novel, Direct NO Donor Regulates Osteoblast and Osteoclast Functions and Increases Bone Mass in Ovariectomized Mice. J. Bone Miner. Res. 2017, 32, 46–59. [Google Scholar]
- Amano, H.; Iwaki, F.; Oki, M.; Aoki, K.; Ohba, S. An osteogenic helioxanthin derivative suppresses the formation of bone-resorbing osteoclasts. Regen. Ther. 2019, 11, 290–296. [Google Scholar]
- Kaneko, K.; Miyamoto, Y.; Tsukuura, R.; Sasa, K.; Akaike, T.; Fujii, S.; Yoshimura, K.; Nagayama, K.; Hoshino, M.; Inoue, S.; et al. 8-Nitro-cGMP is a promoter of osteoclast differentiation induced by RANKL. Nitric Oxide 2018, 72, 46–51. [Google Scholar]
- Mentaverri, R.; Kamel, S.; Wattel, A.; Prouillet, C.; Sevenet, N.; Petit, J.P.; Tordjmann, T.; Brazier, M. Regulation of bone resorption and osteoclast survival by nitric oxide: Possible involvement of NMDA-receptor. J. Cell Biochem. 2003, 88, 1145–1156. [Google Scholar]
- Gloria, M.A.D.; Mouro, M.G.; Geraldini, S.; Higa, E.M.S.; Carvalho, A.B. Cbfa1 expression in vascular smooth muscle cells may be elevated by increased nitric oxide/iNOS. J. Bras. Nefrol. 2020, 42, 300–306. [Google Scholar]
- Cepeda, S.B.; Sandoval, M.J.; Crescitelli, M.C.; Rauschemberger, M.B.; Massheimer, V.L. The isoflavone genistein enhances osteoblastogenesis: Signaling pathways involved. J. Physiol. Biochem. 2020, 76, 99–110. [Google Scholar]
- Ramdani, G.; Schall, N.; Kalyanaraman, H.; Wahwah, N.; Moheize, S.; Lee, J.J.; Sah, R.L.; Pfeifer, A.; Casteel, D.E.; Pilz, R.B. cGMP-dependent protein kinase-2 regulates bone mass and prevents diabetic bone loss. J. Endocrinol. 2018, 238, 203–219. [Google Scholar]
- Gerbarg, P.L.; Brown, R.P. Pause menopause with Rhodiola rosea, a natural selective estrogen receptor modulator. Phytomedicine 2016, 23, 763–769. [Google Scholar]
- Crescitelli, M.C.; Rauschemberger, M.B.; Cepeda, S.; Sandoval, M.; Massheimer, V.L. Role of estrone on the regulation of osteoblastogenesis. Mol. Cell. Endocrinol. 2019, 498, 110582. [Google Scholar]
- Wittkowske, C.; Reilly, G.C.; Lacroix, D.; Perrault, C.M. In Vitro Bone Cell Models: Impact of Fluid Shear Stress on Bone Formation. Front. Bioeng. Biotechnol. 2016, 4, 87. [Google Scholar]
- Maycas, M.; Esbrit, P.; Gortázar, A.R. Molecular mechanisms in bone mechanotransduction. Histol. Histopathol. 2017, 32, 751–760. [Google Scholar]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar]
- Kamm, A.; Przychodzen, P.; Kuban-Jankowska, A.; Jacewicz, D.; Dabrowska, A.M.; Nussberger, S.; Wozniak, M.; Gorska-Ponikowska, M. Nitric oxide and its derivatives in the cancer battlefield. Nitric Oxide 2019, 93, 102–114. [Google Scholar]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediat. Inflamm. 2014, 2014, 561459. [Google Scholar]
- Teixeira, C.C.; Ischiropoulos, H.; Leboy, P.S.; Adams, S.L.; Shapiro, I.M. Nitric oxide-nitric oxide synthase regulates key maturational events during chondrocyte terminal differentiation. Bone 2005, 37, 37–45. [Google Scholar]
- Drissi, H.; Zuscik, M.; Rosier, R.; O'Keefe, R. Transcriptional regulation of chondrocyte maturation: Potential involvement of transcription factors in OA pathogenesis. Mol. Aspects Med. 2005, 26, 169–179. [Google Scholar]
- Ono, T.; Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell Biol. 2018, 149, 325–341. [Google Scholar]
- MacIntyre, I.; Zaidi, M.; Alam, A.S.; Datta, H.K.; Moonga, B.S.; Lidbury, P.S.; Hecker, M.; Vane, J.R. Osteoclastic inhibition: An action of nitric oxide not mediated by cyclic GMP. Proc. Natl. Acad. Sci. USA 1991, 88, 2936–2940. [Google Scholar]
- Löwik, C.W.; Nibbering, P.H.; van de Ruit, M.; Papapoulos, S.E. Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J. Clin. Investig. 1994, 93, 1465–1472. [Google Scholar]
- Ralston, S.H.; Ho, L.P.; Helfrich, M.H.; Grabowski, P.S.; Johnston, P.W.; Benjamin, N. Nitric oxide: A cytokine-induced regulator of bone resorption. J. Bone Miner. Res. 1995, 10, 1040–1049. [Google Scholar]
- Yaroslavskiy, B.B.; Li, Y.; Ferguson, D.J.; Kalla, S.E.; Oakley, J.I.; Blair, H.C. Autocrine and paracrine nitric oxide regulate attachment of human osteoclasts. J. Cell. Biochem. 2004, 91, 962–972. [Google Scholar]
- Ralston, S.H.; Grabowski, P.S. Mechanisms of cytokine induced bone resorption: Role of nitric oxide, cyclic guanosine monophosphate, and prostaglandins. Bone 1996, 19, 29–33. [Google Scholar]
- Helfrich, M.H.; Evans, D.E.; Grabowski, P.S.; Pollock, J.S.; Ohshima, H.; Ralston, S.H. Expression of nitric oxide synthase isoforms in bone and bone cell cultures. J. Bone Miner. Res. 1997, 12, 1108–1115. [Google Scholar]
- Yan, Q.; Feng, Q.; Beier, F. Endothelial nitric oxide synthase deficiency in mice results in reduced chondrocyte proliferation and endochondral bone growth. Arthritis Rheum. 2010, 62, 2013–2022. [Google Scholar]
- Hefler, L.A.; Reyes, C.A.; O'Brien, W.E.; Gregg, A.R. Perinatal development of endothelial nitric oxide synthase-deficient mice. Biol. Reprod. 2001, 64, 666–673. [Google Scholar]
- Afzal, F.; Polak, J.; Buttery, L. Endothelial nitric oxide synthase in the control of osteoblastic mineralizing activity and bone integrity. J. Pathol. 2004, 202, 503–510. [Google Scholar]
- Watanuki, M.; Sakai, A.; Sakata, T.; Tsurukami, H.; Miwa, M.; Uchida, Y.; Watanabe, K.; Ikeda, K.; Nakamura, T. Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J. Bone Miner. Res. 2002, 17, 1015–1025. [Google Scholar]
- Liu, H.; Rosen, C.J. Nitric oxide and bone: The phoenix rises again. J. Clin. Investig. 2021, 131, e147072. [Google Scholar]
