

Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment

 ,
,  ,
,  , , ,
, , , 
Abstract
1. Introduction
2. Structure Variations and Genetics of Lp(a)
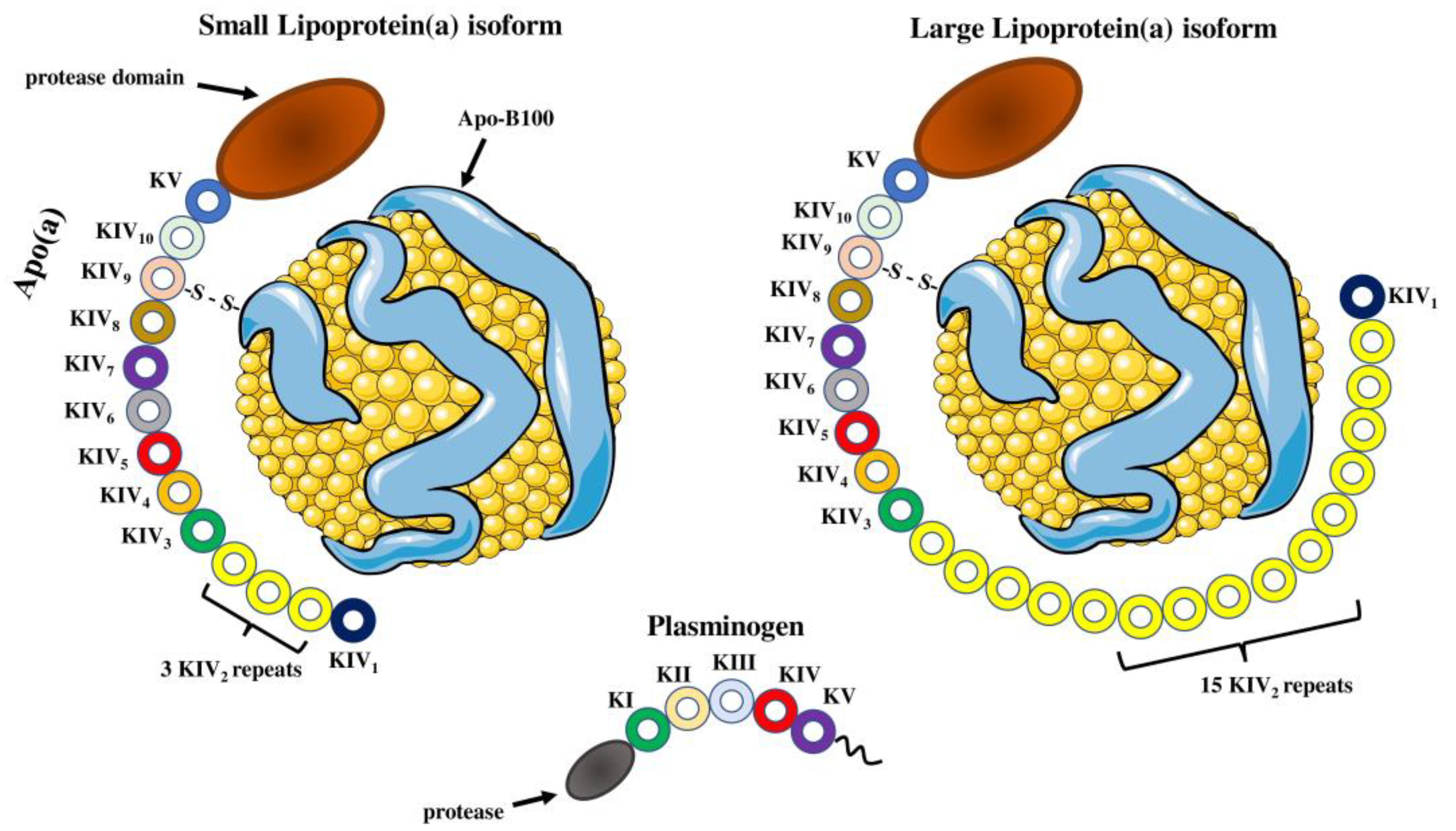
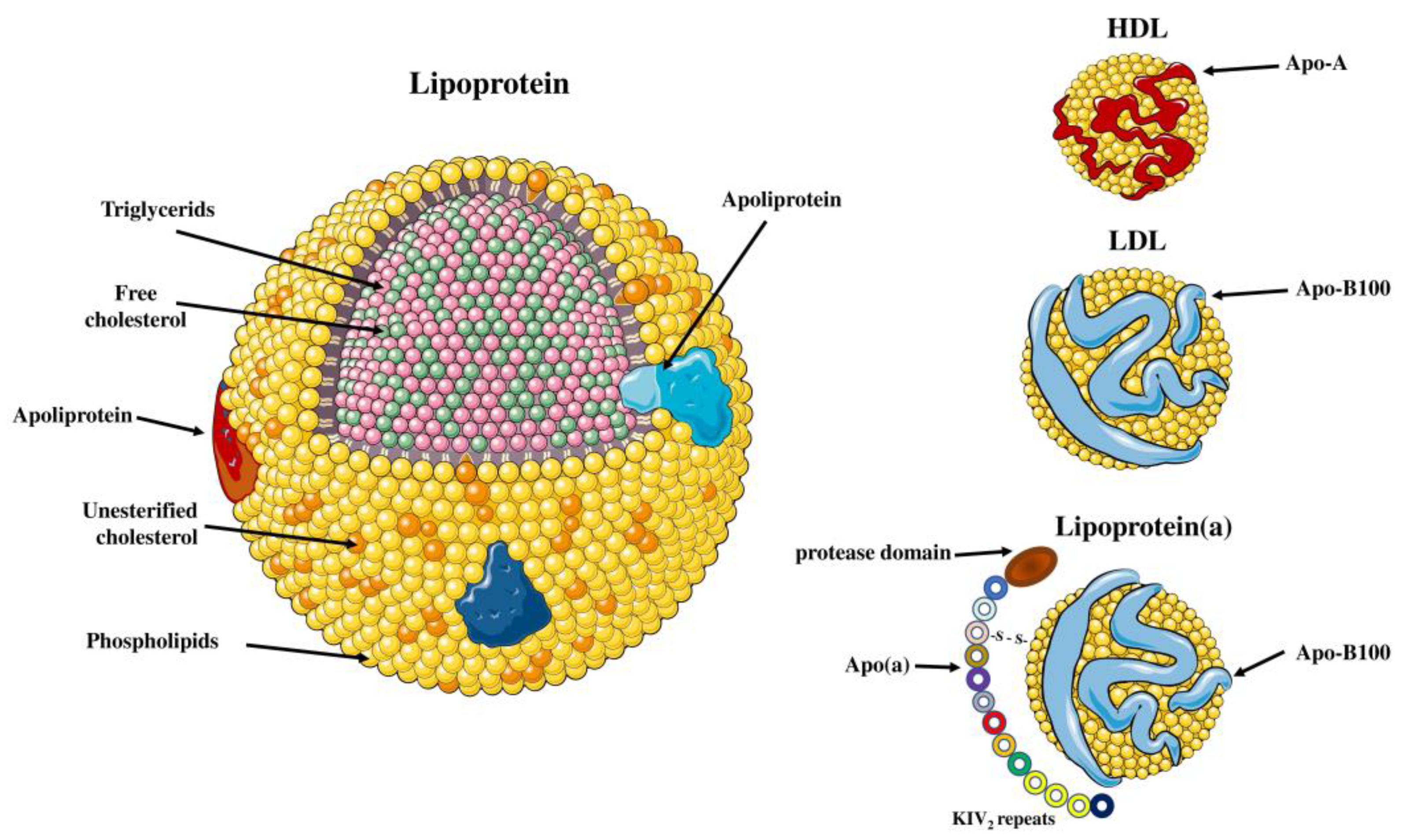
2.1. Structure of the Lp(a) Particle
2.2. Apolipoprotein(a) Structure and Function
2.3. LPA Gene Variations and Atherosclerotic Disease
3. Measurement of Plasma Lp(a) Concentration and Standardization
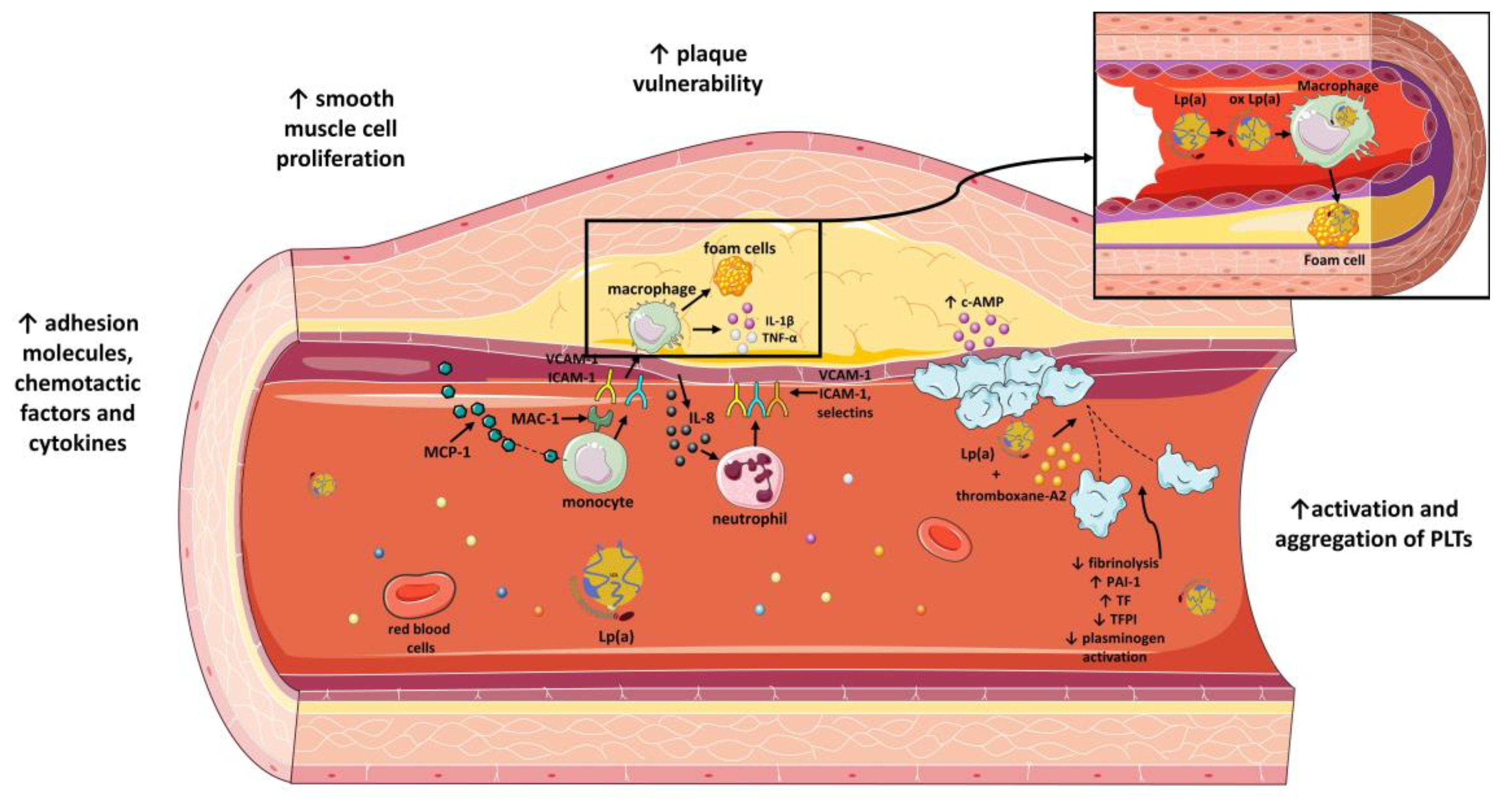
4. Role of Lp(a) on Atherosclerosis
4.1. Attachment of Lp(a) to Arterial Wall
4.2. Effect of Lp(a) on Expression of Adhesion Molecules, Chemotactic Factors and Other Cytokines
4.3. The Impact of Oxidation in Lp(a) Function
4.4. Effect on Vascular Smooth Muscle Cells
4.5. Lp(a) and Plaque Vulnerability
5. Role of Lp(a) in Inflammation
5.1. Oxidized Phospholipids and Lp(a)
5.2. Effect of Lp(a) in Chemotaxis
5.3. Effect of Lp(a) in Monocyte Phenotype
5.4. Lp(a) Induced Changes in the Inflammatory Milieu
6. Role of Lp(a) on Endothelial Function
6.1. Lp(a) Modifies the Properties of Endothelial Cells
6.2. Effect of Lp(a) on Endothelial-Dependent Vascular Tone
7. Role of Lp(a) on Thrombogenicity
7.1. Activation and Aggregation of PLTs
7.2. Interraction of Lp(a) with the Tissue Factor (TF)
7.3. Inhibition of Fibrinolysis by Plasmin
7.4. Increased Expression of Plasminogen Activator Inhibitor
8. Lp(a) and Neointimal Hyperplasia
9. Predictive Value of Lp(a) Levels
10. Lp(a)-Lowering Treatment
11. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kiechl, S.; Willeit, J. The natural course of atherosclerosis. Part I: Incidence and progression. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.J.; Morrisett, J.D.; Sharrett, A.R.; Patsch, W.; Tyroler, H.A.; Wu, K.; Heiss, G. Lipoprotein[a] as a risk factor for preclinical atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 1993, 13, 826–833. [Google Scholar] [CrossRef] [PubMed]
- WHO. Cardiovascular Diseases; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Lechner, K.; von Schacky, C.; McKenzie, A.L.; Worm, N.; Nixdorff, U.; Lechner, B.; Krankel, N.; Halle, M.; Krauss, R.M.; Scherr, J. Lifestyle factors and high-risk atherosclerosis: Pathways and mechanisms beyond traditional risk factors. Eur. J. Prev. Cardiol. 2020, 27, 394–406. [Google Scholar] [CrossRef]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Berg, K. A New Serum Type System in Man—The Lp System. Acta Pathol. Microbiol. Scand. 1963, 59, 369–382. [Google Scholar] [CrossRef]
- Rath, M.; Niendorf, A.; Reblin, T.; Dietel, M.; Krebber, H.J.; Beisiegel, U. Detection and quantification of lipoprotein(a) in the arterial wall of 107 coronary bypass patients. Arteriosclerosis 1989, 9, 579–592. [Google Scholar] [CrossRef]
