
Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design


Abstract
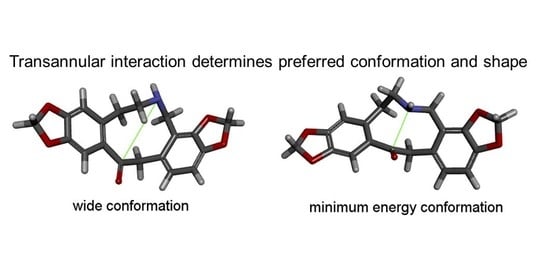
1. Introduction
2. Results and Discussion
2.1. Transannular Interactions in Protopine Analogues
2.2. Conformational Preferencing and Resultant Molecular Shapes in Imino and Azo Group Embedded Rings
2.3. Shape and Electrostatic Potential Comparisons
2.4. Drug Design Implications
3. Materials and Methods
3.1. Software
3.2. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.K.; Unsworth, W.P. A Happy Medium: The Synthesis of Medicinally Important Medium-Sized Rings via Ring Expansion. Chem. Sci. 2020, 11, 2876–2881. [Google Scholar] [PubMed]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [PubMed]
- Basu, A.; Kumar, G.S. Nucleic Acids Binding Strategies of Small Molecules: Lessons from Alkaloids. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1995–2016. [Google Scholar]
- Nakonieczna, S.; Grabarska, A.; Gawel, K.; Wroblewska-Luczka, P.; Czerwonka, A.; Stepulak, A.; Kukula-Koch, W. Isoquinolone Alkaloids from Coptis chinensis Franch: Focus on Coptisine as a Potential Therapeutic Candidate against Gastric Cancer Cells. Int. J. Mol. Sci. 2022, 23, 103330. [Google Scholar]
- Mari, G.; De Crescentini, L.; Benedetti, S.; Palma, F.; Santeusanio, S.; Mantellini, F. Synthesis of New Dihydroberberine and Tetrahydroberberine analogues and Evaluation of their Antiproliferative Activity on NCI-H1975 Cells. Beilstein J. Org. Chem. 2020, 16, 1606–1616. [Google Scholar]
- Fu, L.; Mou, J.; Deng, Y.; Ren, X. Structural Modifications of Berberine and their Binding Effects towards Polymorphic Deoxyribonucleic Acid Structures: A Review. Front. Pharmacol. 2022, 13, 940282. [Google Scholar] [CrossRef]
- Milani, G.; Cavalluzzi, M.M.; Solidoro, R.; Salvagno, L.; Quintieri, L.; Di Somma, A.; Rosato, A.; Corbo, F.; Franchini, C.; Duilio, A.; et al. Molecular Simplification of Natural Products: Synthesis, Antibacterial Activity, and Molecular Docking Studies of Berberine Open Models. Biomedicines 2021, 9, 452. [Google Scholar]
- Bremner, J. Multiple Action-Based Design Approaches to Antibacterials; Springer Nature: Singapore, Singapore, 2021. [Google Scholar]
- Huang, W.; Kong, L.; Cao, Y.; Yan, L. Identification and Quantification, Metabolism and Pharmacokinetics, Pharmacological Activities, and Botanical Preparations of Protopine: A Review. Molecules 2022, 27, 215. [Google Scholar] [CrossRef]
- Sreenivasmurthy, S.G.; Iyaswamy, A.; Krishnamoorthi, S.; Senapati, S.; Malampati, S.; Zhu, Z.; Su, C.-F.; Liu, J.; Guan, X.-J.; Tong, B.C.-K.; et al. Protopine Promotes the Proteasomal Degradation of Pathological Tau in Alzheimer’s Disease Models via HDAC6 Inhibition. Phytomedicine 2022, 96, 153887. [Google Scholar] [CrossRef]
