






Use of Trifluoro-Acetate Derivatives for GC-MS and GC-MS/MS Quantification of Trace Amounts of Stera-3β,5α,6β-Triols (Tracers of Δ5-Sterol Autoxidation) in Environmental Samples


Abstract
:1. Introduction
2. Results and Discussion
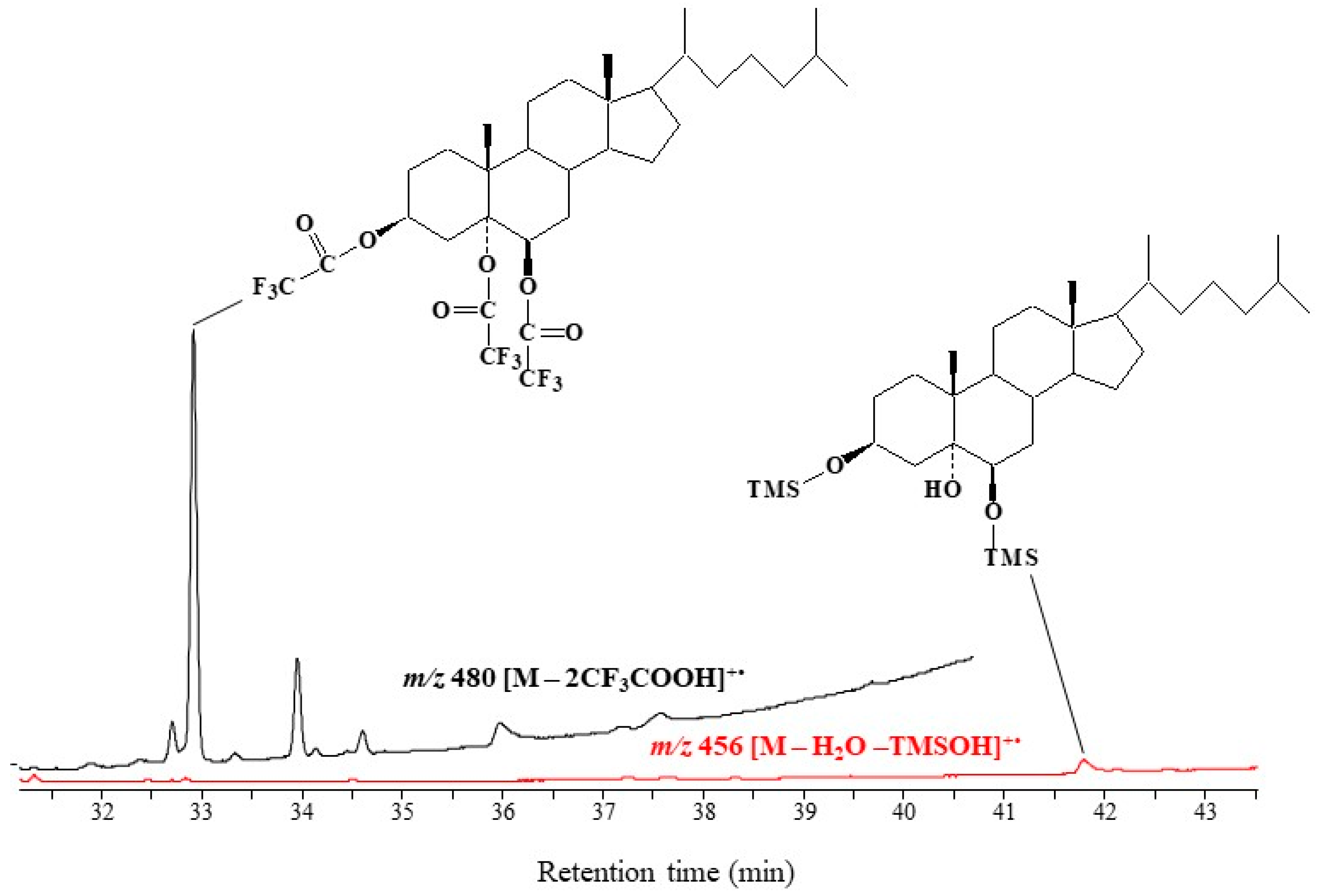
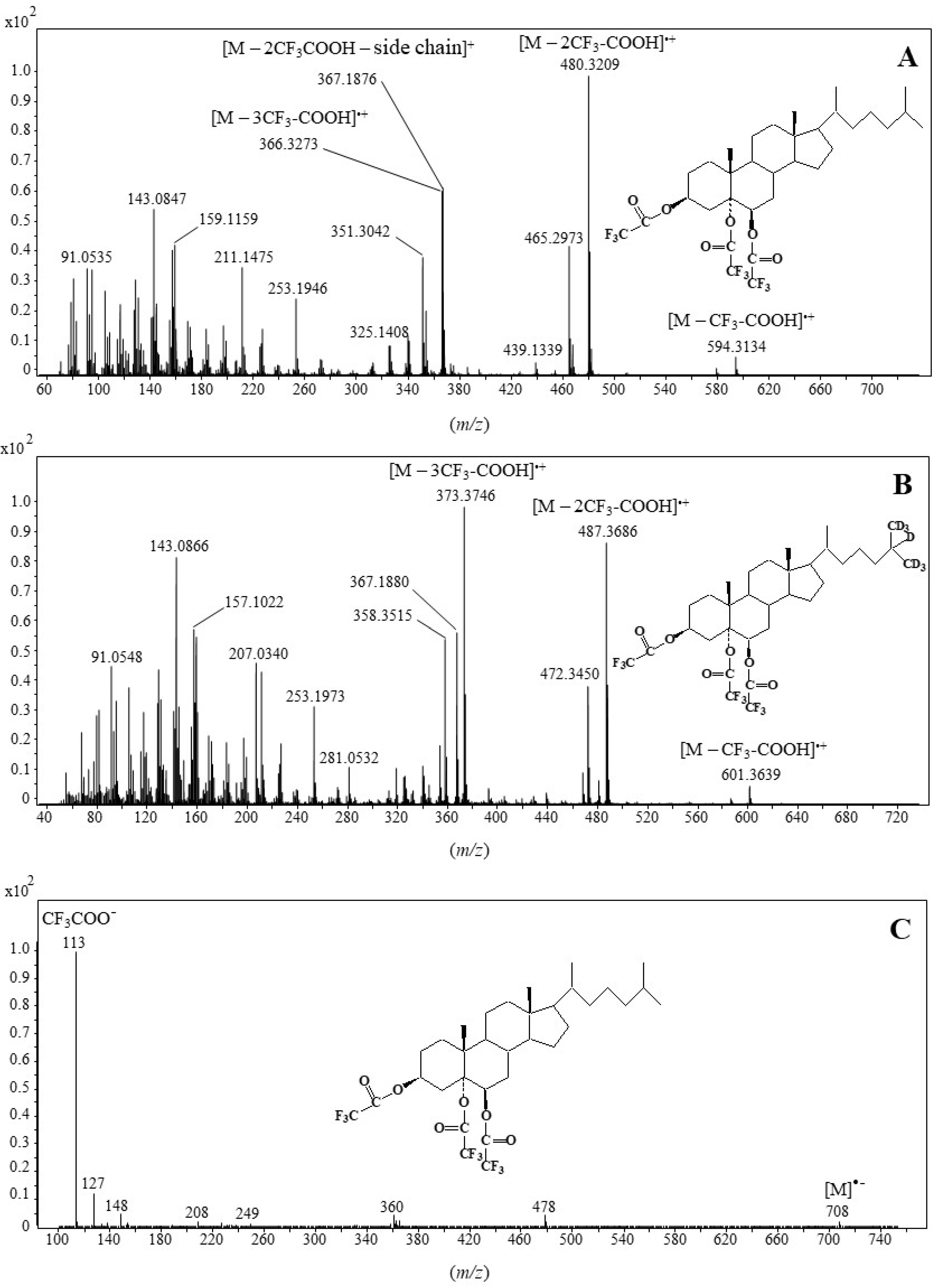
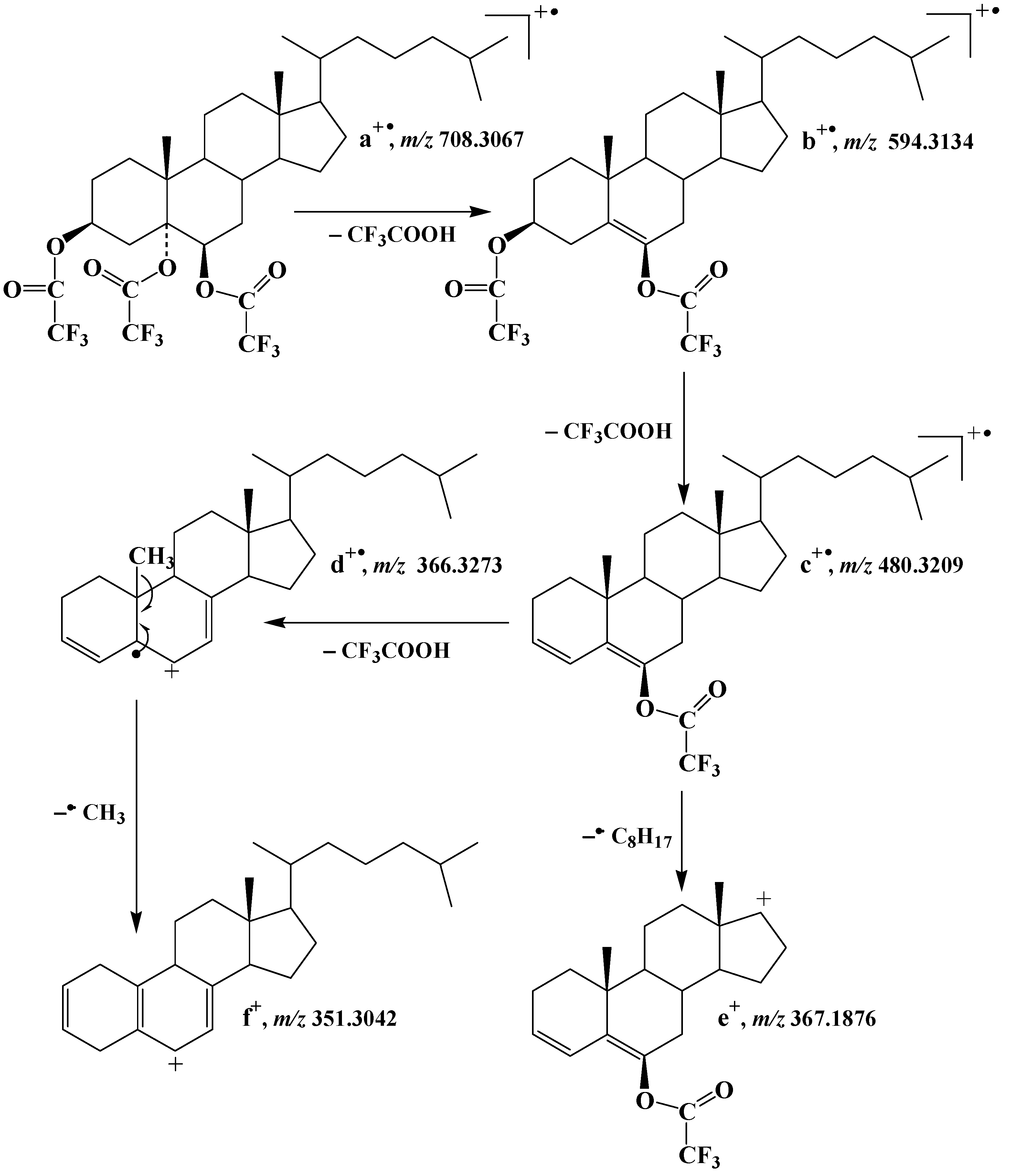
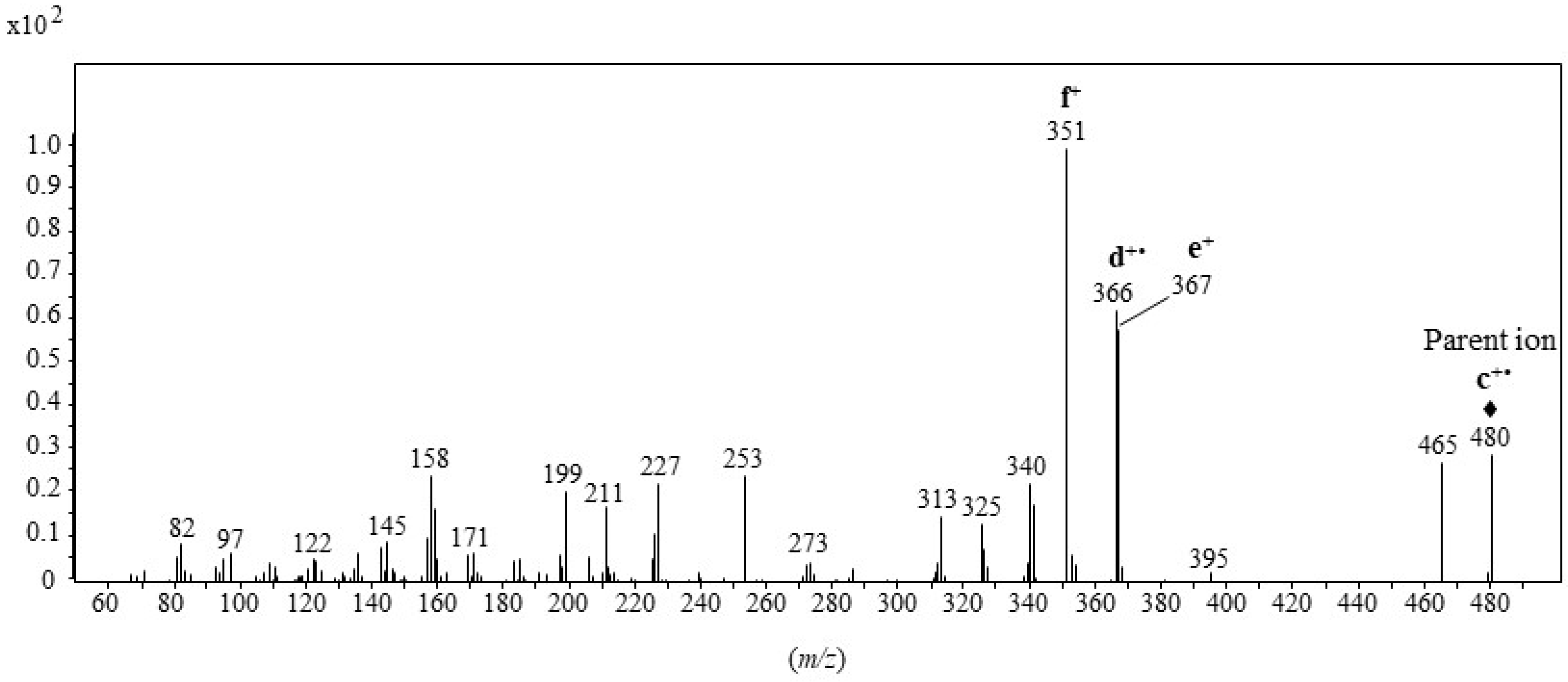
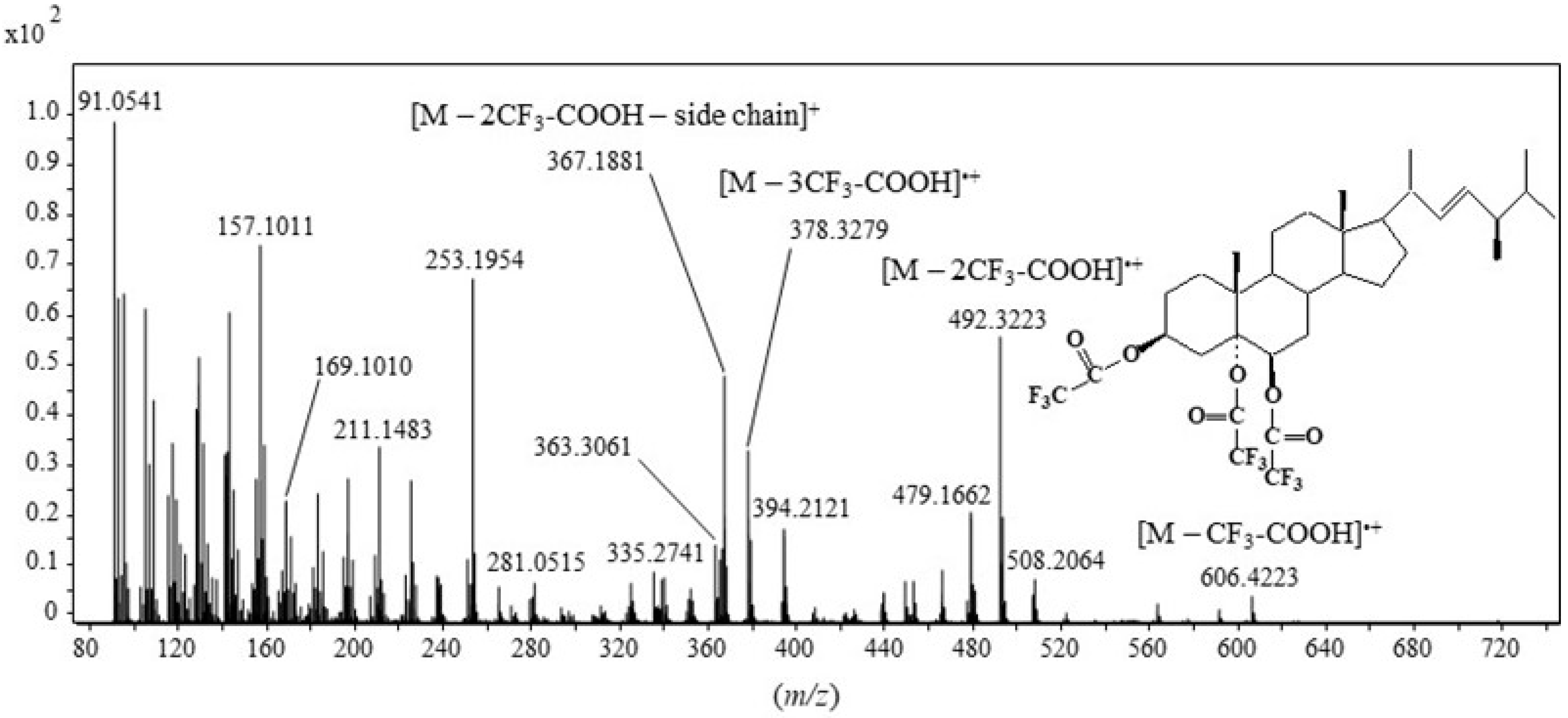
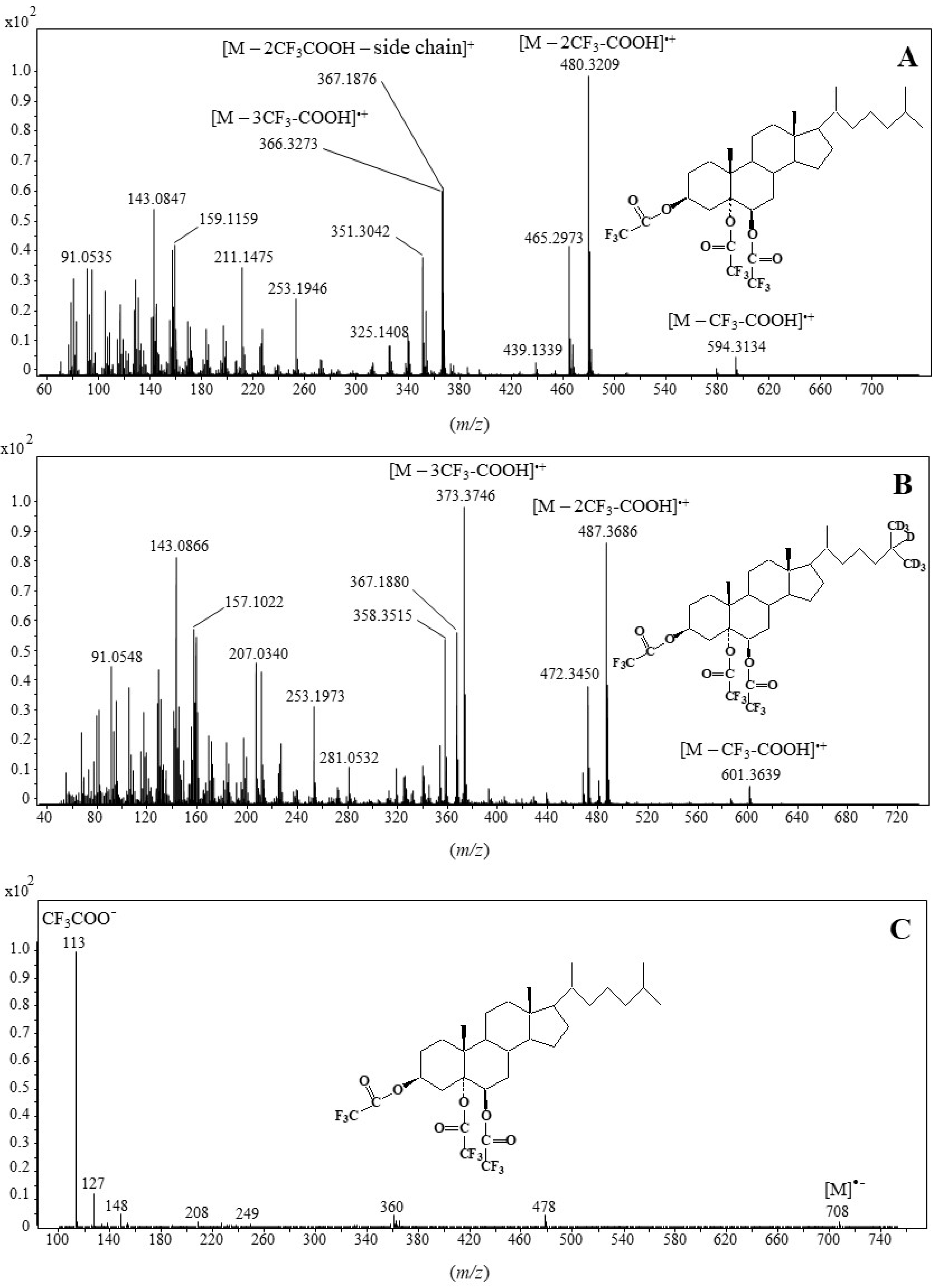
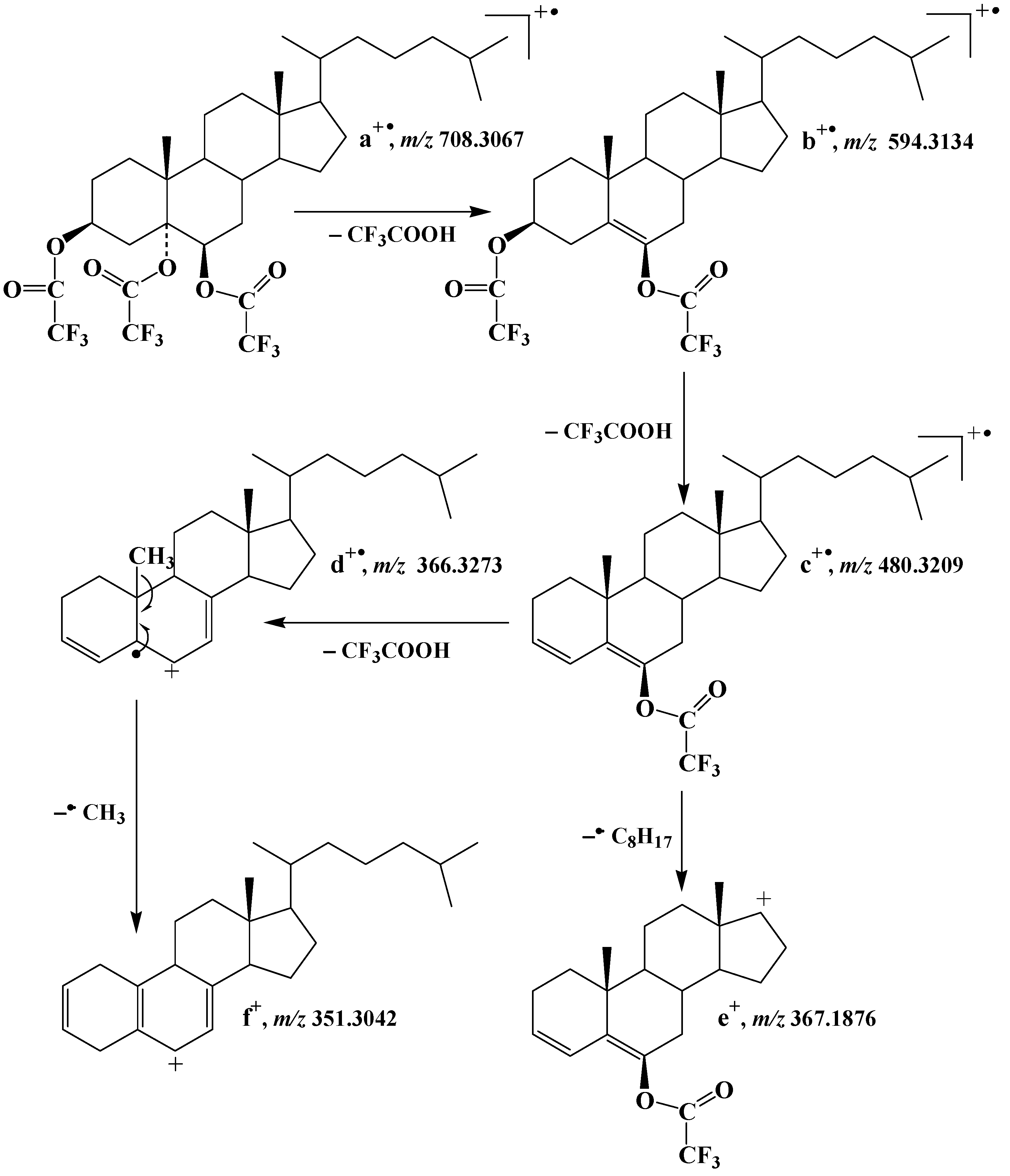
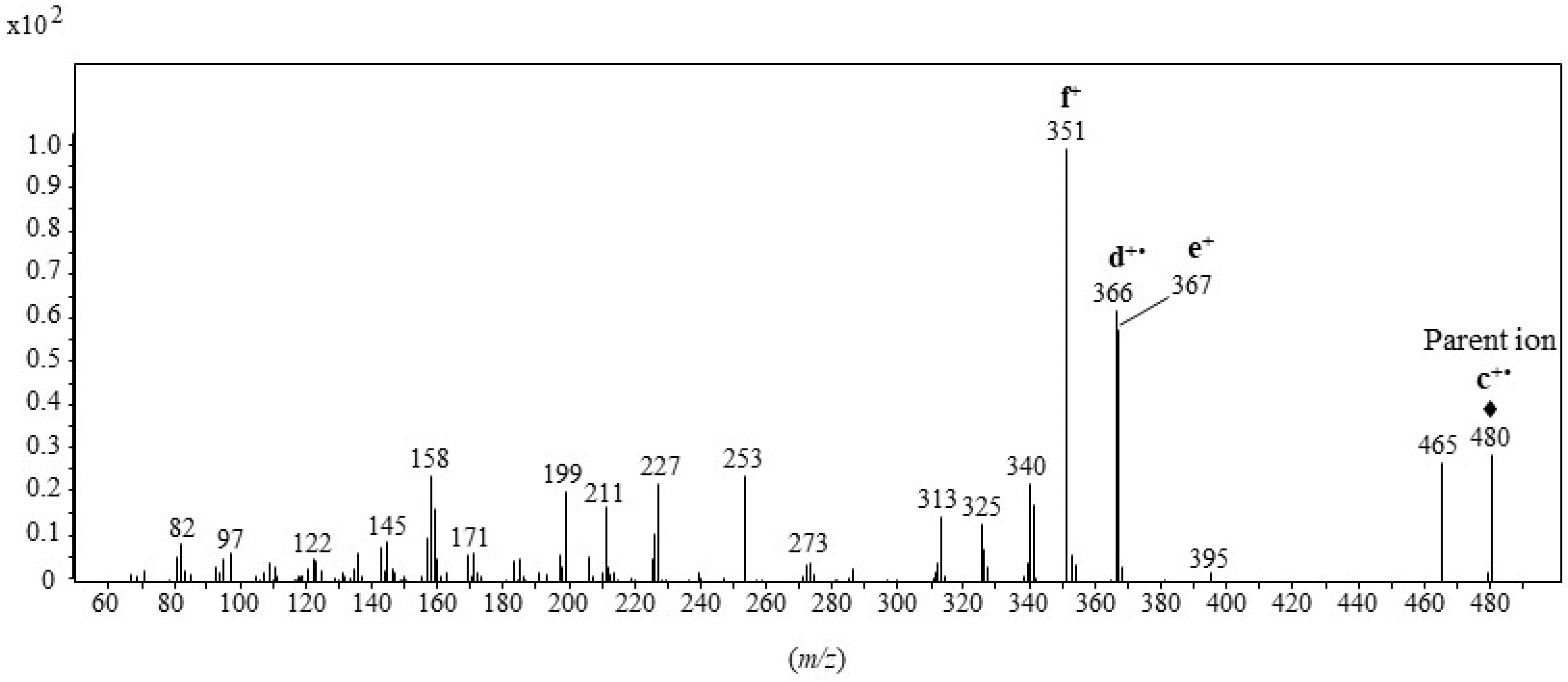
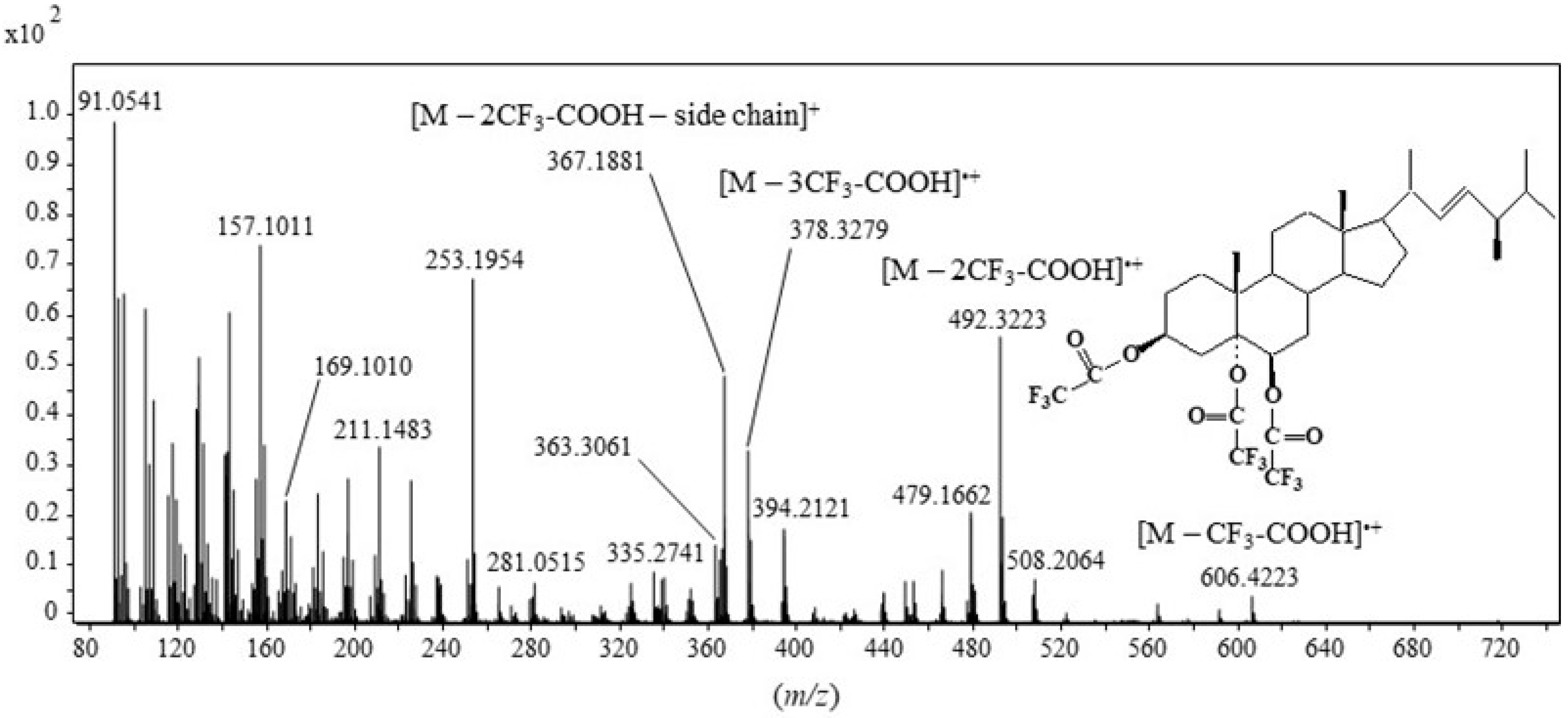
2.1. Formation and Characterization of Trifluoroacetate Derivative of Cholesta-3β,5α,6β-Triol
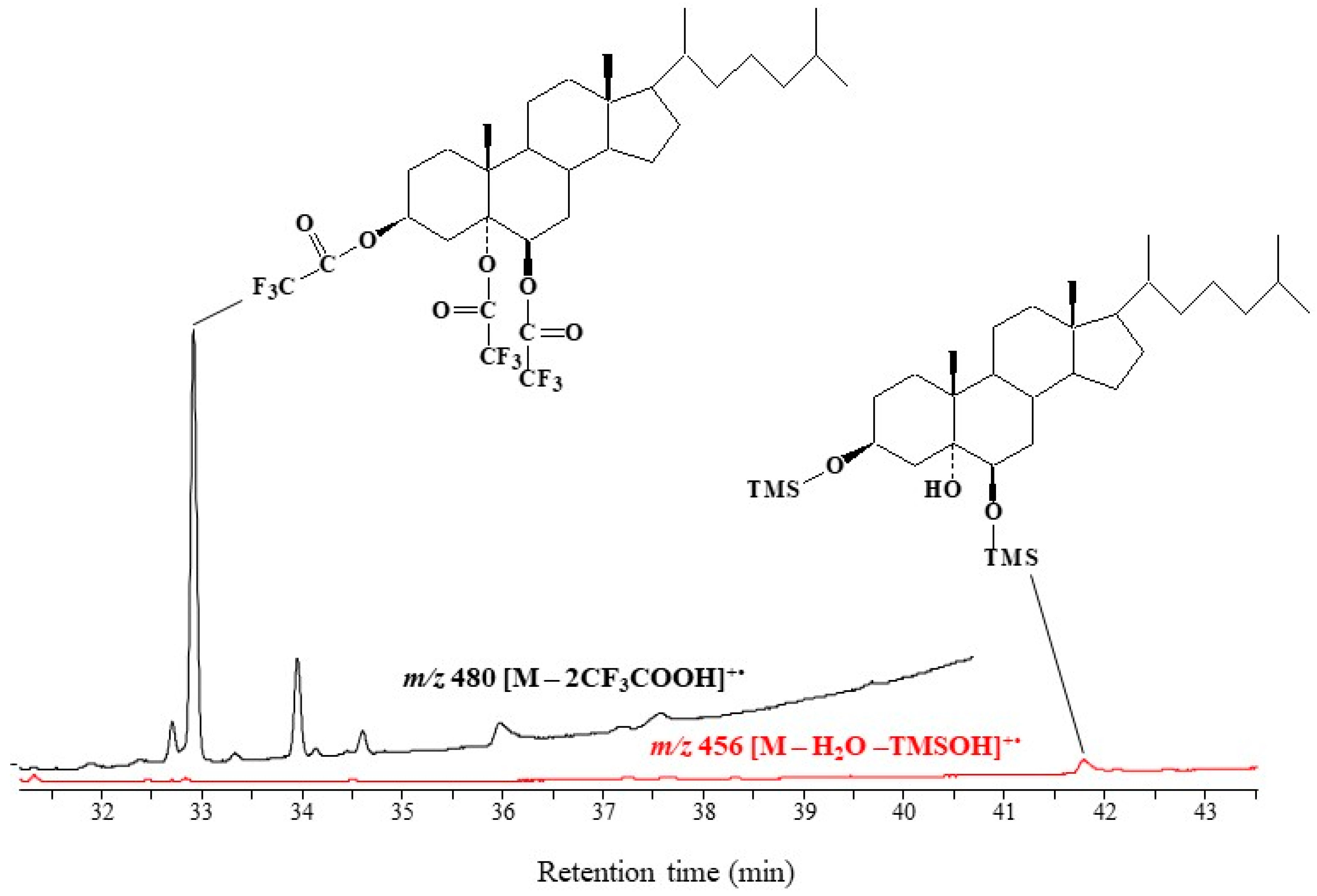
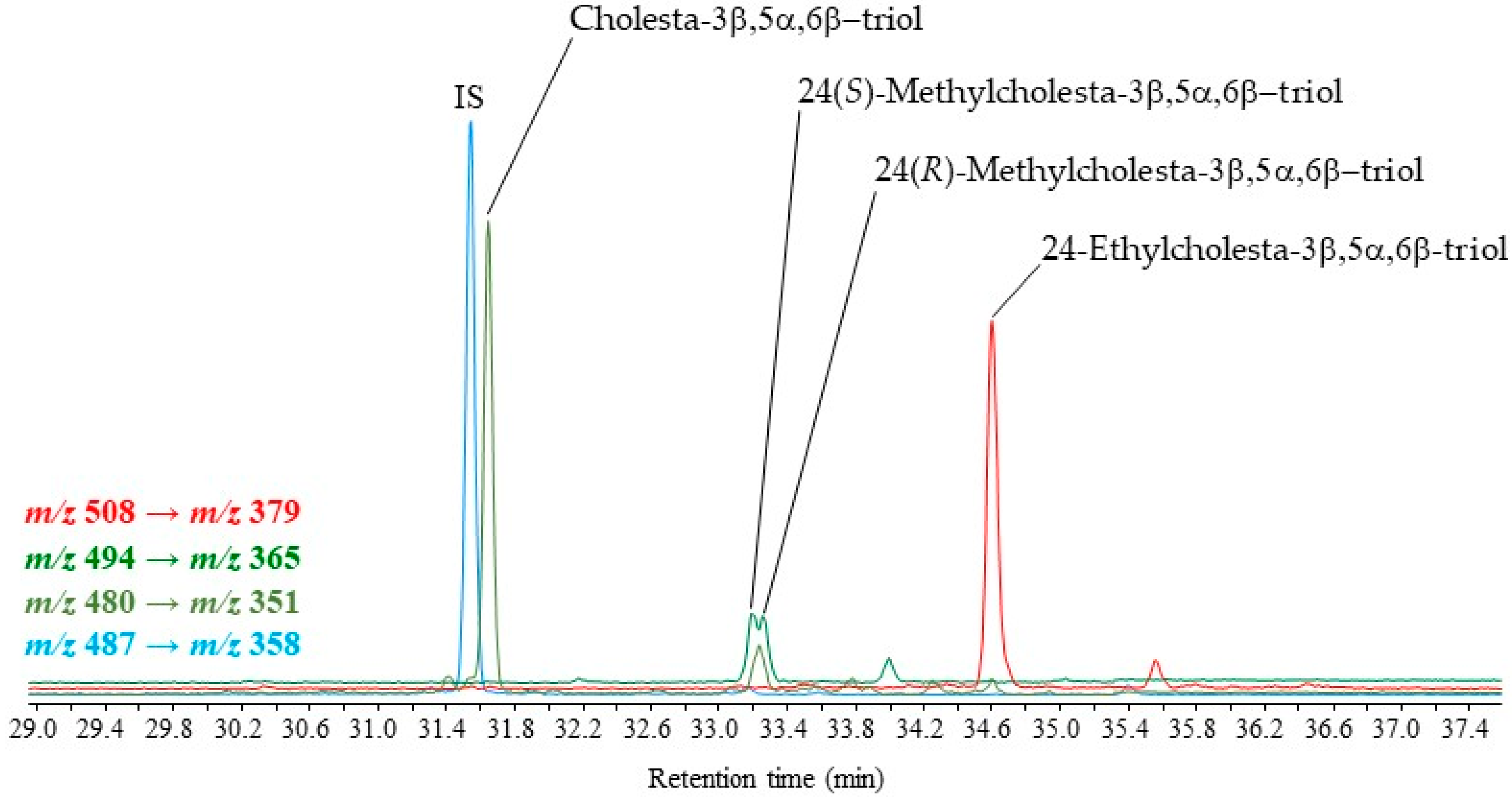
2.2. Validation of the Derivatization Method
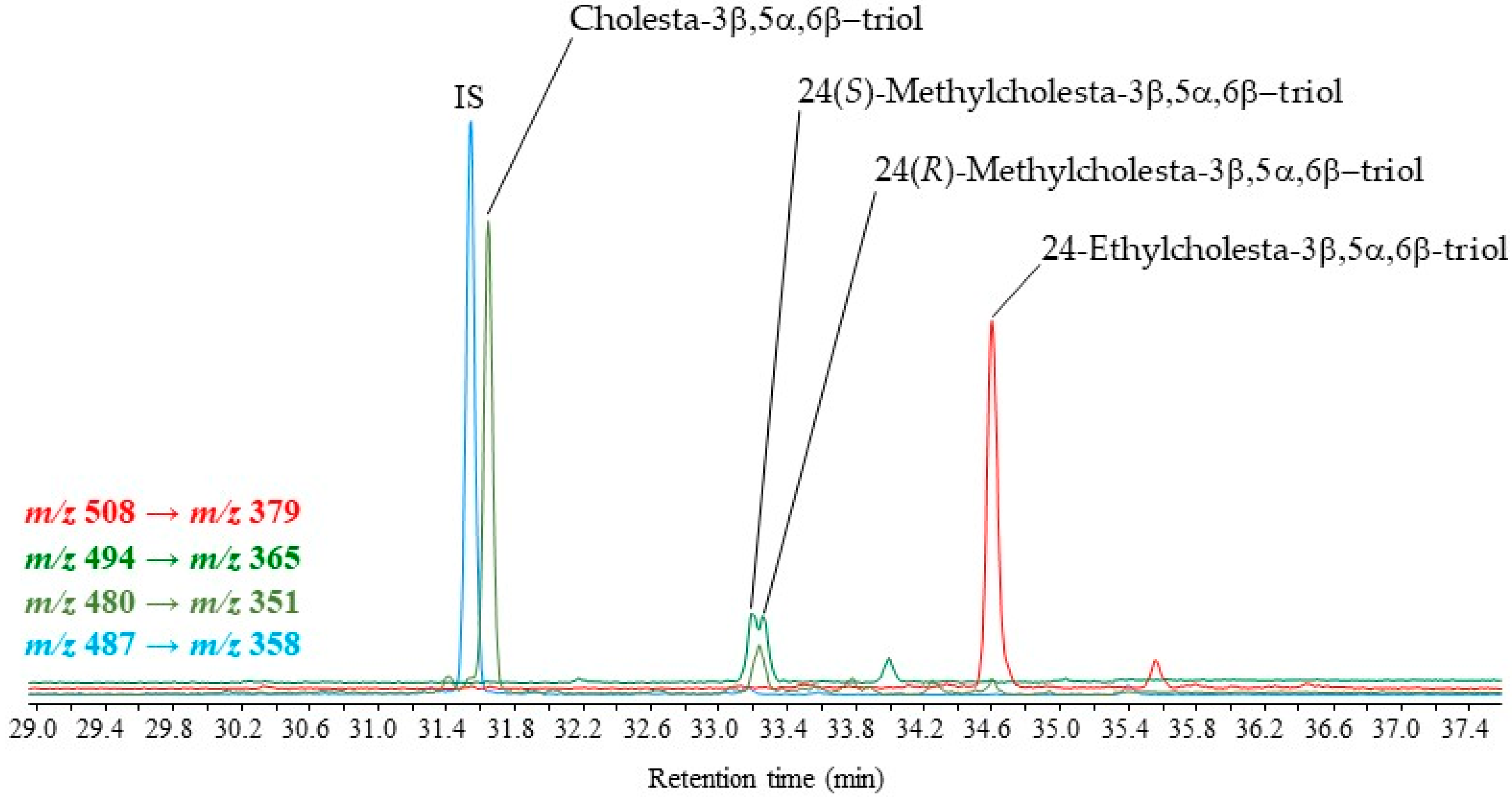
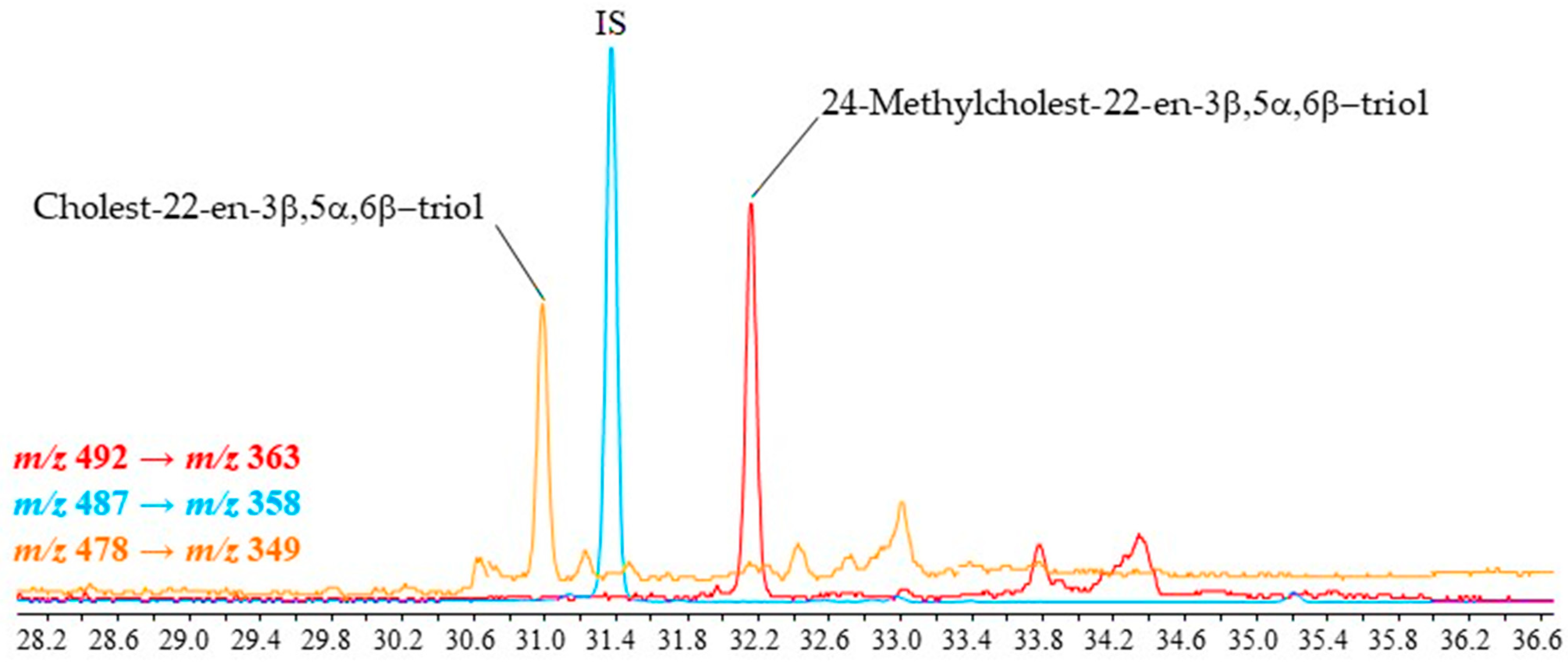
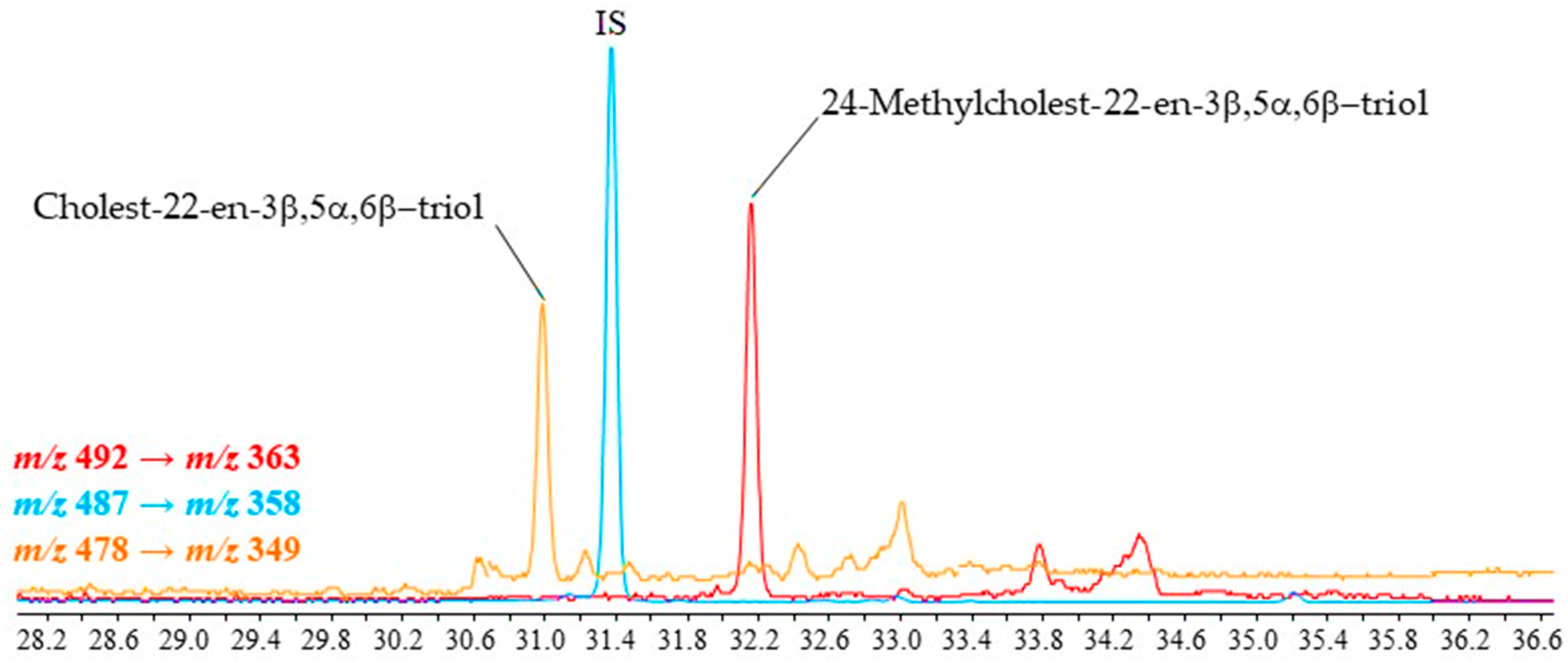
2.3. Application to Different Environmental Samples
3. Materials and Methods
3.1. Chemicals
3.2. Environmental Samples
3.3. Trifluoroacetylation Method
3.4. Silylation
3.5. Gas Chromatography-Tandem Electron Ionization Mass Spectrometry (GC-EIMS/MS)
3.6. Gas Chromatography-EI Quadrupole Time-of-Flight Mass Spectrometry (GC-QTOF)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Smith, L.L. Cholesterol autoxidation 1981–1986. Chem. Phys. Lipids 1981, 44, 87–125. [Google Scholar] [CrossRef]
- Christodoulou, S.; Marty, J.-C.; Miquel, J.-C.; Volkman, J.K.; Rontani, J.-F. Use of lipids and their degradation products as biomarkers for carbon cycling in the northwestern Mediterranean Sea. Mar. Chem. 2009, 113, 25–40. [Google Scholar] [CrossRef]
- Silvente-Poirot, S.; Poirot, M. Cholesterol epoxide hydrolase and cancer. Curr. Opin. Pharmacol. 2012, 12, 696–703. [Google Scholar] [CrossRef]