- Cushing, G.L.; Gaubatz, J.W.; Nava, M.L.; Burdick, B.J.; Bocan, T.M.; Guyton, J.R.; Weilbaecher, D.; DeBakey, M.E.; Lawrie, G.M.; Morrisett, J.D. Quantitation and localization of apolipoproteins [a] and B in coronary artery bypass vein grafts resected at re-operation. Arteriosclerosis 1989, 9, 593–603. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef]
- Lampsas, S.; Oikonomou, E.; Pantelidis, P.; Theofilis, P.; Grammatopoulos, K.; Marathonitis, A.; Vavuranakis, M.A.; Siasos, G.; Tousoulis, D.; Vavuranakis, M. Lipoprotein (a) levels and Abdominal Aortic Aneurysm. A Systematic Review and Meta-analysis. Curr. Pharm. Des. 2022, 28, 3492–3499. [Google Scholar] [CrossRef]
- Ruscica, M.; Sirtori, C.R.; Corsini, A.; Watts, G.F.; Sahebkar, A. Lipoprotein(a): Knowns, unknowns and uncertainties. Pharmacol. Res. 2021, 173, 105812. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) measurements for clinical application. J. Lipid Res. 2016, 57, 526–537. [Google Scholar] [CrossRef]
- Gaubatz, J.W.; Heideman, C.; Gotto, A.M., Jr.; Morrisett, J.D.; Dahlen, G.H. Human plasma lipoprotein [a]. Structural properties. J. Biol. Chem. 1983, 258, 4582–4589. [Google Scholar] [CrossRef]
- Sommer, A.; Gorges, R.; Kostner, G.M.; Paltauf, F.; Hermetter, A. Sulfhydryl-selective fluorescence labeling of lipoprotein(a) reveals evidence for one single disulfide linkage between apoproteins(a) and B-100. Biochemistry 1991, 30, 11245–11249. [Google Scholar] [CrossRef]
- Guevara, J., Jr.; Spurlino, J.; Jan, A.Y.; Yang, C.Y.; Tulinsky, A.; Prasad, B.V.; Gaubatz, J.W.; Morrisett, J.D. Proposed mechanisms for binding of apo[a] kringle type 9 to apo B-100 in human lipoprotein[a]. Biophys. J. 1993, 64, 686–700. [Google Scholar] [CrossRef]
- Tomlinson, J.E.; McLean, J.W.; Lawn, R.M. Rhesus monkey apolipoprotein(a). Sequence, evolution, and sites of synthesis. J. Biol. Chem. 1989, 264, 5957–5965. [Google Scholar] [CrossRef]
- Leibundgut, G.; Scipione, C.; Yin, H.; Schneider, M.; Boffa, M.B.; Green, S.; Yang, X.; Dennis, E.; Witztum, J.L.; Koschinsky, M.L.; et al. Determinants of binding of oxidized phospholipids on apolipoprotein (a) and lipoprotein (a). J. Lipid Res. 2013, 54, 2815–2830. [Google Scholar] [CrossRef]
- Utermann, G.; Weber, W. Protein composition of Lp(a) lipoprotein from human plasma. FEBS Lett. 1983, 154, 357–361. [Google Scholar] [CrossRef]
- Dieplinger, H.; Utermann, G. The seventh myth of lipoprotein(a): Where and how is it assembled? Curr. Opin. Lipidol. 1999, 10, 275–283. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Marcovina, S.M. Structure-function relationships in apolipoprotein(a): Insights into lipoprotein(a) assembly and pathogenicity. Curr. Opin. Lipidol. 2004, 15, 167–174. [Google Scholar] [CrossRef]