- Nie, C.; Wang, B.; Wang, B.; Lv, N.; Yu, R.; Zhang, E. Protopine Triggers Apoptosis via the Intrinsic Pathway and Regulation of ROS/PI3K/Akt Signalling Pathway in Liver Carcinoma. Cancer. Cell Int. 2021, 21, 396. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Fu, Y.; Liu, L.; Lin, K.; Zhao, X.; Zhang, Y.; Chen, X.; Cai, Z.; Huang, Y.; Li, Y. Effect of α-Allocryptopine on Delayed Afterdepolarizations and Triggered Activities in Mice Cardiomyocytes Treated with Isoproterenol. Evid.-Based Complement. Altern. Med. 2015, 2015, 634172. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Liu, Y.-Q.; Xu, B.; Gao, J.-L.; Fu, Y.-C.; Chen, Y.; Xue, Q.; Li, Y. Allocryptopine and Benzyltetrahydropalmatine Block hERG Potassium Channels Expressed in HEK293 Cells. Acta Pharmacol. Sin. 2013, 34, 847–858. [Google Scholar] [PubMed]
- Xiong, R.-G.; Huang, S.-Y.; Wu, S.-X.; Zhou, D.-D.; Yang, Z.-J.; Saimaiti, A.; Zhao, C.-N.; Shang, A.; Zhang, Y.-J.; Gan, R.-Y.; et al. Anticancer Effects and Mechanisms of Berberine from Medicinal Herbs: An Update Review. Molecules 2022, 27, 4523. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Yu, P.; Leng, L.; Luo, L.; Liu, J.; Li, S.; Lai, X.; Luan, F.; Meng, X. Berberine: A Review of its Pharmacokinetics Properties and Therapeutic Potentials in Diverse Vascular Diseases. Front. Pharmacol. 2021, 12, 762654. [Google Scholar] [PubMed]
- Wu, J.; Luo, Y.; Deng, D.; Su, S.; Li, S.; Xiang, L.; Hu, Y.; Wang, P.; Meng, X. Coptisine from Coptis chinensis Exerts Diverse Beneficial Properties: A Concise Review. J. Cell Mol. Med. 2019, 23, 7946–7960. [Google Scholar] [CrossRef]
- Panwaria, P.; Das, A. Understanding the n—π* Non-Covalent Interaction Using Different Experimental and Theoretical Approaches. Phys. Chem. Chem. Phys. 2022, 24, 22371–22389. [Google Scholar]
- Takahashi, H.; Iguchi, M.; Onda, M. Utilization of Protopine and Related Alkaloids. XVII. Spectroscopic Studies on the Ten-Membered Ring Conformations of Protopine and α-Allocryptopine. Chem. Pharm. Bull. 1985, 33, 4475–4782. [Google Scholar]
- Bremner, J.; Griffith, R. Density Functional Theory Assessment of Transannular N…Y Interactions in Some Medium-Sized Heterocycles. Comput. Theor. Chem. 2022, 1208, 113543. [Google Scholar]
- Meanwell, N.A. Synopsis of Some Recent Tactical Application of Bioisosteres in Drug Design. J. Med. Chem. 2011, 54, 2529–2591. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Nonvovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Clark, T.; Murray, J.S.; Lane, P.; Politzer, P. Why are Dimethyl Sulfoxide and Dimethyl Sulfone such Good Solvents? J. Mol. Model. 2018, 14, 689–697. [Google Scholar] [CrossRef]
- Israelov, I.A.; Yunusov, M.S.; Yunusov, S.Y. Structure of Fumaridine and Fumaramine. Khimiya Prir. Soedin. 1970, 6, 588–591. [Google Scholar]
- Shamma, M.; Moniot, J.L. Revised Structures for Fumaridine and Fumaramine. J. Chem. Soc. Chem. Commun. 1975, 89–90. [Google Scholar] [CrossRef]