- Poirot, M.; Silvente-Poirot, S. Cholesterol-5,6-epoxides: Chemistry, biochemistry, metabolic fate and cancer. Biochimie 2013, 95, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Rontani, J.-F.; Zabeti, N.; Wakeham, S.G. The fate of marine lipids: Biotic vs. abiotic degradation of particulate sterols and alkenones in the northwestern Mediterranean Sea. Mar. Chem. 2009, 113, 9–18. [Google Scholar] [CrossRef]
- Koek, M.M.; Jellema, R.H.; van der Greef, J.; Tas, A.C.; Hankemeier, T. Quantitative metabolomics based on gas chromatography mass spectrometry: Status and perspectives. Metabolomics 2011, 7, 307–328. [Google Scholar] [CrossRef]
- Yang, R.; Xue, L.; Zhang, L.; Wang, X.; Qi, X.; Jiang, J.; Yu, L.; Wang, X.; Zhang, W.; Zhang, Q.; et al. Phytosterol contents of edible oils and their contributions to estimated phytosterol intake in the Chinese diet. Foods 2019, 8, 334. [Google Scholar] [CrossRef]
- dos Santos, M.A.Z.; Roehrs, M.; de Pereira, C.M.P.; Freitag, R.A.; de Bairros, A.V. Analysis of phytosterols in plants and derived products by gas chromatography—A short critical review. Austin Chromatogr. 2014, 1, 4. [Google Scholar]
- Benfenati, E.; Cools, E.; Fattore, E.; Fanelli, R. A GC-MS method for the analysis of fecal and plant sterols in sediment samples. Chemosphere 1994, 29, 1393–1405. [Google Scholar] [CrossRef]
- Goad, L.J.; Akihisa, T. Mass Spectrometry of Sterols. In Analysis of Sterols; Goad, L.J., Akihisa, T., Eds.; Springer: Dordrecht, Germany, 1997; pp. 152–196. [Google Scholar]
- Harvey, D.J.; Vouros, P. Mass spectrometric fragmentation of trimethylsilyl and related alkylsilyl derivatives. Mass Spectrom. Rev. 2020, 39, 105–211. [Google Scholar] [CrossRef]
- Wu, J.; Hu, R.; Yue, J.; Yang, Z.; Zhang, L. Determination of fecal ster-12-ols by gas chromatography–mass spectrometry with solid-phase extraction and injection-port derivatization. J. Chromatogr. A 2009, 1216, 1053–1058. [Google Scholar] [CrossRef]
- Balme, S.; Gülaçar, F.O. Rapid screening of phytosterols in orange juice by solid-phase microextraction on polyacrylate fibre derivatisation and gas chromatographic–mass spectrometric. Food Chem. 2012, 132, 613–618. [Google Scholar] [CrossRef]
