
DIA-MS Based Proteomics Combined with RNA-Seq Data to Unveil the Mitochondrial Dysfunction in Human Glioblastoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
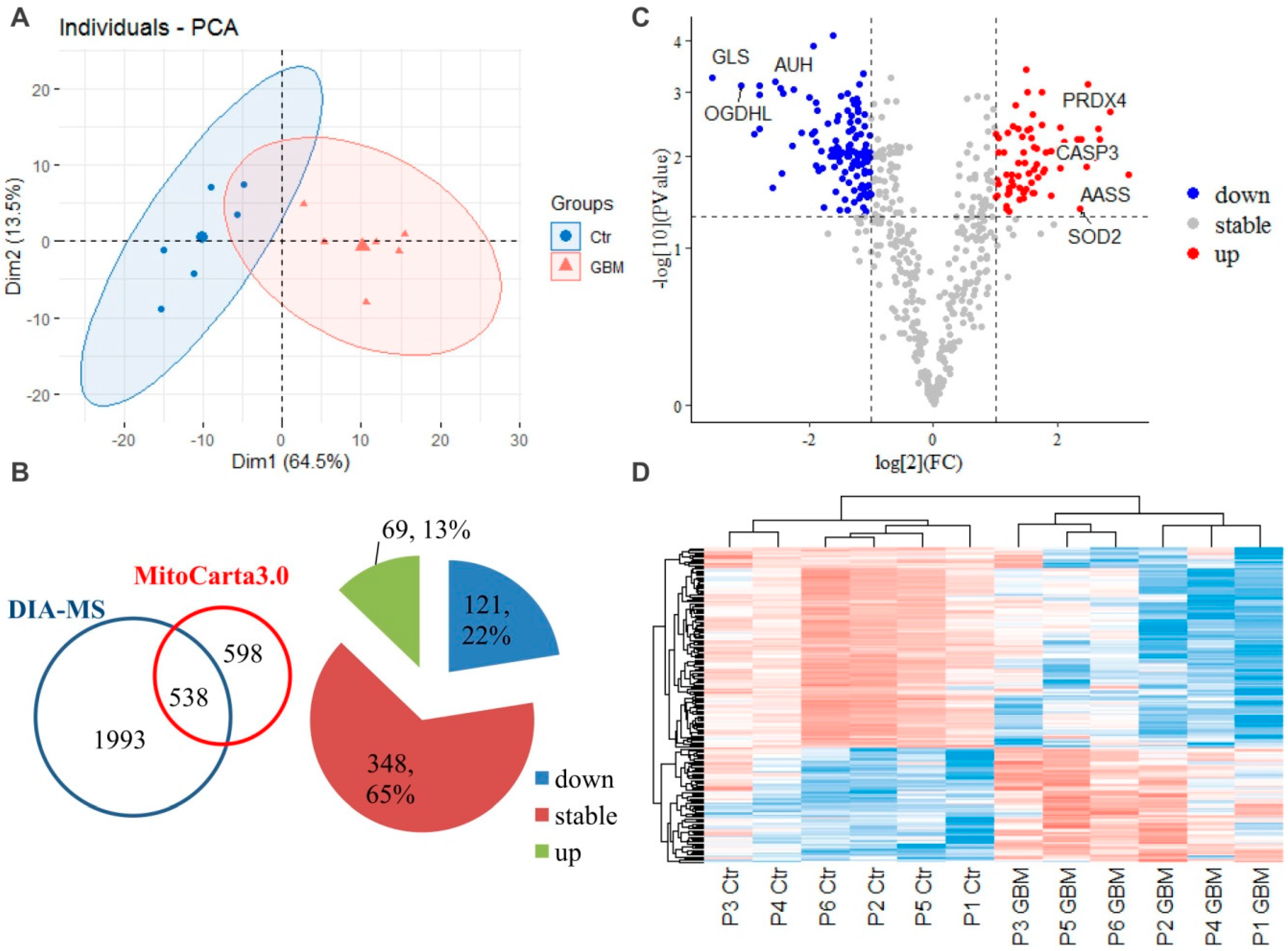
2.1. Mitochondrial Proteome Alterations
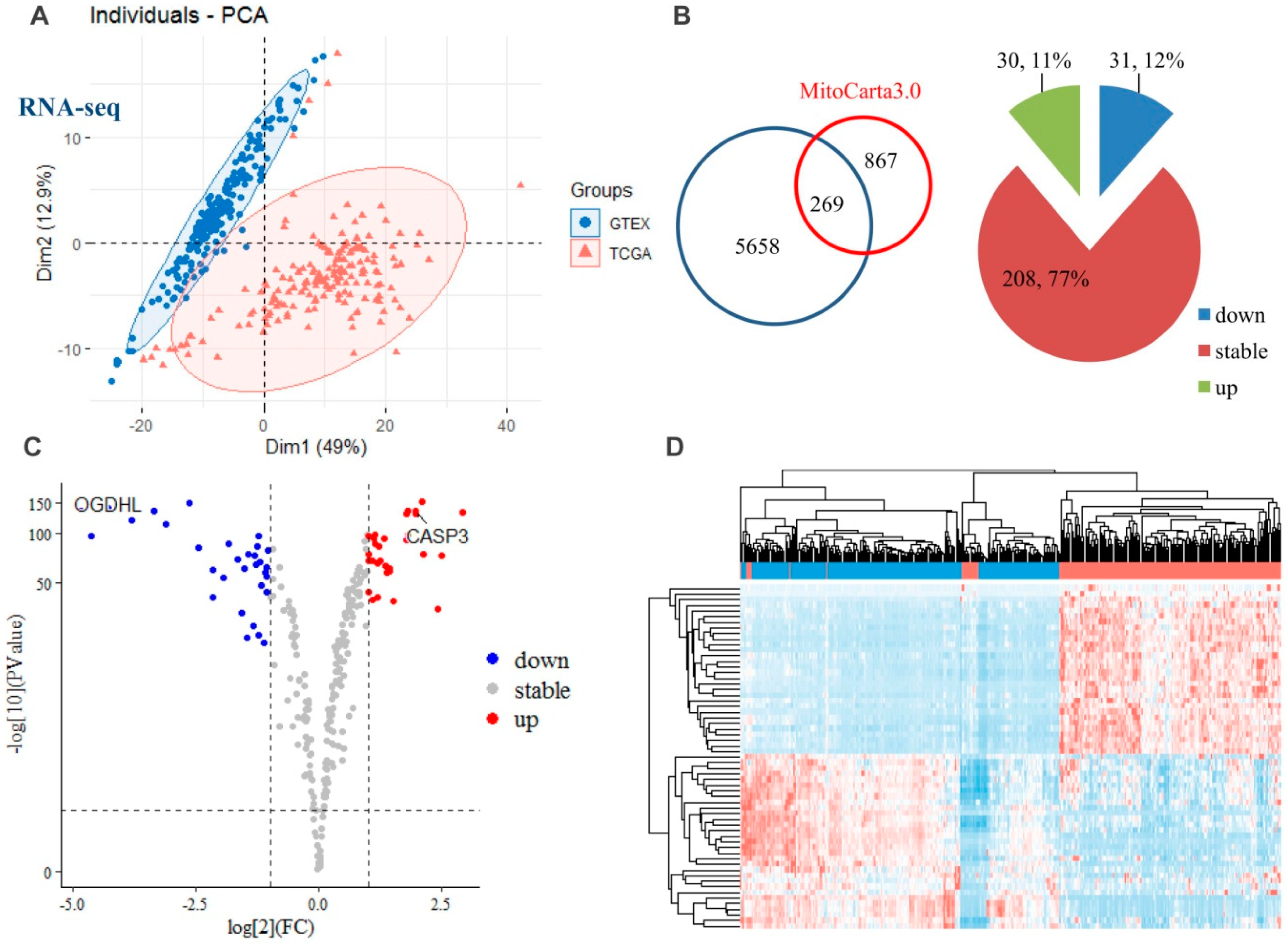
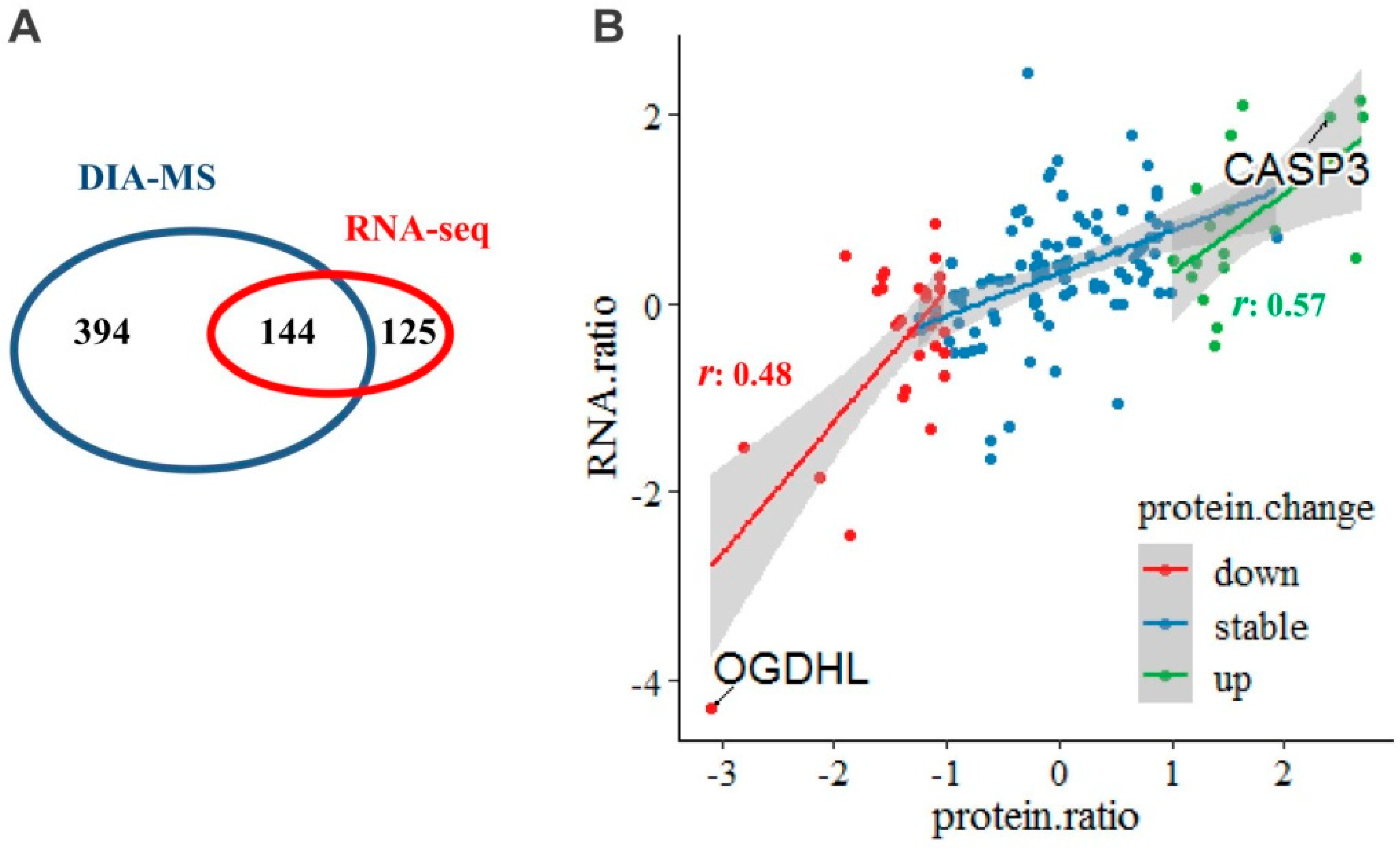
2.2. Transcriptomic Alterations of the Mitochondria-Specific Proteins
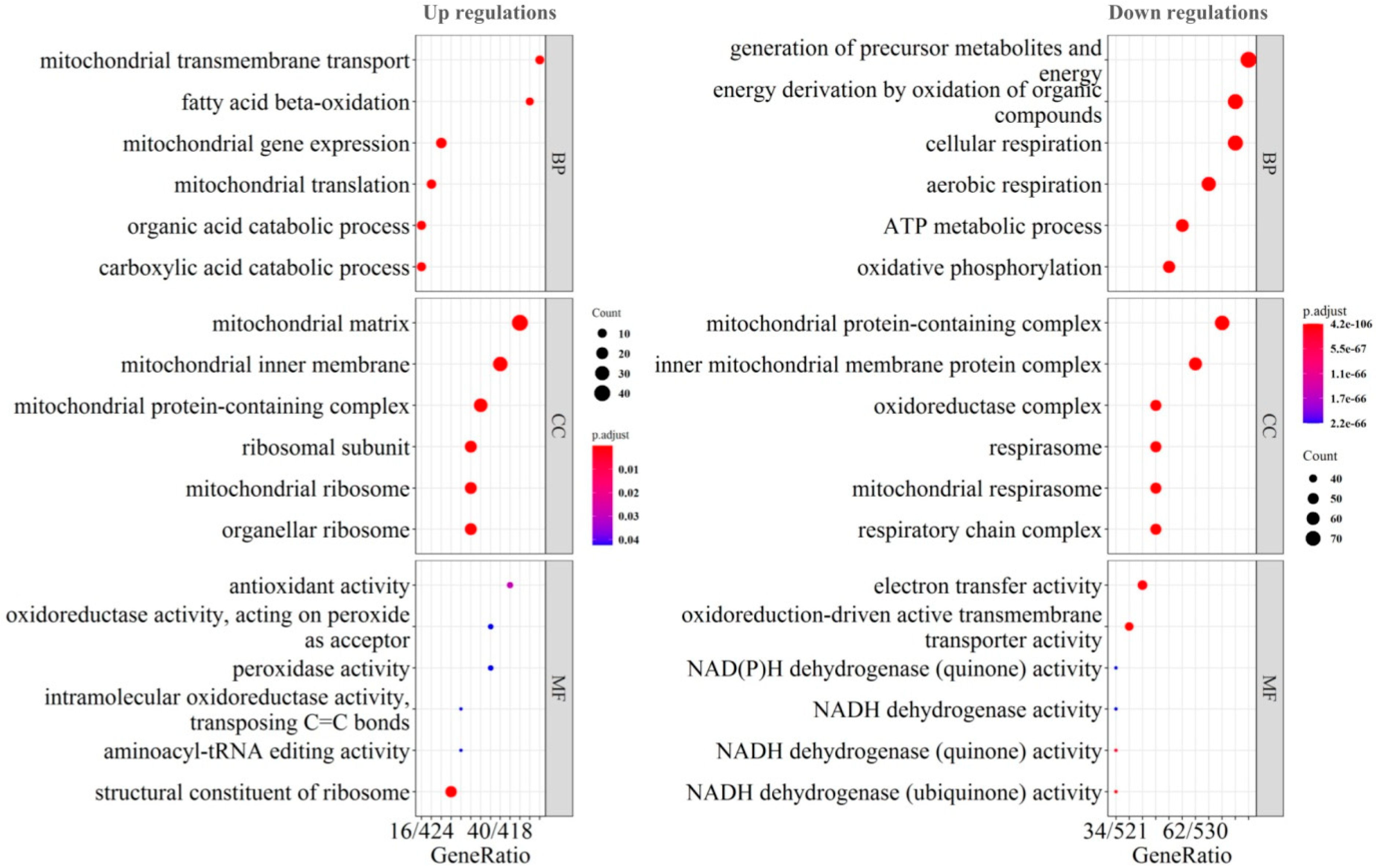
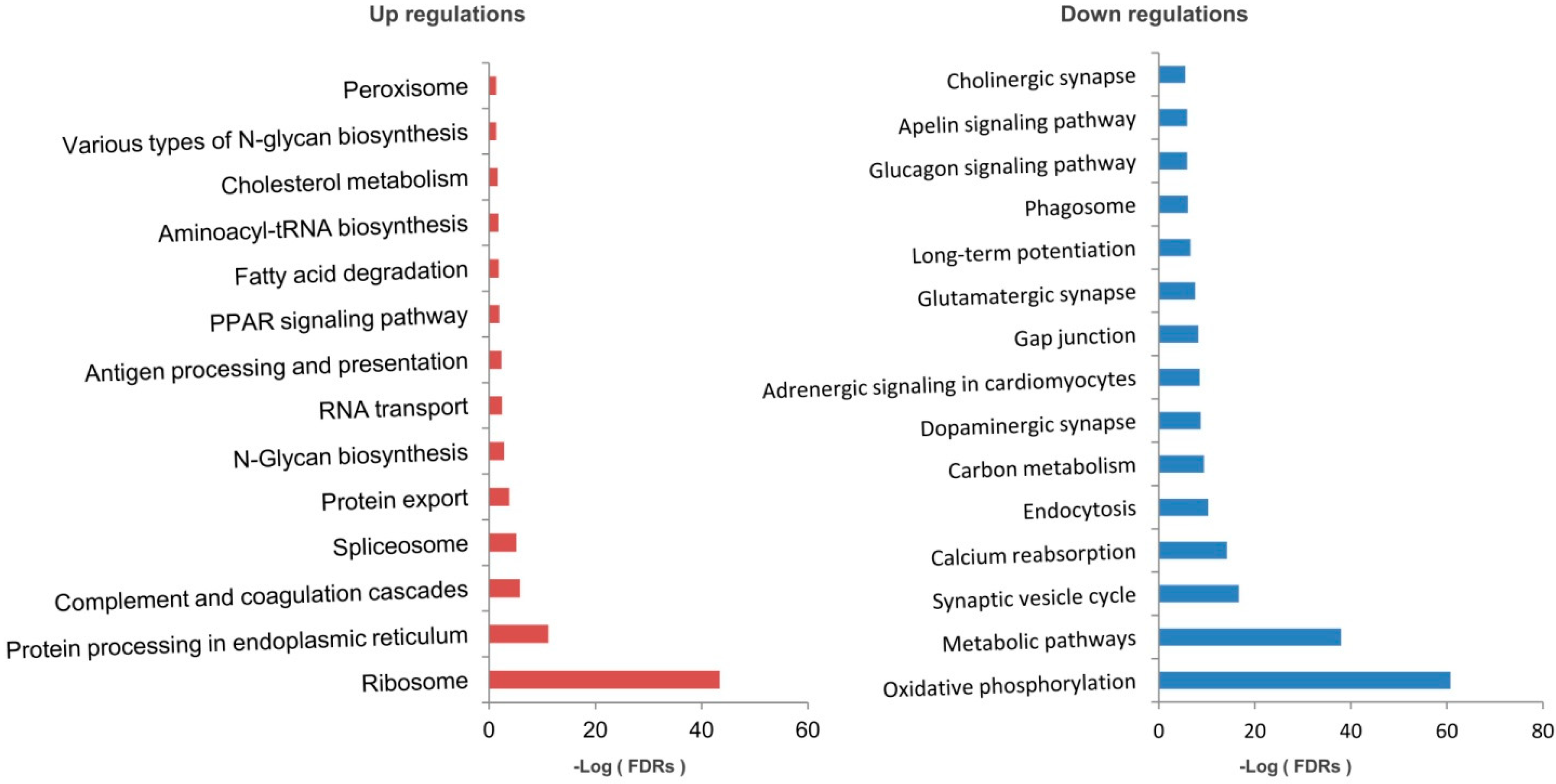
2.3. Mitochondrial Dysfunction by Integrated Proteomic and RNA-Seq Findings
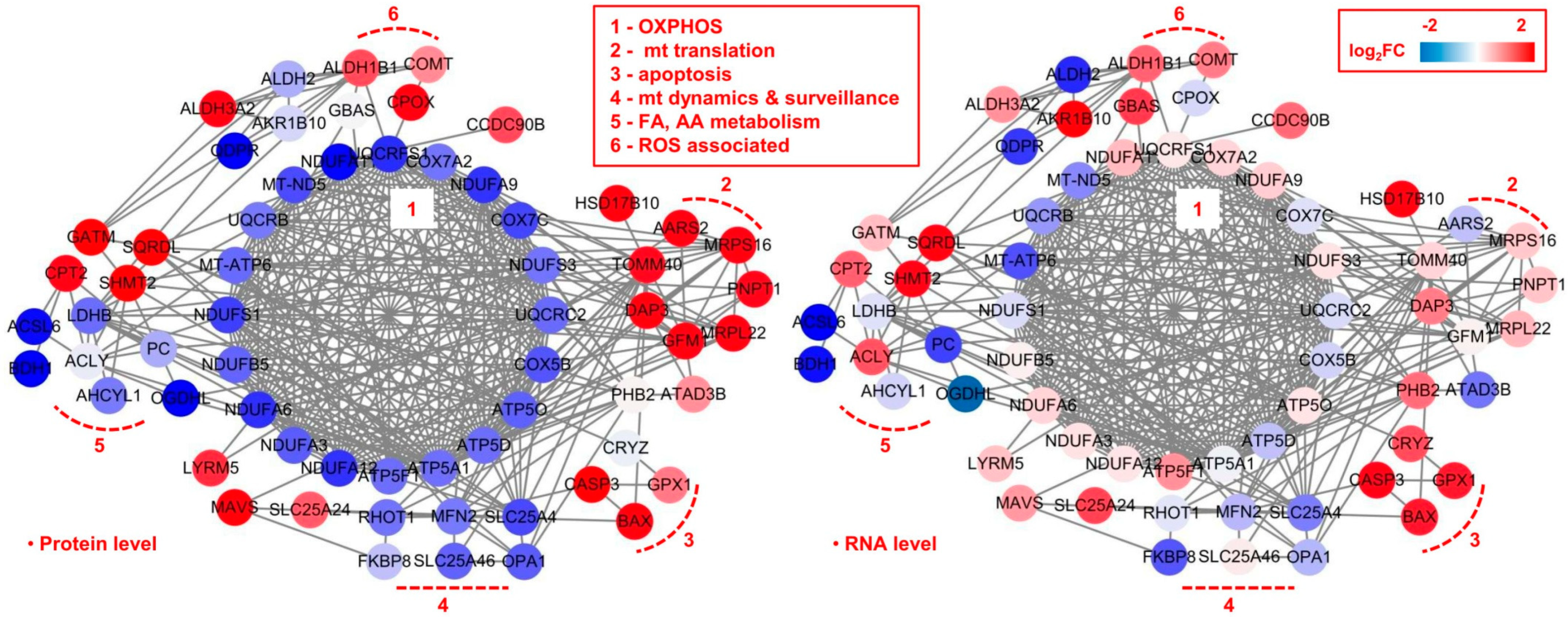
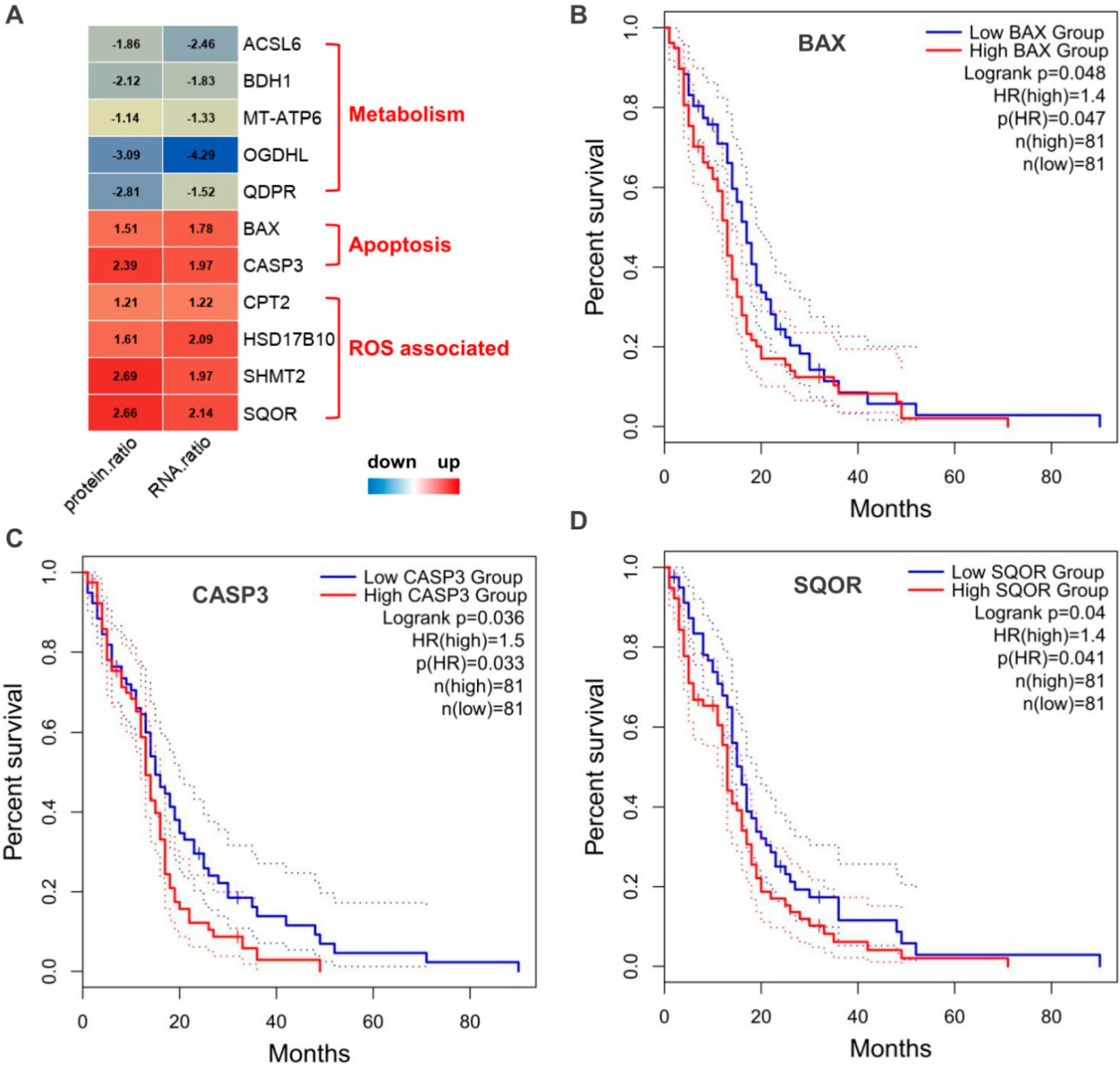
2.4. Possible Signatures in Human GBM Mitochondrion
3. Discussion
3.1. OXPHOS
3.2. Mitochondrial Dynamics
3.3. Mitochondrial Translation
3.4. Apoptosis