- Østby, K.-A.; Gundersen, G.; Haaland, A.; Nöth, H. Dative Sigma- and Pi-bonding in Boron-Nitrogen Compounds: Molecular Structures of (CH3)2NB(CH3)N(CH3)2 and [(CH3)2B]2NN(CH3)2 Determined by Gas Electron Diffraction and Quantum Chemical Calculations. Dalton Trans. 2005, 13, 2284–2291. [Google Scholar] [CrossRef] [PubMed]
- Wangchuk, P.; Bremner, J.B.; Samten; Rattanajak, R.; Kamchonwongpaisan, S. Antiplasmodial Agents from the Butanese Medicinal Plant Corydalis calliantha. Phytother. Res. 2010, 24, 481–485. [Google Scholar]
- Lei, Q.; Liu, H.; Peng, Y.; Xiao, P. In Silico Target Fishing and Pharmacological Profiling for the Isoquinoline Alkaloids of Macleaya cordata (Bo Luo Hui). Chin. Med. 2015, 10, 37. [Google Scholar]
- Fu, H.; Yang, P.; Hai, J.; Li., H. Utilization of Circular Dichroism and Electrospray Ionization Mass Spectrometry to Understand the Formation and Conversion of G-quadruplex DNA at the Human c-myb Proto-Oncogene. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2018, 203, 70–76. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev. 1997, 23, 4–25. [Google Scholar]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties that Influence the Oral Bioavailability of Drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Johnson, B.G.; Gill, P.M.W.; People, J.A.; Fox, D.J. Computing Molecular Electrostatic Potentials with the PRISM algorithm. Chem. Phys. Lett. 1993, 206, 239–246. [Google Scholar] [CrossRef]
- Weinhold, F. Natural Bond Critical Point Analysis: Quantitative Relationships between NBO-based and QTAIM-based Topological Descriptors of Chemical Bonding. J. Comp. Chem. 2012, 33, 2440–2449. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Area as a Sum of Fragment Based Contributions and its Applications to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Y,R | Charge N | Charge Y1 1 | Charge Y2 2 | Density 3 |
|---|---|---|---|---|
| CO,CH3 | 0.104 | 0.835 | −0.579 | 0.023 |
| CO,H | −0.396 | 0.790 | −0.555 | 0.019 |
| CO,OH | 0.168 | 0.870 | −0.534 | 0.018 |
| BOH,CH3 | 0.348 | 0.910 | −0.891 | 0.109 |
| BOH,H | −0.240 | 0.805 | −0.806 | 0.098 |
| BOH,OH | −0.059 | 0.834 | −0.730 | 0.073 |
| CS,CH3 | −0.042 | 0.248 | −0.293 | 0.021 |
| CS,H | −0.345 | 0.211 | −0.280 | 0.019 |
| CS,OH | 0.053 | 0.250 | −0.229 | 0.019 |
| SO,CH3 | −0.027 | 0.366 | −0.523 | 0.022 |
| SO,H | −0.416 | 0.297 | −0.494 | 0.021 |
| SO,OH | −0.041 | 0.475 | −0.540 | 0.022 |
| S,CH3 | 0.132 | −0.202 | NA | 0.015 |
| S,H | −0.265 | −0.220 | NA | 0.014 |
| S,OH | 0.180 | −0.135 | NA | 0.014 |
| O,CH3 | 0.019 | −0.254 | NA | 0.014 |
| O,H | −0.521 | −0.281 | NA | 0.014 |
| O,OH | 0.156 | −0.235 | NA | 0.015 |
| CH2,CH3 | 0.204 | 0.287 | −0.010, −0.103 | 0.019 (N…H) |
| CH2,H | −0.321 | 0.314 | −0.023, −0.092 | 0.018 (N…H) |
| CH2,OH | 0.233 | 0.167 | −0.009, −0.009 | 0.019 (N…H) |
| Y 1 | Charge N | Charge Y1 2 | Charge Y2 3 | Density 4 | Shape |
|---|---|---|---|---|---|
| CO trans min | −0.420 | 0.712 | −0.529 | 0.016 | U with twist |