- Birk, J.J.; Dippold, M.; Wiesenberg, G.L.; Glaser, B. Combined quantification of faecal sterols, stanols, stanones and bile acids in soils and terrestrial sediments by gas chromatography–mass spectrometry. J. Chromatogr. A 2012, 1242, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kloos, D.P.; Gay, E.; Lingeman, H.; Bracher, F.; Müller, C.; Mayboroda, O.A.; Deelder, A.M.; Neessen, W.M.A.; Giera, M. Comprehensive gas chromatography–electron ionisation mass spectrometric analysis of fatty acids and sterols using sequential one-pot silylation: Quantification and isotopologue analysis. Rapid Commun. Mass Spectrom. 2014, 28, 1507–1514. [Google Scholar] [CrossRef]
- Junker, J.; Chong, I.; Kamp, F.; Steiner, H.; Giera, M.; Müller, C.; Bracher, F. Comparison of strategies for the determination of sterol sulfates via GC-MS leading to a novel deconjugation-derivatization protocol. Molecules 2019, 24, 2353. [Google Scholar] [CrossRef] [PubMed]
- Park, P.S.W.; Addis, P.W. Derivatization of 5 -cholestane-3β,5,6β-triol into trimethylsilyl ether sterol for GC analysis. J. Am. Oil Chem. Soc. 1989, 66, 1632–1634. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Aubert, C. Characterization of isomeric allylic diols resulting from chlorophyll phytyl side-chain photo- and autoxidation by electron ionization gas chromatography/mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 637–646. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Charrière, B.; Sempéré, R.; Doxaran, D.; Vaultier, F.; Vonk, J.E.; Volkman, J.K. Degradation of sterols and terrigenous organic matter in waters of the Mackenzie Shelf, Canadian Arctic. Org. Geochem. 2014, 75, 61–73. [Google Scholar] [CrossRef]
- Angelis, Y.S.; Kioussi, M.K.; Kiousi, P.; Brenna, J.T.; Georgakopoulos, C.G. Examination of the kinetic isotopic effect to the acetylation derivatization for the gas chromatographic-combustion-isotope ratio mass spectrometric doping control analysis of endogenous steroids. Drug Test. Anal. 2012, 4, 923–927. [Google Scholar] [CrossRef]