- Cao, W.; Helder, M.N.; Bravenboer, N.; Wu, G.; Jin, J.; Ten Bruggenkate, C.M.; Klein-Nulend, J.; Schulten, E. Is There a Governing Role of Osteocytes in Bone Tissue Regeneration? Curr. Osteoporos. Rep. 2020, 18, 541–550. [Google Scholar]
- Deng, S.; Dai, G.; Chen, S.; Nie, Z.; Zhou, J.; Fang, H.; Peng, H. Dexamethasone induces osteoblast apoptosis through ROS-PI3K/AKT/GSK3β signaling pathway. Biomed. Pharmacother. 2019, 110, 602–608. [Google Scholar]
- Nishimura, R.; Hata, K.; Kida, J. Regulation of osteoblasts and chondrocytes by Wnt signaling. Clin. Calcium 2019, 29, 299–307. [Google Scholar]
- Otsuka, E.; Hirano, K.; Matsushita, S.; Inoue, A.; Hirose, S.; Yamaguchi, A.; Hagiwara, H. Effects of nitric oxide from exogenous nitric oxide donors on osteoblastic metabolism. Eur. J. Pharmacol. 1998, 349, 345–350. [Google Scholar]
- Mancini, L.; Moradi-Bidhendi, N.; Becherini, L.; Martineti, V.; MacIntyre, I. The biphasic effects of nitric oxide in primary rat osteoblasts are cGMP dependent. Biochem. Biophys. Res. Commun. 2000, 274, 477–481. [Google Scholar]
- Hikiji, H.; Shin, W.S.; Oida, S.; Takato, T.; Koizumi, T.; Toyo-Oka, T. Direct action of nitric oxide on osteoblastic differentiation. FEBS Lett. 1997, 410, 238–242. [Google Scholar]
- Damoulis, P.D.; Hauschka, P.V. Cytokines induce nitric oxide production in mouse osteoblasts. Biochem. Biophys. Res. Commun. 1994, 201, 924–931. [Google Scholar]
- Ralston, S.H.; Todd, D.; Helfrich, M.; Benjamin, N.; Grabowski, P.S. Human osteoblast-like cells produce nitric oxide and express inducible nitric oxide synthase. Endocrinology 1994, 135, 330–336. [Google Scholar]
- Minne, H.W.; Pfeilschifter, J.; Scharla, S.; Mutschelknauss, S.; Schwarz, A.; Krempien, B.; Ziegler, R. Inflammation-mediated osteopenia in the rat: A new animal model for pathological loss of bone mass. Endocrinology 1984, 115, 50–54. [Google Scholar]
- Sacitharan, P.K. Ageing and Osteoarthritis. Subcell Biochem. 2019, 91, 123–159. [Google Scholar]
- Grässel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel Molecular Mechanisms Increase Our Understanding of the Disease Pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar]
- Akaraphutiporn, E.; Sunaga, T.; Bwalya, E.C.; Yanlin, W.; Carol, M.; Okumura, M. An Insight into the Role of Apoptosis and Autophagy in Nitric Oxide-Induced Articular Chondrocyte Cell Death. Cartilage 2020, 13, 826S–838S. [Google Scholar]
- Krishnan, Y.; Grodzinsky, A.J. Cartilage diseases. Matrix Biol. 2018; 71–72, 51–69. [Google Scholar]
- Charlier, E.; Deroyer, C.; Ciregia, F.; Malaise, O.; Neuville, S.; Plener, Z.; Malaise, M.; de Seny, D. Chondrocyte dedifferentiation and osteoarthritis (OA). Biochem. Pharmacol. 2019, 165, 49–65. [Google Scholar]
- Casal-Beiroa, P.; Balboa-Barreiro, V.; Oreiro, N.; Pértega-Díaz, S.; Blanco, F.J.; Magalhães, J. Optical Biomarkers for the Diagnosis of Osteoarthritis through Raman Spectroscopy: Radiological and Biochemical Validation Using Ex Vivo Human Cartilage Samples. Diagnostics 2021, 11, 546. [Google Scholar]
- Wu, Y.; Lin, Z.; Yan, Z.; Wang, Z.; Fu, X.; Yu, K. Sinomenine contributes to the inhibition of the inflammatory response and the improvement of osteoarthritis in mouse-cartilage cells by acting on the Nrf2/HO-1 and NF-κB signaling pathways. Int. Immunopharmacol. 2019, 75, 105715. [Google Scholar]
- Moon, S.M.; Lee, S.A.; Han, S.H.; Park, B.R.; Choi, M.S.; Kim, J.S.; Kim, S.G.; Kim, H.J.; Chun, H.S.; Kim, D.K.; et al. Aqueous extract of Codium fragile alleviates osteoarthritis through the MAPK/NF-κB pathways in IL-1β-induced rat primary chondrocytes and a rat osteoarthritis model. Biomed. Pharmacother. 2018, 97, 264–270. [Google Scholar]
- Brown, S.B.; Hornyak, J.A.; Jungels, R.R.; Shah, Y.Y.; Yarmola, E.G.; Allen, K.D.; Sharma, B. Characterization of Post-Traumatic Osteoarthritis in Rats Following Anterior Cruciate Ligament Rupture by Non-Invasive Knee Injury (NIKI). J. Orthop. Res. 2020, 38, 356–367. [Google Scholar]
- Sun, E.Y.; Fleck, A.K.M.; Abu-Hakmeh, A.E.; Kotsakis, A.; Leonard, G.R.; Wan, L.Q. Cartilage Metabolism is Modulated by Synovial Fluid Through Metalloproteinase Activity. Ann. Biomed. Eng. 2018, 46, 810–818. [Google Scholar]
- Goldring, M.B.; Berenbaum, F. The regulation of chondrocyte function by proinflammatory mediators: Prostaglandins and nitric oxide. Clin. Orthop. Relat. Res. 2004, 427, S37–S46. [Google Scholar]
- Stefanovic-Racic, M.; Möllers, M.O.; Miller, L.A.; Evans, C.H. Nitric oxide and proteoglycan turnover in rabbit articular cartilage. J. Orthop. Res. 1997, 15, 442–449. [Google Scholar]
- Ho, Y.-J.; Lu, J.-W.; Ho, L.-J.; Lai, J.-H.; Huang, H.-S.; Lee, C.-C.; Lin, T.-Y.; Lien, S.-B.; Lin, L.-C.; Chen, L.W.; et al. Anti-inflammatory and anti-osteoarthritis effects of Cm-02 and Ck-02. Biochem. Biophys. Res. Commun. 2019, 517, 155–163. [Google Scholar]
- Fei, J.; Liang, B.; Jiang, C.; Ni, H.; Wang, L. Luteolin inhibits IL-1β-induced inflammation in rat chondrocytes and attenuates osteoarthritis progression in a rat model. Biomed. Pharmacother. 2019, 109, 1586–1592. [Google Scholar]
- O’Sullivan, S.; Medina, C.; Ledwidge, M.; Radomski, M.W.; Gilmer, J.F. Nitric oxide-matrix metaloproteinase-9 interactions: Biological and pharmacological significance--NO and MMP-9 interactions. Biochim. Biophys. Acta 2014, 1843, 603–617. [Google Scholar]
- Studer, R.; Jaffurs, D.; Stefanovic-Racic, M.; Robbins, P.D.; Evans, C.H. Nitric oxide in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 377–379. [Google Scholar]