3.5. ROS
3.6. FA and AA Metabolism
4. Limitations
5. Conclusions
6. Materials and Methods
6.1. Patients and Tissue Samples
6.2. Mitochondrial Isolation
6.3. Protein Digestion
6.4. LC-MS/MS (DIA) Detection
6.5. Proteomic Data Processing
6.6. RNA-Seq Datasets
6.7. Catalogue of Gene Symbols for Human Mitochondrion
6.8. Bioinformatics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.R.; Laperriere, N.; O’Callaghan, C.J.; Brandes, A.A.; Menten, J.; Phillips, C.; Fay, M.; Nishikawa, R.; Cairncross, J.G.; Roa, W.; et al. Short-Course Radiation Plus Temozolomide in Elderly Patients with Glioblastoma. N. Engl. J. Med. 2017, 376, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. APJCP 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Kumar, S.; Ashraf, R.; K, A.C. Mitochondrial dynamics regulators: Implications for therapeutic intervention in cancer. Cell Biol. Toxicol. 2022, 38, 377–406. [Google Scholar] [CrossRef]
- Zong, W.-X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Ho, W.S.; Lu, R. Targeting Mitochondrial Oxidative Phosphorylation in Glioblastoma Therapy. Neuromol. Med. 2022, 24, 18–22. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Sighel, D.; Notarangelo, M.; Aibara, S.; Re, A.; Ricci, G.; Guida, M.; Soldano, A.; Adami, V.; Ambrosini, C.; Broso, F.; et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep. 2021, 35, 109024. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Maiti, P.; Barrientos, A. Mitochondrial ribosomes in cancer. Semin. Cancer Biol. 2017, 47, 67–81. [Google Scholar] [CrossRef]