- Choi, M.A.; Kim, J.Y.; Chung, B.C. Detection of anabolic steroids by GC/SIM/MS with trifluoroacetylation in equine plasma and urine. Anal. Lett. 1999, 32, 1301–1312. [Google Scholar] [CrossRef]
- Harnik, M.; Hürzeler, E.; Jensen, E.V. Acetylation and trifluoroacetylation of steroids at carbon 16. Tetrahedron 1967, 23, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Marcos, J.; Pozo, O.J. Derivatization of steroids in biological samples for GC–MS and LC–MS analyses. Bioanalysis 2015, 7, 2515–2536. [Google Scholar] [CrossRef]
- Söderhölm, S.L.; Damm, M.; Kappe, C.O. Microwave-assisted derivatization procedures for gas chromatography/mass spectrometry analysis. Mol. Divers. 2010, 14, 869–888. [Google Scholar] [CrossRef]
- Orata, F. Derivatization reactions and reagents for gas chromatography analysis. Adv. Gas Chromatogr. Prog. Agric. Biomed. Ind. Appl. 2012, 91, 83–108. [Google Scholar]
- Tredget, E.E.; Falk, N.; Scott, P.G.; Hogg, A.M.; Burke, J.F. Determination of 4-hydroxyproline in collagen by gas chromatography/mass spectrometry. Anal. Biochem. 1990, 190, 259–265. [Google Scholar] [CrossRef]
- Lin, D.L.; Wang, S.M.; Wu, C.H.; Chen, B.G.; Liu, R.H. Chemical derivatization for the analysis of drugs by GC-MS-A conceptual review. J. Food Drug Anal. 2008, 16, 1–10. [Google Scholar] [CrossRef]
- Mellon, F.A. Mass spectrometry principles and applications. In Encyclopedia of Food Sciences and Nutrition, 2nd ed.; Caballero, B., Ed.; Academic Press: Cambridge, MA, USA, 2003; pp. 3739–3749. [Google Scholar]
- Benveniste, P. Sterol biosynthesis. Annu. Rev. Plant Physiol. 1986, 37, 275–308. [Google Scholar] [CrossRef]
- Nes, W.R. The biochemistry of plant sterols. Adv. Lipid Res. 1977, 15, 233–324. [Google Scholar]
- Atwood, A.R.; Volkman, J.K.; Sachs, J.P. Characterization of unusual sterols and long chain diols, triols, keto-ols and n-alkenols in El Junco Lake, Galápagos. Org. Geochem. 2014, 66, 80–89. [Google Scholar] [CrossRef]