- Charlier, E.; Relic, B.; Deroyer, C.; Malaise, O.; Neuville, S.; Collée, J.; Malaise, M.G.; De Seny, D. Insights on Molecular Mechanisms of Chondrocytes Death in Osteoarthritis. Int. J. Mol. Sci. 2016, 17, 2146. [Google Scholar]
- Ren, H.; Yang, H.; Xie, M.; Wen, Y.; Liu, Q.; Li, X.; Liu, J.; Xu, H.; Tang, W.; Wang, M. Chondrocyte apoptosis in rat mandibular condyles induced by dental occlusion due to mitochondrial damage caused by nitric oxide. Arch. Oral Biol. 2019, 101, 108–121. [Google Scholar]
- He, B.; Wu, F.; Li, X.; Liu, Y.; Fan, L.; Li, H. Mitochondrial dependent pathway is involved in the protective effects of carboxymethylated chitosan on nitric oxide-induced apoptosis in chondrocytes. BMC Complement. Med. Ther. 2020, 20, 23. [Google Scholar]
- Del Carlo, M., Jr.; Loeser, R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002, 46, 394–403. [Google Scholar]
- Whiteman, M.; Armstrong, J.S.; Cheung, N.S.; Siau, J.L.; Rose, P.; Schantz, J.T.; Jones, D.P.; Halliwell, B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004, 18, 1395–1397. [Google Scholar]
- Abramson, S.B.; Attur, M.; Amin, A.R.; Clancy, R. Nitric oxide and inflammatory mediators in the perpetuation of osteoarthritis. Curr. Rheumatol. Rep. 2001, 3, 535–541. [Google Scholar]
- Fermor, B.; Weinberg, J.B.; Pisetsky, D.S.; Misukonis, M.A.; Banes, A.J.; Guilak, F. The effects of static and intermittent compression on nitric oxide production in articular cartilage explants. J. Orthop. Res. 2001, 19, 729–737. [Google Scholar]
- Lou, Y.; Wang, C.; Zheng, W.; Tang, Q.; Chen, Y.; Zhang, X.; Guo, X.; Wang, J. Salvianolic acid B inhibits IL-1β-induced inflammatory cytokine production in human osteoarthritis chondrocytes and has a protective effect in a mouse osteoarthritis model. Int. Immunopharmacol. 2017, 46, 31–37. [Google Scholar]
- Kitaura, H.; Fujimura, Y.; Yoshimatsu, M.; Kohara, H.; Morita, Y.; Aonuma, T.; Fukumoto, E.; Masuyama, R.; Yoshida, N.; Takano-Yamamoto, T. IL-12- and IL-18-mediated, nitric oxide-induced apoptosis in TNF-α-mediated osteoclastogenesis of bone marrow cells. Calcif. Tissue Int. 2011, 89, 65–73. [Google Scholar]
- Boileau, C.; Martel-Pelletier, J.; Moldovan, F.; Jouzeau, J.Y.; Netter, P.; Manning, P.T.; Pelletier, J.P. The in situ up-regulation of chondrocyte interleukin-1-converting enzyme and interleukin-18 levels in experimental osteoarthritis is mediated by nitric oxide. Arthritis Rheum. 2002, 46, 2637–2647. [Google Scholar]
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995, 146, 75–85. [Google Scholar]
- Chen, A.F.; Davies, C.M.; De Lin, M.; Fermor, B. Oxidative DNA damage in osteoarthritic porcine articular cartilage. J. Cell Physiol. 2008, 217, 828–833. [Google Scholar]
- Copp, M.E.; Flanders, M.C.; Gagliardi, R.; Gilbertie, J.M.; Sessions, G.A.; Chubinskaya, S.; Loeser, R.F.; Schnabel, L.V.; Diekman, B.O. The combination of mitogenic stimulation and DNA damage induces chondrocyte senescence. Osteoarthr. Cartil. 2021, 29, 402–412. [Google Scholar]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113.
- Clancy, R.M.; Gomez, P.F.; Abramson, S.B. Nitric oxide sustains nuclear factor kappaB activation in cytokine-stimulated chondrocytes. Osteoarthr. Cartil. 2004, 12, 552–558. [Google Scholar]
- Morgan, M.; Nazemian, V.; Harrington, K.; Ivanusic, J.J. Mini review: The role of sensory innervation to subchondral bone in osteoarthritis pain. Front. Endocrinol. 2022, 13, 1047943. [Google Scholar]
- Coaccioli, S.; Sarzi-Puttini, P.; Zis, P.; Rinonapoli, G.; Varrassi, G. Osteoarthritis: New Insight on Its Pathophysiology. J. Clin. Med. 2022, 11, 6013. [Google Scholar]
- Balaganur, V.; Pathak, N.N.; Lingaraju, M.C.; More, A.S.; Latief, N.; Kumari, R.R.; Kumar, D.; Tandan, S.K. Effect of S-methylisothiourea, an inducible nitric oxide synthase inhibitor, in joint pain and pathology in surgically induced model of osteoarthritis. Connect. Tissue Res. 2014, 55, 367–377. [Google Scholar]
- Aley, K.O.; McCarter, G.; Levine, J.D. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J. Neurosci. 1998, 18, 7008–7014. [Google Scholar]
- Hancock, C.M.; Riegger-Krugh, C. Modulation of pain in osteoarthritis: The role of nitric oxide. Clin. J. Pain 2008, 24, 353–365. [Google Scholar]
- Weng, Q.; Goh, S.L.; Wu, J.; Persson, M.S.M.; Wei, J.; Sarmanova, A.; Li, X.; Hall, M.; Doherty, M.; Jiang, T.; et al. Comparative efficacy of exercise therapy and oral non-steroidal anti-inflammatory drugs and paracetamol for knee or hip osteoarthritis: A network meta-analysis of randomised controlled trials. Br. J. Sports Med 2023, bjsports-2022-105898.
- Bindu, S.; Mazumder, S.; Bandyopadhyay, U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: A current perspective. Biochem. Pharmacol. 2020, 180, 114147. [Google Scholar]
- Panchal, N.K.; Prince Sabina, E. Non-steroidal anti-inflammatory drugs (NSAIDs): A current insight into its molecular mechanism eliciting organ toxicities. Food Chem. Toxicol. 2023, 172, 113598. [Google Scholar]
- Richards, M.M.; Maxwell, J.S.; Weng, L.; Angelos, M.G.; Golzarian, J. Intra-articular treatment of knee osteoarthritis: From anti-inflammatories to products of regenerative medicine. Phys. Sportsmed. 2016, 44, 101–108. [Google Scholar]
- Richard, M.J.; Driban, J.B.; McAlindon, T.E. Pharmaceutical treatment of osteoarthritis. Osteoarthr. Cartil. 2022, S1063-4584(1022)00928-00921.