- White, A.L.; Lanford, R.E. Cell surface assembly of lipoprotein(a) in primary cultures of baboon hepatocytes. J. Biol. Chem. 1994, 269, 28716–28723. [Google Scholar] [CrossRef] [PubMed]
- Jenner, J.L.; Seman, L.J.; Millar, J.S.; Lamon-Fava, S.; Welty, F.K.; Dolnikowski, G.G.; Marcovina, S.M.; Lichtenstein, A.H.; Barrett, P.H.; deLuca, C.; et al. The metabolism of apolipoproteins (a) and B-100 within plasma lipoprotein (a) in human beings. Metabolism 2005, 54, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Demant, T.; Seeberg, K.; Bedynek, A.; Seidel, D. The metabolism of lipoprotein(a) and other apolipoprotein B-containing lipoproteins: A kinetic study in humans. Atherosclerosis 2001, 157, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Reblin, T.; Rader, D.J.; Beisiegel, U.; Greten, H.; Brewer, H.B., Jr. Correlation of apolipoprotein(a) isoproteins with Lp(a) density and distribution in fasting plasma. Atherosclerosis 1992, 94, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lindahl, M.; Lhomme, M.; Calabresi, L.; Chapman, M.J.; Davidson, W.S. Structure of HDL: Particle subclasses and molecular components. In High Density Lipoproteins; Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2015; Volume 224, pp. 3–51. [Google Scholar] [CrossRef]
- Ponsin, G. Relationship between structure and metabolism of HDL apolipoproteins: Study with synthetic peptides. Adv. Exp. Med. Biol. 1988, 243, 139–147. [Google Scholar] [CrossRef]
- Utermann, G.; Menzel, H.J.; Kraft, H.G.; Duba, H.C.; Kemmler, H.G.; Seitz, C. Lp(a) glycoprotein phenotypes. Inheritance and relation to Lp(a)-lipoprotein concentrations in plasma. J. Clin. Investig. 1987, 80, 458–465. [Google Scholar] [CrossRef]
- McLean, J.W.; Tomlinson, J.E.; Kuang, W.J.; Eaton, D.L.; Chen, E.Y.; Fless, G.M.; Scanu, A.M.; Lawn, R.M. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987, 330, 132–137. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Beisiegel, U.; Henne-Bruns, D.; Eaton, D.L.; Lawn, R.M. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry 1990, 29, 640–644. [Google Scholar] [CrossRef]
- Gabel, B.R.; Koschinsky, M.I. Analysis of the proteolytic activity of a recombinant form of apolipoprotein(a). Biochemistry 1995, 34, 15777–15784. [Google Scholar] [CrossRef]
- Kalaivani, V.; Jaleel, A. Apolipoprotein(a), an enigmatic anti-angiogenic glycoprotein in human plasma: A curse or cure? Pharmacol. Res. 2020, 158, 104858. [Google Scholar] [CrossRef]
- Guevara, J., Jr.; Jan, A.Y.; Knapp, R.; Tulinsky, A.; Morrisett, J.D. Comparison of ligand-binding sites of modeled apo[a] kringle-like sequences in human lipoprotein[a]. Arterioscler. Thromb. Vasc. Biol. 1993, 13, 758–770. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Zhang, Z.H.; Gaur, V.P.; Albers, J.J. Identification of 34 apolipoprotein(a) isoforms: Differential expression of apolipoprotein(a) alleles between American blacks and whites. Biochem. Biophys. Res. Commun. 1993, 191, 1192–1196. [Google Scholar] [CrossRef]