- Rampen, S.W.; Abbas, B.A.; Schouten, S.; Sinninghe Damste, J.S. A comprehensive study of sterols in marine diatoms (Bacillariophyta): Implications for their use as tracers for diatom productivity. Limnol. Oceanogr. 2010, 55, 91–105. [Google Scholar] [CrossRef]
- Volkman, J.K. Sterols and other triterpenoids: Source specificity and evolution of biosynthetic pathways. Org. Geochem. 2005, 36, 139–159. [Google Scholar] [CrossRef]
- Mackenzie, A.S.; Brassell, S.C.; Eglinton, G.; Maxwell, J.R. Chemical fossils: The geological fate of steroids. Science 1982, 217, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Volkman, J.K. A review of sterol markers for marine and terrigenous organic matter. Org. Geochem. 1986, 9, 83–99. [Google Scholar]
- Wakeham, S.G.; Beier, J.A. Fatty acid and sterol biomarkers as indicators of particulate matter source and alteration processes in the Black Sea. Deep Sea Res. Part A 1991, 38, S943–S968. [Google Scholar] [CrossRef]
- Ea, S.; Aubert, C.; Rontani, J.-F.; Teral, Y.; Campredon, M. Elucidation of electron ionization mass spectrometric fragmentation pathways of trimethylsilyl ether derivatives of vicinal diols deriving from haplamine by collision-induced dissociation gas chromatography/tandem mass spectrometry and 18O labelling. Rapid Commun. Mass Spectrom. 2014, 28, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, C. Quantitative and stereospecific dihydroxylations of Δ5-steroids: A green synthesis of plant growth hormone intermediates. J. Agric. Food Chem. 2013, 61, 12522–12530. [Google Scholar] [PubMed]
- Aghnatios, C.; Losno, R.; Dulac, F. A fine fraction of soil used as an aerosol analogue during the DUNE experiment: Sequential solubility in water, decreasing pH step-by-step. Biogeosciences 2014, 11, 4627–4633. [Google Scholar]