- Schjerning, A.M.; McGettigan, P.; Gislason, G. Cardiovascular effects and safety of (non-aspirin) NSAIDs. Nat. Rev. Cardiol. 2020, 17, 574–584. [Google Scholar]
- Szponder, T.; Latalski, M.; Danielewicz, A.; Krać, K.; Kozera, A.; Drzewiecka, B.; Nguyen Ngoc, D.; Dobko, D.; Wessely-Szponder, J. Osteoarthritis: Pathogenesis, Animal Models, and New Regenerative Therapies. J. Clin. Med. 2022, 12, 5. [Google Scholar]
- Yu, S.M.; Han, Y.; Kim, S.J. Simvastatin abolishes nitric oxide- and reactive oxygen species-induced cyclooxygenase-2 expression by blocking the nuclear factor κB pathway in rabbit articular chondrocytes. Cell Biol. Int. 2020, 44, 2153–2162. [Google Scholar]
- Tsai, K.L.; Huang, Y.H.; Kao, C.L.; Yang, D.M.; Lee, H.C.; Chou, H.Y.; Chen, Y.C.; Chiou, G.Y.; Chen, L.H.; Yang, Y.P.; et al. A novel mechanism of coenzyme Q10 protects against human endothelial cells from oxidative stress-induced injury by modulating NO-related pathways. J. Nutr. Biochem. 2012, 23, 458–468. [Google Scholar]
- Eitner, A.; Müller, S.; König, C.; Wilharm, A.; Raab, R.; Hofmann, G.O.; Kamradt, T.; Schaible, H.G. Inhibition of Inducible Nitric Oxide Synthase Prevents IL-1β-Induced Mitochondrial Dysfunction in Human Chondrocytes. Int. J. Mol. Sci. 2021, 22, 2477. [Google Scholar]
- Bentz, M.; Zaouter, C.; Shi, Q.; Fahmi, H.; Moldovan, F.; Fernandes, J.C.; Benderdour, M. Inhibition of inducible nitric oxide synthase prevents lipid peroxidation in osteoarthritic chondrocytes. J. Cell Biochem. 2012, 113, 2256–2267. [Google Scholar]
- Castro, R.R.; Cunha, F.Q.; Silva, F.S., Jr.; Rocha, F.A. A quantitative approach to measure joint pain in experimental osteoarthritis—Evidence of a role for nitric oxide. Osteoarthr. Cartil. 2006, 14, 769–776. [Google Scholar]
- Järvinen, K.; Vuolteenaho, K.; Nieminen, R.; Moilanen, T.; Knowles, R.G.; Moilanen, E. Selective iNOS inhibitor 1400W enhances anti-catabolic IL-10 and reduces destructive MMP-10 in OA cartilage. Survey of the effects of 1400W on inflammatory mediators produced by OA cartilage as detected by protein antibody array. Clin. Exp. Rheumatol. 2008, 26, 275–282. [Google Scholar]
- Hellio le Graverand, M.P.; Clemmer, R.S.; Redifer, P.; Brunell, R.M.; Hayes, C.W.; Brandt, K.D.; Abramson, S.B.; Manning, P.T.; Miller, C.G.; Vignon, E. A 2-year randomised, double-blind, placebo-controlled, multicentre study of oral selective iNOS inhibitor, cindunistat (SD-6010), in patients with symptomatic osteoarthritis of the knee. Ann. Rheum. Dis. 2013, 72, 187–195. [Google Scholar]
- Ma, Y.; Song, X.; Ma, T.; Li, Y.; Bai, H.; Zhang, Z.; Hu, H.; Yuan, R.; Wen, Y.; Gao, L. Aminoguanidine inhibits IL-1β-induced protein expression of iNOS and COX-2 by blocking the NF-κB signaling pathway in rat articular chondrocytes. Exp. Ther. Med. 2020, 20, 2623–2630. [Google Scholar]
- Lee, J.; Hong, Y.S.; Jeong, J.H.; Yang, E.J.; Jhun, J.Y.; Park, M.K.; Jung, Y.O.; Min, J.K.; Kim, H.Y.; Park, S.H.; et al. Coenzyme Q10 ameliorates pain and cartilage degradation in a rat model of osteoarthritis by regulating nitric oxide and inflammatory cytokines. PLoS ONE 2013, 8, e69362. [Google Scholar]
- Park, C.; Jeong, J.W.; Lee, D.S.; Yim, M.J.; Lee, J.M.; Han, M.H.; Kim, S.; Kim, H.S.; Kim, G.Y.; Park, E.K.; et al. Sargassum serratifolium Extract Attenuates Interleukin-1β-Induced Oxidative Stress and Inflammatory Response in Chondrocytes by Suppressing the Activation of NF-κB, p38 MAPK, and PI3K/Akt. Int. J. Mol. Sci. 2018, 19, 2308. [Google Scholar]
- Khan, N.M.; Haseeb, A.; Ansari, M.Y.; Devarapalli, P.; Haynie, S.; Haqqi, T.M. Wogonin, a plant derived small molecule, exerts potent anti-inflammatory and chondroprotective effects through the activation of ROS/ERK/Nrf2 signaling pathways in human Osteoarthritis chondrocytes. Free Radic. Biol. Med. 2017, 106, 288–301. [Google Scholar]