- Lackner, C.; Cohen, J.C.; Hobbs, H.H. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum. Mol. Genet. 1993, 2, 933–940. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Hobbs, H.H.; Albers, J.J. Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: Basis for a standardized isoform nomenclature. Clin. Chem. 1996, 42, 436–439. [Google Scholar] [CrossRef]
- Hixson, J.E.; Britten, M.L.; Manis, G.S.; Rainwater, D.L. Apolipoprotein(a) (Apo(a)) glycoprotein isoforms result from size differences in Apo(a) mRNA in baboons. J. Biol. Chem. 1989, 264, 6013–6016. [Google Scholar] [CrossRef]
- Knight, B.L. Gene structure of apolipoprotein(a) and the regulation of its expression. Biochem. Soc. Trans. 1999, 27, 447–452. [Google Scholar] [CrossRef]
- Zysow, B.R.; Lindahl, G.E.; Wade, D.P.; Knight, B.L.; Lawn, R.M. C/T polymorphism in the 5’ untranslated region of the apolipoprotein(a) gene introduces an upstream ATG and reduces in vitro translation. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 58–64. [Google Scholar] [CrossRef]
- Noureen, A.; Fresser, F.; Utermann, G.; Schmidt, K. Sequence variation within the KIV-2 copy number polymorphism of the human LPA gene in African, Asian, and European populations. PLoS ONE 2015, 10, e0121582. [Google Scholar] [CrossRef]
- Yano, Y.; Shimokawa, K.; Okada, Y.; Noma, A. Immunolocalization of lipoprotein(a) in wounded tissues. J. Histochem. Cytochem. 1997, 45, 559–568. [Google Scholar] [CrossRef]
- Van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef]
- Sabarinath, P.S.; Appukuttan, P.S. Immunopathology of desialylation: Human plasma lipoprotein(a) and circulating anti-carbohydrate antibodies form immune complexes that recognize host cells. Mol. Cell. Biochem. 2015, 403, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Erqou, S.; Thompson, A.; Di Angelantonio, E.; Saleheen, D.; Kaptoge, S.; Marcovina, S.; Danesh, J. Apolipoprotein(a) isoforms and the risk of vascular disease: Systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol. 2010, 55, 2160–2167. [Google Scholar] [CrossRef] [PubMed]
- Zeljkovic, A.; Bogavac-Stanojevic, N.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; Vekic, J.; Spasic, S. Combined effects of small apolipoprotein (a) isoforms and small, dense LDL on coronary artery disease risk. Arch. Med. Res. 2009, 40, 29–35. [Google Scholar] [CrossRef]
- Frank, S.L.; Klisak, I.; Sparkes, R.S.; Mohandas, T.; Tomlinson, J.E.; McLean, J.W.; Lawn, R.M.; Lusis, A.J. The apolipoprotein(a) gene resides on human chromosome 6q26-27, in close proximity to the homologous gene for plasminogen. Hum. Genet. 1988, 79, 352–356. [Google Scholar] [CrossRef]