- Ray, S.K.; Patel, S.J.; Welsh, C.T.; Wilford, G.G.; Hogan, E.L.; Banik, N.L. Molecular evidence of apoptotic death in malignant brain tumors including glioblastoma multiforme: Upregulation of calpain and caspase-3. J. Neurosci. Res. 2002, 69, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Dokic, I.; Hartmann, C.; Herold-Mende, C.; Regnier-Vigouroux, A. Glutathione peroxidase 1 activity dictates the sensitivity of glioblastoma cells to oxidative stress. Glia 2012, 60, 1785–1800. [Google Scholar] [CrossRef]
- Park, J.; Shim, J.K.; Kang, J.H.; Choi, J.; Chang, J.H.; Kim, S.Y.; Kang, S.G. Regulation of bioenergetics through dual inhibition of aldehyde dehydrogenase and mitochondrial complex I suppresses glioblastoma tumorspheres. Neuro-Oncology 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Soehngen, E.; Schaefer, A.; Koeritzer, J.; Huelsmeyer, V.; Zimmer, C.; Ringel, F.; Gempt, J.; Schlegel, J. Hypoxia upregulates aldehyde dehydrogenase isoform 1 (ALDH1) expression and induces functional stem cell characteristics in human glioblastoma cells. Brain Tumor Pathol. 2014, 31, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Xie, B.; Xiao, W.; Fan, M.; Xu, S.; Duan, Y.; Hamsafar, Y.; Evans, A.C.; Huang, J.; Zhou, W.; et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun. 2022, 13, 1511. [Google Scholar] [CrossRef]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, Z.; Yan, H.; Wang, W.; Wu, Z.; Zhang, F.; Zhang, Q.; Shi, G.; Du, J.; Cai, H.; et al. Creatine promotes cancer metastasis through activation of Smad2/3. Cell Metab. 