- Yan, Z.; Lin, Z.; Wu, Y.; Zhan, J.; Qi, W.; Lin, J.; Shen, J.; Xue, X.; Pan, X. The protective effect of myricitrin in osteoarthritis: An in vitro and in vivo study. Int. Immunopharmacol. 2020, 84, 106511. [Google Scholar]
- Leonidou, A.; Lepetsos, P.; Mintzas, M.; Kenanidis, E.; Macheras, G.; Tzetis, M.; Potoupnis, M.; Tsiridis, E. Inducible nitric oxide synthase as a target for osteoarthritis treatment. Expert. Opin. Ther. Targets 2018, 22, 299–318. [Google Scholar]
- Kakita, H.; Aoyama, M.; Nagaya, Y.; Asai, H.; Hussein, M.H.; Suzuki, M.; Kato, S.; Saitoh, S.; Asai, K. Diclofenac enhances proinflammatory cytokine-induced phagocytosis of cultured microglia via nitric oxide production. Toxicol. Appl. Pharmacol. 2013, 268, 99–105. [Google Scholar]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar]
- Abusarah, J.; Bentz, M.; Benabdoune, H.; Rondon, P.E.; Shi, Q.; Fernandes, J.C.; Fahmi, H.; Benderdour, M. An overview of the role of lipid peroxidation-derived 4-hydroxynonenal in osteoarthritis. Inflamm. Res. 2017, 66, 637–651. [Google Scholar]
- Kaur, J.; Dhaunsi, G.S.; Turner, R.B. Interleukin-1 and nitric oxide increase NADPH oxidase activity in human coronary artery smooth muscle cells. Med. Princ. Pract. 2004, 13, 26–29. [Google Scholar]
- Vuolteenaho, K.; Moilanen, T.; Al-Saffar, N.; Knowles, R.G.; Moilanen, E. Regulation of the nitric oxide production resulting from the glucocorticoid-insensitive expression of iNOS in human osteoarthritic cartilage. Osteoarthr. Cartil. 2001, 9, 597–605. [Google Scholar]
- Hsu, C.C.; Lin, C.L.; Jou, I.M.; Wang, P.H.; Lee, J.S. The protective role of nitric oxide-dependent innate immunosuppression in the early stage of cartilage damage in rats: Role of nitric oxide in cartilage damage. Bone Jt. Res. 2017, 6, 253–258. [Google Scholar]
- Zhong, L.; Schivo, S.; Huang, X.; Leijten, J.; Karperien, M.; Post, J.N. Nitric Oxide Mediates Crosstalk between Interleukin 1β and WNT Signaling in Primary Human Chondrocytes by Reducing DKK1 and FRZB Expression. Int. J. Mol. Sci. 2017, 18, 2491. [Google Scholar]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of Pharmacological Treatments with Long-term Pain Control in Patients With Knee Osteoarthritis: A Systematic Review and Meta-analysis. JAMA 2018, 320, 2564–2579. [Google Scholar]
- Di Naso, F.C.; Rodrigues, G.; Simões Dias, A.; Porawski, M.; Fillmann, H.; Marroni, N.P. Hepatic nitrosative stress in experimental diabetes. J. Diabetes Complicat. 2012, 26, 378–381. [Google Scholar]
- Wang, J.; Kalhor, A.; Lu, S.; Crawford, R.; Ni, J.D.; Xiao, Y. iNOS expression and osteocyte apoptosis in idiopathic, non-traumatic osteonecrosis. Acta Orthop. 2015, 86, 134–141. [Google Scholar]
- Lee, W.; Yang, S.; Lee, C.; Park, E.K.; Kim, K.M.; Ku, S.K.; Bae, J.S. Aloin reduces inflammatory gene iNOS via inhibition activity and p-STAT-1 and NF-κB. Food Chem. Toxicol. 2019, 126, 67–71. [Google Scholar]
- Maghsoudi, H.; Hallajzadeh, J.; Rezaeipour, M. Evaluation of the effect of polyphenol of escin compared with ibuprofen and dexamethasone in synoviocyte model for osteoarthritis: An in vitro study. Clin. Rheumatol. 2018, 37, 2471–2478. [Google Scholar]
- Impellizzeri, D.; Siracusa, R.; Cordaro, M.; Peritore, A.F.; Gugliandolo, E.; D’Amico, R.; Fusco, R.; Crupi, R.; Rizzarelli, E.; Cuzzocrea, S.; et al. Protective effect of a new hyaluronic acid-carnosine conjugate on the modulation of the inflammatory response in mice subjected to collagen-induced arthritis. Biomed. Pharmacother. 2020, 125, 110023. [Google Scholar]
- Li, X.; Guo, Y.; Huang, S.; He, M.; Liu, Q.; Chen, W.; Liu, M.; Xu, D.; He, P. Coenzyme Q10 Prevents the Interleukin-1 Beta Induced Inflammatory Response via Inhibition of MAPK Signaling Pathways in Rat Articular Chondrocytes. Drug Dev. Res. 2017, 78, 403–410. [Google Scholar]