- Utermann, G. The mysteries of lipoprotein(a). Science 1989, 246, 904–910. [Google Scholar] [CrossRef]
- Erdel, M.; Hubalek, M.; Lingenhel, A.; Kofler, K.; Duba, H.C.; Utermann, G. Counting the repetitive kringle-IV repeats in the gene encoding human apolipoprotein(a) by fibre-FISH. Nat. Genet. 1999, 21, 357–358. [Google Scholar] [CrossRef]
- Sandholzer, C.; Hallman, D.M.; Saha, N.; Sigurdsson, G.; Lackner, C.; Csaszar, A.; Boerwinkle, E.; Utermann, G. Effects of the apolipoprotein(a) size polymorphism on the lipoprotein(a) concentration in 7 ethnic groups. Hum. Genet. 1991, 86, 607–614. [Google Scholar] [CrossRef]
- Coassin, S.; Kronenberg, F. Lipoprotein(a) beyond the kringle IV repeat polymorphism: The complexity of genetic variation in the LPA gene. Atherosclerosis 2022, 349, 17–35. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjaerg-Hansen, A.; Steffensen, R.; Nordestgaard, B.G. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA 2009, 301, 2331–2339. [Google Scholar] [CrossRef]
- Tregouet, D.A.; Konig, I.R.; Erdmann, J.; Munteanu, A.; Braund, P.S.; Hall, A.S.; Grosshennig, A.; Linsel-Nitschke, P.; Perret, C.; DeSuremain, M.; et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat. Genet. 2009, 41, 283–285. [Google Scholar] [CrossRef]
- Tsimikas, S.; Hall, J.L. Lipoprotein(a) as a potential causal genetic risk factor of cardiovascular disease: A rationale for increased efforts to understand its pathophysiology and develop targeted therapies. J. Am. Coll. Cardiol. 2012, 60, 716–721. [Google Scholar] [CrossRef]
- Kivimaki, M.; Magnussen, C.G.; Juonala, M.; Kahonen, M.; Kettunen, J.; Loo, B.M.; Lehtimaki, T.; Viikari, J.; Raitakari, O.T. Conventional and Mendelian randomization analyses suggest no association between lipoprotein(a) and early atherosclerosis: The Young Finns Study. Int. J. Epidemiol. 2011, 40, 470–478. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Back, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef]
- Ali, S.; Bunker, C.H.; Aston, C.E.; Ukoli, F.A.; Kamboh, M.I. Apolipoprotein A kringle 4 polymorphism and serum lipoprotein (a) concentrations in African blacks. Hum. Biol. 1998, 70, 477–490. [Google Scholar]
- Kraft, H.G.; Lingenhel, A.; Pang, R.W.; Delport, R.; Trommsdorff, M.; Vermaak, H.; Janus, E.D.; Utermann, G. Frequency distributions of apolipoprotein(a) kringle IV repeat alleles and their effects on lipoprotein(a) levels in Caucasian, Asian, and African populations: The distribution of null alleles is non-random. Eur. J. Hum. Genet. 1996, 4, 74–87. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Viney, N.J.; Xia, S.; Witztum, J.L.; Marcovina, S.M. Relationship of lipoprotein(a) molar concentrations and mass according to lipoprotein(a) thresholds and apolipoprotein(a) isoform size. J. Clin. Lipidol. 2018, 12, 1313–1323. [Google Scholar] [CrossRef]