- Rontani, J.-F.; Amiraux, R.; Smik, L.; Wakeham, S.G.; Paulmier, A.; Vaultier, F.; Sun-Yong, H.; Jun-Oh, M.; Belt, S.T. Type II photosensitized oxidation in senescent microalgal cells at different latitudes: Does low under-ice irradiance in polar regions enhance efficiency? Sci. Total Environ. 2021, 779, 146363. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Lalande, C.; Vilgrain, L.; Vaultier, F.; Amiraux, R. Control of the preservation of sympagic algal material in surficial sediments of central and eastern Baffin Bay by bactericidal hydroperoxides and free fatty acids. Mar. Chem. 2022, 247, 104177. [Google Scholar] [CrossRef]
- Rontani, J.-F.; Charriere, B.; Menniti, C.; Katra, I.; Aubert, D. Effects of dry and wet Negev dust deposition on the induction of autoxidation of dust lipid components in seawater. Water 2022, 14, 4092. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 3β,5α,6β-Steratriols | c+• Parent Ion [M—2CF3COOH]+• | f+ Product Ion [M—2CF3COOH—CH3]+ | CE * (eV) |
|---|---|---|---|
| Cholesta-3β,5α,6β-triol | 480 | 351 | 14 |
| Cholest-22-en3β,5α,6β-triol | 478 | 349 | 14 |
| 24-Methylcholest-22-en3β,5α,6β-triol | 492 | 363 | 14 |
| 24-Methylcholest-24(28)-en3β,5α,6β-triol | 492 | 363 | 14 |
| 24-Methylcholesta-3β,5α,6β-triol | 494 | 365 | 14 |
| 24-Ethylcholest-22-en3β,5α,6β-triol | 506 | 377 | 14 |
| 24-Ethylcholesta-3β,5α,6β-triol | 508 | 379 | 14 |
| Mode | Concentration Range (ng/mL) | Linear Regression Equation | Coefficient of Determination (R2) |
|---|---|---|---|
| SIM | |||
| Ion m/z 480 | 2.3–46.5 a | y = 0.0315x − 0.0202 | 0.9952 |
| Ion m/z 594 | 2.3–46.5 | y = 0.0324x − 0.0029 | 0.9995 |
| MRM | |||
| m/z 480 → m/z 351 | 2.3–46.5 | y = 0.0358x − 0.0194 | 0.9974 |
| Mode | Concentration (ng/mL) | Relative Standard Deviation * (%) | Difference between Obtained and Expected Concentration (%) | ||
|---|---|---|---|---|---|
| Expected | Obtained | n | |||
| SIM | |||||
| Ion m/z 480 | 46.5 | 44.7 | 6 | 4.4 | −3.9 |
| 18.6 | 18.4 | 9 | 8.7 | −1.1 | |
| 9.3 | 8.9 | 8 | 3.8 | −4.3 | |
| 2.325 | 2.618 | 6 | 9.9 | 12.6 | |