| CO trans | −0.321 | 0.787 | −0.534 | 0.023 | flat with step |
| CO trans2 | −0.430 | 0.764 | −0.519 | 0.016 | boomerang |
| CO cis | −0.674 | 0.820 | −0.512 | none | boomerang |
| CO cis pinched | −0.598 | 0.607 | −0.494 | none | U with twist |
| BOH trans | −0.253 | 0.585 | −0.746 | 0.112 | flat with step |
| BOH cis | −0.648 | 0.600 | −0.689 | none | U with twist |
| CS trans | −0.251 | 0.271 | −0.275 | 0.022 | flat with step |
| CS cis | −0.522 | 0.165 | −0.193 | none | U with twist |
| SO cis | −0.564 | 0.137 | −0.408 | none | U with twist |
| SO trans | −0.265 | 0.326 | −0.511 | 0.031 | flat with step |
| S trans | −0.330 | −0.164 | NA | 0.015 | boomerang |
| O trans | −0.409 | −0.225 | NA | 0.017 | flat with step |
| CH2 trans | −0.291 | 0.237 | −0.035/−0.013 | 0.019 (N…H) | boomerang |
| CF2 trans | −0.250 | 0.747 | −0.267/−0.290 | 0.019 0.014 (N…F) | boomerang |
| CCH2 trans | −0.279 | 0.361 | −0.628 | 0.017 | flat with step |
| CCF2 trans | −0.280 | −0.012 | 0.279 | 0.018 | flat with step |
| Y 1 | Charge N | Charge Y1 2 | Charge Y2 3 | Density 4 | Shape |
|---|---|---|---|---|---|
| CO trans | −0.009 | 0.584 | −0.497 | 0.019 (N33) | boomerang |
| CO trans2 | −0.154 | 0.729 | −0.477 | 0.016 (N33) | boomerang |
| CO trans min | −0.089 | 0.534 | −0.481 | 0.016 (N33) | U with twist |
| CO cis | −0187 | 0.553 | −0.486 | none | U with twist |
| CO cis2 | −0.145 | 0.635 | −0.470 | none | U with twist |
| BOH trans | 0.225 | 0.896 | −0.878 | 0.108 (N32) | flat with step |
| BOH cis | −0.158 | 0.617 | −0.656 | none | U with twist |
| CS trans | 0.039 | 0.081 | −0.239 | 0.018 (N33) | flat with twist |
| CS cis | −0.192 | 0.086 | −0.150 | none | U with twist |
| SO trans | −0.045 | 0.246 | −0.469 | 0.023 | flat with step |
| SO cis | −0.185 | 0.214 | −0.467 | none | U with twist |
| Compound | Charge N | Charge C | Charge H 1 | Density 2 | Shape |
|---|---|---|---|---|---|
| Coptisine | 0.262 | 0.230 | NA | 0.288 | Flat |
| Dihydrocoptisine | −0.204 | 0.334 | NA | 0.296 | Flat with twist |
| Tetrahydrocoptisine | −0.405 | 0.389 | 0.033 | 0.265 | Flat with step |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffith, R.; Bremner, J.B. Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design. Molecules 2023, 28, 1631. https://doi.org/10.3390/molecules28041631
Griffith R, Bremner JB. Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design. Molecules. 2023; 28(4):1631. https://doi.org/10.3390/molecules28041631
Chicago/Turabian StyleGriffith, Renate, and John B. Bremner. 2023. "Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design" Molecules 28, no. 4: 1631. https://doi.org/10.3390/molecules28041631
APA StyleGriffith, R., & Bremner, J. B. (2023). Computational Evaluation of N-Based Transannular Interactions in Some Model Fused Medium-Sized Heterocyclic Systems and Implications for Drug Design. Molecules, 28(4), 1631. https://doi.org/10.3390/molecules28041631