- Lim, S.; Choi, A.H.; Kwon, M.; Joung, E.J.; Shin, T.; Lee, S.G.; Kim, N.G.; Kim, H.R. Evaluation of antioxidant activities of various solvent extract from Sargassum serratifolium and its major antioxidant components. Food Chem. 2019, 278, 178–184. [Google Scholar]
- Joung, E.J.; Kwon, M.; Gwon, W.G.; Cao, L.; Lee, S.G.; Utsuki, T.; Wakamatsu, N.; Kim, J.I.; Kim, H.R. Meroterpenoid-Rich Fraction of the Ethanol Extract of Sargassum Serratifolium Suppresses Collagen-Induced Rheumatoid Arthritis in DBA/1J Mice Via Inhibition of Nuclear Factor κB Activation. Mol. Nutr. Food Res. 2020, 64, e1900373. [Google Scholar]
- Park, J.S.; Lee, H.J.; Lee, D.Y.; Jo, H.S.; Jeong, J.H.; Kim, D.H.; Nam, D.C.; Lee, C.J.; Hwang, S.C. Chondroprotective Effects of Wogonin in Experimental Models of Osteoarthritis in vitro and in vivo. Biomol. Ther. 2015, 23, 442–448. [Google Scholar]
- Abusarah, J.; Benabdoune, H.; Shi, Q.; Lussier, B.; Martel-Pelletier, J.; Malo, M.; Fernandes, J.C.; de Souza, F.P.; Fahmi, H.; Benderdour, M. Elucidating the Role of Protandim and 6-Gingerol in Protection Against Osteoarthritis. J. Cell Biochem. 2017, 118, 1003–1013. [Google Scholar]
- Lei, Y. Myricitrin decreases traumatic injury of the spinal cord and exhibits antioxidant and anti‑inflammatory activities in a rat model via inhibition of COX‑2, TGF‑β1, p53 and elevation of Bcl‑2/Bax signaling pathway. Mol Med Rep 2017, 16, 7699–7705. [Google Scholar]
- Wang, X.D.; Wan, X.C.; Liu, A.F.; Li, R.; Wei, Q. Effects of umbilical cord mesenchymal stem cells loaded with graphene oxide granular lubrication on cytokine levels in animal models of knee osteoarthritis. Int. Orthop. 2021, 45, 381–390. [Google Scholar]
- Shah, S.; Esdaille, C.J.; Bhattacharjee, M.; Kan, H.M.; Laurencin, C.T. The synthetic artificial stem cell (SASC): Shifting the paradigm of cell therapy in regenerative engineering. Proc. Natl. Acad. Sci. USA 2022, 119, e2116865118. [Google Scholar]
- Kandiah, K.; Duraisamy, N.; Amirthalingam, V.; Ramasamy, B. Scavenging free radicals and soaring osteoinduction by extra cellular matrix protein-based nanocomposites on degenerative bone treatments. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 77, 1189–1195. [Google Scholar]
- Zhang, M.; Hu, W.; Cai, C.; Wu, Y.; Li, J.; Dong, S. Advanced application of stimuli-responsive drug delivery system for inflammatory arthritis treatment. Mater. Today Bio 2022, 14, 100223. [Google Scholar]
- Yuan, L.J.; Ueng, S.W.; Lin, S.S.; Yeh, W.L.; Yang, C.Y.; Lin, P.Y. Attenuation of apoptosis and enhancement of proteoglycan synthesis in rabbit cartilage defects by hyperbaric oxygen treatment are related to the suppression of nitric oxide production. J. Orthop. Res. 2004, 22, 1126–1134. [Google Scholar]
- Tilwani, R.K.; Vessillier, S.; Pingguan-Murphy, B.; Lee, D.A.; Bader, D.L.; Chowdhury, T.T. Oxygen tension modulates the effects of TNFα in compressed chondrocytes. Inflamm. Res. 2017, 66, 49–58. [Google Scholar]
- Yuan, L.J.; Niu, C.C.; Lin, S.S.; Yang, C.Y.; Chan, Y.S.; Chen, W.J.; Ueng, S.W. Effects of low-intensity pulsed ultrasound and hyperbaric oxygen on human osteoarthritic chondrocytes. J. Orthop. Surg. Res. 2014, 9, 5. [Google Scholar]
- Huang, T.H.; Lu, Y.C.; Kao, C.T. Low-level diode laser therapy reduces lipopolysaccharide (LPS)-induced bone cell inflammation. Lasers Med. Sci. 2012, 27, 621–627. [Google Scholar]
- Zhao, Z.; Ji, H.; Jing, R.; Liu, C.; Wang, M.; Zhai, L.; Bai, X.; Xing, G. Extracorporeal shock-wave therapy reduces progression of knee osteoarthritis in rabbits by reducing nitric oxide level and chondrocyte apoptosis. Arch. Orthop. Trauma Surg. 2012, 132, 1547–1553. [Google Scholar]
- Melikian, N.; Seddon, M.D.; Casadei, B.; Chowienczyk, P.J.; Shah, A.M. Neuronal nitric oxide synthase and human vascular regulation. Trends. Cardiovasc. Med. 2009, 19, 256–262. [Google Scholar]