| Ion m/z 594 | 46.5 | 44.4 | 6 | 5.4 | −4.5 |
| 18.6 | 18.3 | 9 | 8.5 | −1.6 | |
| 9.3 | 9.2 | 8 | 3.0 | −1.1 | |
| 2.325 | 2.696 | 6 | 4.2 | 15.9 | |
| MRM | |||||
| m/z 480 → m/z 351 | 46.5 | 45.6 | 6 | 3.1 | −1.9 |
| 18.6 | 18.63 | 9 | 4.1 | 0.2 | |
| 9.3 | 9.0 | 8 | 3.7 | −3.2 | |
| 2.325 | 2.863 | 6 | 4.2 | 10.2 | |
| Cholesta-3β,5α,6β-Triol | 24-Methylcholesta- 3β,5α,6β-Triols d | 24-Ethylcholesta-3β,5α,6β-Triol | ||||
|---|---|---|---|---|---|---|
| SIM m/z 480 | MRM m/z 480 → m/z 351 | SIM m/z 494 | MRM m/z 494 → m/z 365 | SIM m/z 508 | MRM m/z 508 → m/z 379 | |
| Negev loess sample 1 a | 0.21 | 0.21 | 0.09 | 0.06 | 0.24 | 0.17 |
| Negev loess sample 2 a | 0.24 | 0.19 | 0.07 | 0.05 | 0.19 | 0.12 |
| Negev loess sample 3 a | 0.23 | 0.22 | 0.12 | 0.09 | 0.35 | 0.25 |
| Particles Antarctica st 4 b | 11.4 | 11.73 | 0.80 | nd c | 1.95 | 1.25 |
| Particles Antarctica st 13 b | 7.11 | 8.10 | 0.85 | 1.00 | 2.00 | 1.25 |
| Particles Antarctica st 28 b | 6.22 | 7.40 | 0.95 | 0.95 | 2.00 | 1.10 |
| Particles Antarctica st 42 b | 6.08 | 6.45 | 1.00 | nd c | 1.70 | 0.90 |
| Particles Antarctica st 46 b | 11.73 | 11.88 | 1.05 | 1.00 | 1.85 | 1.15 |
| Sediment Baffin Bay st 600 a | 20.30 | 21.05 | 5.63 | 4.73 | 20.27 | 17.91 |
| Sediment Baffin Bay st 605 a | 22.98 | 23.56 | 8.67 | 5.33 | 37.67 | 24.11 |
| Sediment Baffin Bay st 615 a | 19.32 | 19.32 | 4.11 | 3.47 | 26.68 | 17.21 |
| Sediment Baffin Bay st 707 a | 45.60 | 49.28 | 2.20 | 2.20 | 37.60 | 40.00 |
| Sediment Baffin Bay st 719 a | 18.19 | 17.87 | 5.00 | 3.27 | 24.33 | 13.5 |
| Cholest-22E-en-3β,5α,6β-Triol | 24-Ethylcholest-22E-en-3β,5α,6β-Triol | 24-Methylcholest-22E-en-3β,5α,6β-Triol | ||||
|---|---|---|---|---|---|---|
| SIM m/z 506 | MRM m/z 506 → m/z 377 | SIM m/z 478 | MRM m/z 478 → m/z 349 | SIM m/z 492 | MRM m/z 492 → m/z 363 | |
| Negev loess sample 1 a | - d | 0.11 | nd c | 0.04 | nd c | 0.02 |
| Negev loess sample 2 a | - d | 0.02 | nd c | nd c | nd c | nd c |