2021, 33, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Meng, F.; Wang, J.; Liu, M.; Yang, G.; Song, R.; Zheng, T.; Liang, Y.; Zhang, S.; Yin, D.; et al. A Novel Oxoglutarate Dehydrogenase-Like Mediated miR-214/TWIST1 Negative Feedback Loop Inhibits Pancreatic Cancer Growth and Metastasis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5407–5421. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.L.; Chen, D.; Yan, J.; Yang, Q.; Han, Q.Q.; Li, S.S.; Cheng, L. Proteomic characteristics of bronchoalveolar lavage fluid in critical COVID-19 patients. FEBS J. 2021, 288, 5190–5200. [Google Scholar] [CrossRef]
- Zeng, H.L.; Yu, F.L.; Zhang, Z.; Yang, Q.; Jin, S.; He, X.; Chen, X.; Shen, Y.; Cheng, L.; Guo, L.; et al. Quantitative proteomics study of host response to virulent and attenuated pseudorabies virus infection in mouse brain. Biochim. Biophys. Acta 2018, 1866, 307–315. [Google Scholar] [CrossRef]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q Exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef]
- Demichev, V.; Messner, C.B.; Vernardis, S.I.; Lilley, K.S.; Ralser, M. DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 2020, 17, 41–44. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H.W.; et al. MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.-L.; Hu, L.; Chen, X.; Han, Q.-Q.; Li, H.; Cheng, L.; Li, C.-X. DIA-MS Based Proteomics Combined with RNA-Seq Data to Unveil the Mitochondrial Dysfunction in Human Glioblastoma. Molecules 2023, 28, 1595. https://doi.org/10.3390/molecules28041595
Zeng H-L, Hu L, Chen X, Han Q-Q, Li H, Cheng L, Li C-X. DIA-MS Based Proteomics Combined with RNA-Seq Data to Unveil the Mitochondrial Dysfunction in Human Glioblastoma. Molecules. 2023; 28(4):1595. https://doi.org/10.3390/molecules28041595
Chicago/Turabian StyleZeng, Hao-Long, Lizhi Hu, Xi Chen, Qiang-Qiang Han, Huijun Li, Liming Cheng, and Chao-Xi Li. 2023. "DIA-MS Based Proteomics Combined with RNA-Seq Data to Unveil the Mitochondrial Dysfunction in Human Glioblastoma" Molecules 28, no. 4: 1595. https://doi.org/10.3390/molecules28041595
APA StyleZeng, H.-L., Hu, L., Chen, X., Han, Q.-Q., Li, H., Cheng, L., & Li, C.-X. (2023). DIA-MS Based Proteomics Combined with RNA-Seq Data to Unveil the Mitochondrial Dysfunction in Human Glioblastoma. Molecules, 28(4), 1595. https://doi.org/10.3390/molecules28041595