- McConnell, J.P.; Guadagno, P.A.; Dayspring, T.D.; Hoefner, D.M.; Thiselton, D.L.; Warnick, G.R.; Harris, W.S. Lipoprotein(a) mass: A massively misunderstood metric. J. Clin. Lipidol. 2014, 8, 550–553. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Navabi, N.; Allen, S.; Gonen, A.; Witztum, J.L.; Tsimikas, S. Development and validation of an isoform-independent monoclonal antibody-based ELISA for measurement of lipoprotein(a). J. Lipid Res. 2022, 63, 100239. [Google Scholar] [CrossRef]
- De Boer, L.M.; Oorthuys, A.O.J.; Wiegman, A.; Langendam, M.W.; Kroon, J.; Spijker, R.; Zwinderman, A.H.; Hutten, B.A. Statin therapy and lipoprotein(a) levels: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2022, 29, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Guddeti, R.R.; Patil, S.; Ahmed, A.; Sharma, A.; Aboeata, A.; Lavie, C.J.; Alla, V.M. Lipoprotein(a) and calcific aortic valve stenosis: A systematic review. Prog. Cardiovasc. Dis. 2020, 63, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Lewis, B. Influx in vivo of low density, intermediate density, and very low density lipoproteins into aortic intimas of genetically hyperlipidemic rabbits. Roles of plasma concentrations, extent of aortic lesion, and lipoprotein particle size as determinants. Arterioscler. Thromb. Vasc. Biol. 1992, 12, 6–18. [Google Scholar] [CrossRef]
- Miles, L.A.; Fless, G.M.; Scanu, A.M.; Baynham, P.; Sebald, M.T.; Skocir, P.; Curtiss, L.K.; Levin, E.G.; Hoover-Plow, J.L.; Plow, E.F. Interaction of Lp(a) with plasminogen binding sites on cells. Thromb. Haemost. 1995, 73, 458–465. [Google Scholar] [CrossRef]
- Angles-Cano, E.; Rojas, G. Apolipoprotein(a): Structure-function relationship at the lysine-binding site and plasminogen activator cleavage site. Biol. Chem. 2002, 383, 93–99. [Google Scholar] [CrossRef]
- Kreuzer, J.; Lloyd, M.B.; Bok, D.; Fless, G.M.; Scanu, A.M.; Lusis, A.J.; Haberland, M.E. Lipoprotein (a) displays increased accumulation compared with low-density lipoprotein in the murine arterial wall. Chem. Phys. Lipids 1994, 67–68, 175–190. [Google Scholar] [CrossRef]
- Smith, E.B.; Cochran, S. Factors influencing the accumulation in fibrous plaques of lipid derived from low density lipoprotein. II. Preferential immobilization of lipoprotein (a) (Lp(a)). Atherosclerosis 1990, 84, 173–181. [Google Scholar] [CrossRef]
- van der Hoek, Y.Y.; Sangrar, W.; Cote, G.P.; Kastelein, J.J.; Koschinsky, M.L. Binding of recombinant apolipoprotein(a) to extracellular matrix proteins. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 1792–1798. [Google Scholar] [CrossRef]