| Negev loess sample 3 a | - d | 0.01 | nd c | nd c | nd c | nd c |
| Particles Antarctica st 4 b | - d | 1.42 | 1.11 | nd c | 1.11 | 1.80 |
| Particles Antarctica st 13 b | - d | 1.50 | nd c | nd c | nd c | 1.40 |
| Particles Antarctica st 28 b | - d | 1.60 | nd c | nd c | nd c | 3.70 |
| Particles Antarctica st 42 b | - d | 1.85 | nd c | nd c | nd c | 2.20 |
| Particles Antarctica st 46 b | - d | 1.95 | nd c | nd c | nd c | 1.70 |
| Sediment Baffin Bay st 600 a | - d | 4.09 | 3.80 | 1.45 | 1.64 | 1.63 |
| Sediment Baffin Bay st 605 a | - d | 6.22 | 4.67 | 1.78 | 1.78 | 1.78 |
| Sediment Baffin Bay st 615 a | - d | 2.68 | nd c | 1.26 | 1.84 | 1.84 |
| Sediment Baffin Bay st 707 a | - d | 6.20 | nd c | 3.40 | nd c | 5.40 |
| Sediment Baffin Bay st 719 a | - d | 1.07 | nd c | 1.07 | 1.40 | 1.40 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aubert, C.; Rontani, J.-F. Use of Trifluoro-Acetate Derivatives for GC-MS and GC-MS/MS Quantification of Trace Amounts of Stera-3β,5α,6β-Triols (Tracers of Δ5-Sterol Autoxidation) in Environmental Samples. Molecules 2023, 28, 1547. https://doi.org/10.3390/molecules28041547
Aubert C, Rontani J-F. Use of Trifluoro-Acetate Derivatives for GC-MS and GC-MS/MS Quantification of Trace Amounts of Stera-3β,5α,6β-Triols (Tracers of Δ5-Sterol Autoxidation) in Environmental Samples. Molecules. 2023; 28(4):1547. https://doi.org/10.3390/molecules28041547
Chicago/Turabian StyleAubert, Claude, and Jean-François Rontani. 2023. "Use of Trifluoro-Acetate Derivatives for GC-MS and GC-MS/MS Quantification of Trace Amounts of Stera-3β,5α,6β-Triols (Tracers of Δ5-Sterol Autoxidation) in Environmental Samples" Molecules 28, no. 4: 1547. https://doi.org/10.3390/molecules28041547
APA StyleAubert, C., & Rontani, J.-F. (2023). Use of Trifluoro-Acetate Derivatives for GC-MS and GC-MS/MS Quantification of Trace Amounts of Stera-3β,5α,6β-Triols (Tracers of Δ5-Sterol Autoxidation) in Environmental Samples. Molecules, 28(4), 1547. https://doi.org/10.3390/molecules28041547