- Felson, D.T.; Kim, Y.J. The futility of current approaches to chondroprotection. Arthritis Rheum. 2007, 56, 1378–1383. [Google Scholar]
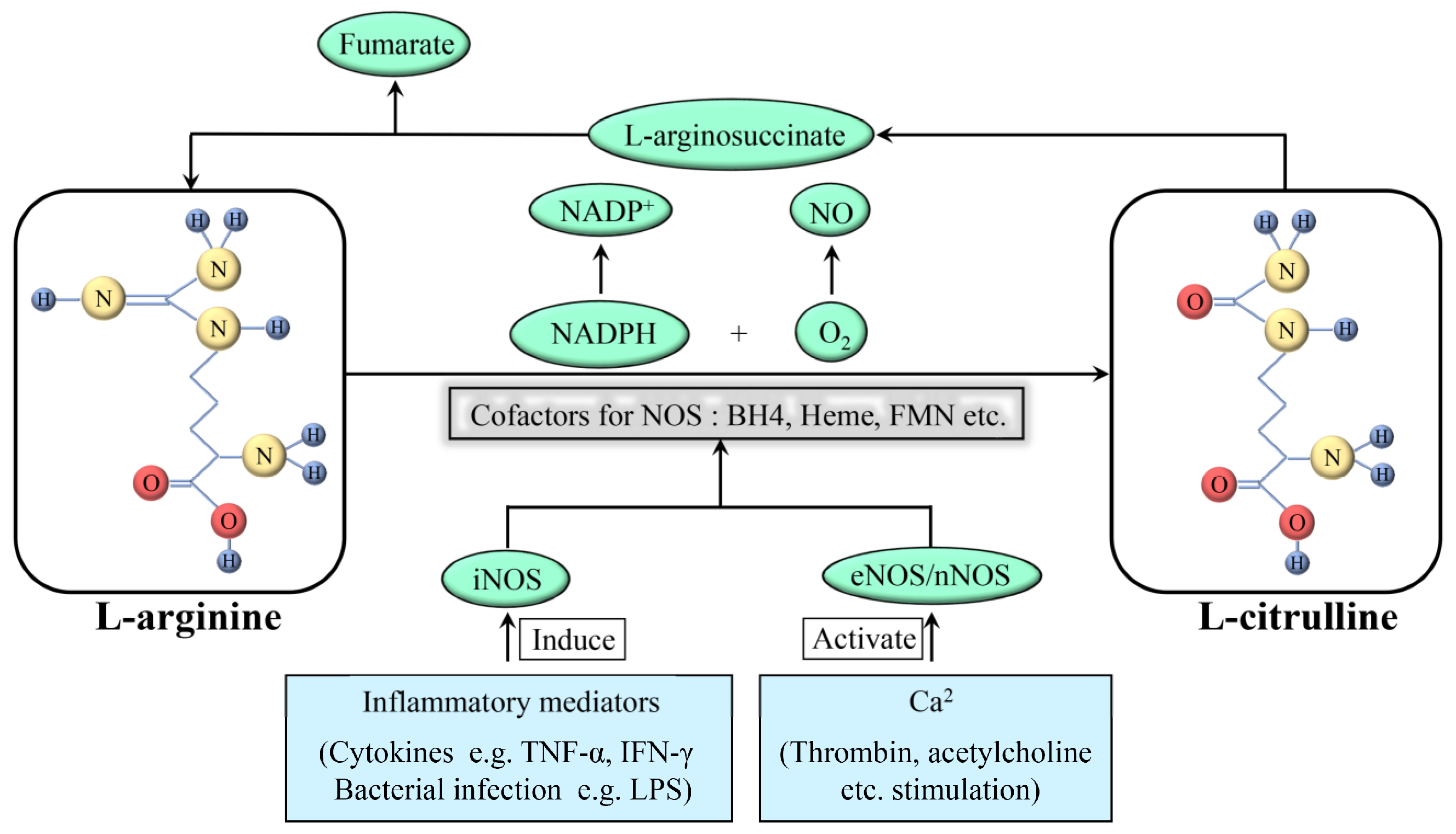
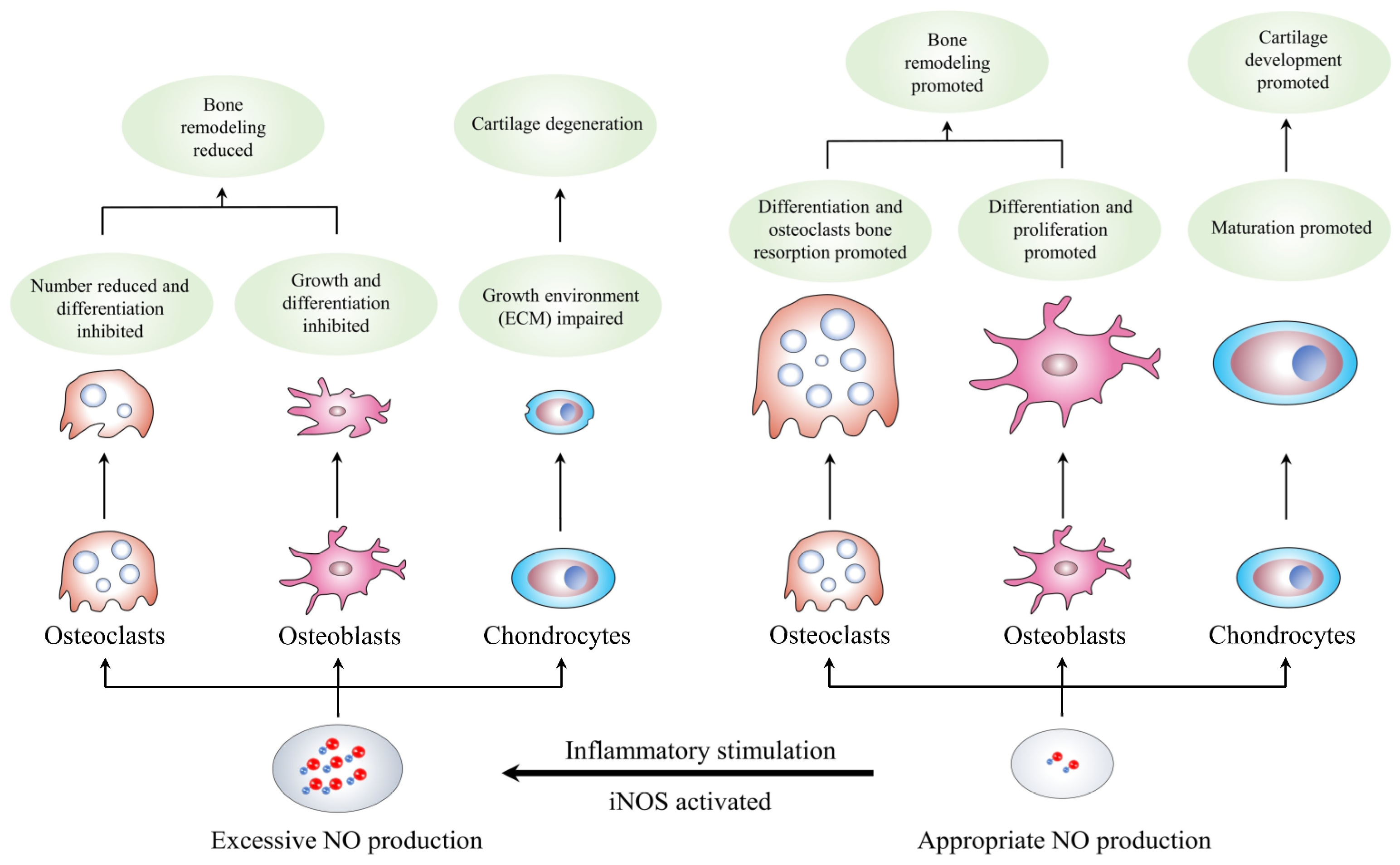


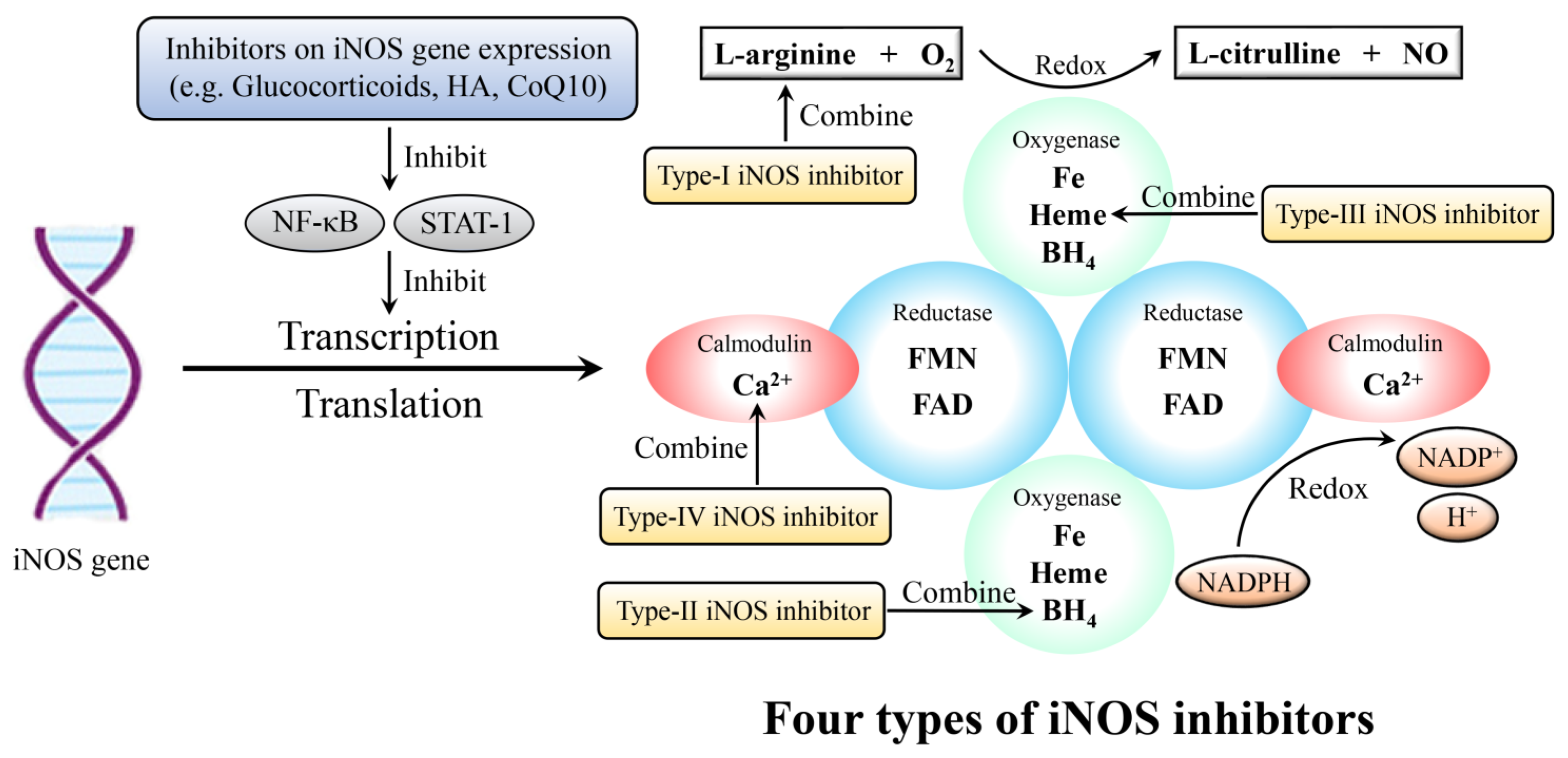
| Targeted-Cell | Type of Study | NO Concentration | Related Mechanisms | Effect of NO | Reference |
|---|---|---|---|---|---|
| Osteoclasts | In vivo and in vitro | High | Reduces the number of osteoclasts and inhibits its spread | Inhibits mature osteoclasts bone resorption | Kalyanaraman et al., 2017 [20] |
| Osteoclasts | In vitro | High | Induced by inhibited cGMP-degrading activity of PDE | Inhibits precellular osteoclasts differentiation | Amano et al., 2019 [21] |
| Osteoclasts | In vitro | Low | Induced by RANKL and produces downstream molecular 8-nitro-cGMP | Promotes osteoclasts differentiation | Kaneko et al., 2018 [22] |
| Osteoclasts | In vitro | Low | Stimulated by cytokines and other mediators such as PG | Promotes osteoclasts bone resorption | Mentaverri et al., 2003 [23] |
| Osteoblasts | In vitro | Low | Induces osteoblast differentiation factor (Cbfa1) expression | Promotes osteoblasts differentiation | Gloria et al., 2020 [24] |
| Osteoblasts | In vivo and in vitro | Low | Activates Src, Erk-1/2 and Akt signaling pathway through sGC and PKG2 | Promotes osteoblasts proliferation and anti-apoptotic effects | Cepeda et al., 2020 [25] Ramdani et al., 2018 [26] |
| Osteoblasts | In vivo and in vitro | Low | Stimulated by estrogen | Promotes osteoblasts growth and development | Gerbarg et al., 2016 [27] Crescitelli et al., 2019 [28] |
| Osteoblasts | In vitro | Low | Responses to mechanical stimulation | Promotes osteoblasts proliferation and survival | Wittkowske et al., 2016 [29] Maycas et al., 2017 [30] |
| Chondrocytes | In vitro | High | Induces caspase expression upregulation | Induces chondrocytes apoptosis | Poderoso et al., 2019 [31] Kamm et al., 2019 [32] |
| Chondrocytes | In vivo and in vitro | High | Stimulated by inflammatory mediators | Affects numerous biomolecular processes in chondrocytes | Wojdasiewicz et al., 2014 [33] |
| Chondrocytes | In vitro | Low | Stimulated chondrocytes hypertrophy and increased the expression of alkaline phosphatase and type X collagen | Promotes chondrocytes maturation | Teixeira et al., 2005 [34] Drissi et al., 2005 [35] |
| Author | Type of Study | Model | Agent | Dose | Method | Outcome | Conclusion |
|---|---|---|---|---|---|---|---|
| Yu et al., 2020 [100] | In vitro | New Zealand White rabbits | N-Monomethyl-L-arginine (L-NMMA) | Not mentioned | L-NMMA on inhibiting NO to accelerate the influence of simvastatin | L-NMMA inhibited NO and COX-2 production and NF-κB activation | L-NMMA enhanced the blocking effect of simvastatin on NF-κB activation by inhibiting NO production |
| Lee et al., 2012 [101] | In vitro | New Zealand White rabbits | L-NMMA | 0.5 mM/ 24 h | L-NMMA on chondrocyte apoptosis | L-NMMA inhibited NO production and NF-kB binding activity | L-NMMA blocks PCB-initiated apoptosis effect |
| Eitner et al., 2021 [102] | In vitro | Human end-stage knee OA chondrocytes | N-Iminoethyl-L-lysine (L-NIL) | 1, 10, or 20 µM/48 h | L-NIL on preventing release of NO, IL-6, PGE2, and iNOS | L-NIL prevented NO release and mitochondrial dysfunction | L-NIL improves the impairment of mitochondrial respiration |
| Bentz et al., 2012 [103] | In vitro | Human OA chondrocytes | L-NIL | 0-20 µM/ 24 h | L-NIL on chondrocyte oxidative stress, apoptosis, inflammation, and catabolism | L-NIL stifled NO release, iNOS activity, nitrated proteins, and HNE generation and restored both HNE and GSTA4-4 levels | L-NIL prevents LPO process and ROS production and attenuates cell death, inflammation, and catabolism |