- Moser, T.L.; Enghild, J.J.; Pizzo, S.V.; Stack, M.S. The extracellular matrix proteins laminin and fibronectin contain binding domains for human plasminogen and tissue plasminogen activator. J. Biol. Chem. 1993, 268, 18917–18923. [Google Scholar] [CrossRef]
- Boonmark, N.W.; Lou, X.J.; Yang, Z.J.; Schwartz, K.; Zhang, J.L.; Rubin, E.M.; Lawn, R.M. Modification of apolipoprotein(a) lysine binding site reduces atherosclerosis in transgenic mice. J. Clin. Investig. 1997, 100, 558–564. [Google Scholar] [CrossRef]
- Klezovitch, O.; Edelstein, C.; Zhu, L.; Scanu, A.M. Apolipoprotein(a) binds via its C-terminal domain to the protein core of the proteoglycan decorin. Implications for the retention of lipoprotein(a) in atherosclerotic lesions. J. Biol. Chem. 1998, 273, 23856–23865. [Google Scholar] [CrossRef][Green Version]
- Riessen, R.; Isner, J.M.; Blessing, E.; Loushin, C.; Nikol, S.; Wight, T.N. Regional differences in the distribution of the proteoglycans biglycan and decorin in the extracellular matrix of atherosclerotic and restenotic human coronary arteries. Am. J. Pathol. 1994, 144, 962–974. [Google Scholar]
- Bdeir, K.; Cane, W.; Canziani, G.; Chaiken, I.; Weisel, J.; Koschinsky, M.L.; Lawn, R.M.; Bannerman, P.G.; Sachais, B.S.; Kuo, A.; et al. Defensin promotes the binding of lipoprotein(a) to vascular matrix. Blood 1999, 94, 2007–2019. [Google Scholar]
- Ehnholm, C.; Jauhiainen, M.; Metso, J. Interaction of lipoprotein(a) with fibronectin and its potential role in atherogenesis. Eur. Heart J. 1990, 11 (Suppl. E), 190–195. [Google Scholar] [CrossRef]
- Falcone, D.J.; Salisbury, B.G. Fibronectin stimulates macrophage uptake of low density lipoprotein-heparin-collagen complexes. Arteriosclerosis 1988, 8, 263–273. [Google Scholar] [CrossRef]
- Salonen, E.M.; Jauhiainen, M.; Zardi, L.; Vaheri, A.; Ehnholm, C. Lipoprotein(a) binds to fibronectin and has serine proteinase activity capable of cleaving it. EMBO J. 1989, 8, 4035–4040. [Google Scholar] [CrossRef]
- Bottalico, L.A.; Keesler, G.A.; Fless, G.M.; Tabas, I. Cholesterol loading of macrophages leads to marked enhancement of native lipoprotein(a) and apoprotein(a) internalization and degradation. J. Biol. Chem. 1993, 268, 8569–8573. [Google Scholar] [CrossRef]
- Tabas, I.; Li, Y.; Brocia, R.W.; Xu, S.W.; Swenson, T.L.; Williams, K.J. Lipoprotein lipase and sphingomyelinase synergistically enhance the association of atherogenic lipoproteins with smooth muscle cells and extracellular matrix. A possible mechanism for low density lipoprotein and lipoprotein(a) retention and macrophage foam cell formation. J. Biol. Chem. 1993, 268, 20419–20432. [Google Scholar]
- Haberland, M.E.; Fless, G.M.; Scanu, A.M.; Fogelman, A.M. Malondialdehyde modification of lipoprotein(a) produces avid uptake by human monocyte-macrophages. J. Biol. Chem. 1992, 267, 4143–4151. [Google Scholar] [CrossRef]