| Castro et al., 2006 [104] | In vivo | OA rats induced by ligament transection surgery | NG-nitro-L-arginine methyl ester (L-NAME) | 30 mg/kg/bid | L-NAME on joint pain, cell influx, nitrite levels and iNOS expression | L-NAME reduced the time of rats’ right hind paw fails to touch the surface while walking | Prophylactic L-NAME can reduce joint pain |
| Järvinen et al., 2008 [105] | In vitro | Cartilage tissue from OA patients | 1400W | 1 mM/120 h | 1400W on production of inflammatory mediators | Treatment with 1400W enhanced the production of anti-catabolic IL-10 and reduced MMP-10 | The inhibiting effects of 1400W may point to its anti-inflammatory mechanisms for OA |
| Graverand et al., 2013 [106] | Clinical human study | Kellgren and Lawrence Grade (KLG) 2 or 3 knee OA patients | SD-6010 | 50 or 200 mg/day | A 2-year multicenter RCT of SD-6010 in patients with symptomatic knee OA | In KLG2 patients, JSN after 48 weeks was lower with SD-6010 50 mg/day versus placebo. No improvement in KLG3 patients | SD-6010 may become effective only in “mild to moderate” OA patients, but it cannot slow the rate of JSN |
| Ma et al., 2020 [107] | In vitro | IL-1β induced Sprague-Dawley (SD) rat chondrocytes | Aminoguanidine (AG) | 0.3, 1 or 3 mM/24,48 or 72 h | AG on COX-2, iNOS, phosphorylated (p)-p65 and NF-κB translocation | AG downregulated iNOS and COX-2 expression by blocking the NF-κB signaling pathway | AG may protect chondrocytes and serve as a potential therapeutic for OA |
| Lee et al., 2013 [108] | In vivo | MIA-induced Wistar rat OA model | Coenzyme Q10 (CoQ10) | 100 mg/kg/qd | CoQ10 on inflammatory mediators production and cartilage degradation | CoQ10 had anti-nociceptive effect and attenuated cartilage degeneration in rat OA model | CoQ10 exerts a therapeutic effect of OA by inhibiting inflammation |
| Park et al., 2018 [109] | In vitro | SW1353 cells and SD rats | Ethanol extract of sargassum serratifolium (EESS) | Extract that hard to measure precise concentrations | EESS on inflammatory mediators production and signaling pathways activation | EESS blocked ROS generation, attenuated NO production, and inhibited MAPK and PI3K/Akt pathways | EESS may have the potential chondroprotection in the prevention and treatment of OA |
| Khan et al., 2017 [110] | In vitro | IL-1β-stimulated human OA chondrocytes and cartilage explants | Wogonin | 10–50 µM/24 h | Wogonin on inflammatory mediators production and MMPs, s-GAG and COL2A1 levels | Wogonin mediated Nrf2/ARE pathways, inhibited matrix degradation and suppressed the expression and production of COX-2 and iNOS | Wogonin exert chondro- and cartilage protection through the suppression of key molecular events |
| Yan et al., 2020 [111] | In vivo and in vitro | C57BL/6 wild-type (WT) rats | Myricitrin (Myr) | 0-100µM/24h in vitro, dose in vivo not mentioned | Myr on inflammatory mediators production and signaling pathway | Myr suppressed the NF-κB and MAPK signaling pathways and decreased OARSI scores in OA rat models. | Myr may have therapeutic potential in the treatment of OA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Ji, P.; Shang, X.; Zhou, Y. Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target. Molecules 2023, 28, 1683. https://doi.org/10.3390/molecules28041683
Jiang H, Ji P, Shang X, Zhou Y. Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target. Molecules. 2023; 28(4):1683. https://doi.org/10.3390/molecules28041683
Chicago/Turabian StyleJiang, Huanyu, Piyao Ji, Xiaobin Shang, and Yan Zhou. 2023. "Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target" Molecules 28, no. 4: 1683. https://doi.org/10.3390/molecules28041683
APA StyleJiang, H., Ji, P., Shang, X., & Zhou, Y. (2023). Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target. Molecules, 28(4), 1683. https://doi.org/10.3390/molecules28041683