- Oikonomou, E.; Souvaliotis, N.; Lampsas, S.; Siasos, G.; Poulakou, G.; Theofilis, P.; Papaioannou, T.G.; Haidich, A.B.; Tsaousi, G.; Ntousopoulos, V.; et al. Endothelial dysfunction in acute and long standing COVID-19: A prospective cohort study. Vascul. Pharmacol. 2022, 144, 106975. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.; Khan, S.; Tam, S.; Koschinsky, M.; Taylor, P.; Yacoub, M. Expression of adhesion molecules by lp(a): A potential novel mechanism for its atherogenicity. FASEB J. 1998, 12, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Takami, S.; Yamashita, S.; Kihara, S.; Ishigami, M.; Takemura, K.; Kume, N.; Kita, T.; Matsuzawa, Y. Lipoprotein(a) enhances the expression of intercellular adhesion molecule-1 in cultured human umbilical vein endothelial cells. Circulation 1998, 97, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, S.N.; Orlova, V.V.; Al-Fakhri, N.; Ihanus, E.; Economopoulou, M.; Isermann, B.; Bdeir, K.; Nawroth, P.P.; Preissner, K.T.; Gahmberg, C.G.; et al. Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J. 2006, 20, 559–561. [Google Scholar] [CrossRef]
- Syrovets, T.; Thillet, J.; Chapman, M.J.; Simmet, T. Lipoprotein(a) is a potent chemoattractant for human peripheral monocytes. Blood 1997, 90, 2027–2036. [Google Scholar] [CrossRef]
- Labudovic, D.; Kostovska, I.; Tosheska Trajkovska, K.; Cekovska, S.; Brezovska Kavrakova, J.; Topuzovska, S. Lipoprotein(a)—Link between Atherogenesis and Thrombosis. Prague Med. Rep. 2019, 120, 39–51. [Google Scholar] [CrossRef]
- Klezovitch, O.; Edelstein, C.; Scanu, A.M. Stimulation of interleukin-8 production in human THP-1 macrophages by apolipoprotein(a). Evidence for a critical involvement of elements in its C-terminal domain. J. Biol. Chem. 2001, 276, 46864–46869. [Google Scholar] [CrossRef]
- Baggiolini, M.; Walz, A.; Kunkel, S.L. Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Investig. 1989, 84, 1045–1049. [Google Scholar] [CrossRef]
- Scipione, C.A.; Sayegh, S.E.; Romagnuolo, R.; Tsimikas, S.; Marcovina, S.M.; Boffa, M.B.; Koschinsky, M.L. Mechanistic insights into Lp(a)-induced IL-8 expression: A role for oxidized phospholipid modification of apo(a). J. Lipid Res. 2015, 56, 2273–2285. [Google Scholar] [CrossRef]
- Oikonomou, E.; Tsaplaris, P.; Anastasiou, A.; Xenou, M.; Lampsas, S.; Siasos, G.; Pantelidis, P.; Theofilis, P.; Tsatsaragkou, A.; Katsarou, O.; et al. Interleukin-1 in Coronary Artery Disease. Curr. Top. Med. Chem. 2022, 22, 2368–2389. [Google Scholar] [CrossRef]
- Tsimikas, S.; Bergmark, C.; Beyer, R.W.; Patel, R.; Pattison, J.; Miller, E.; Juliano, J.; Witztum, J.L. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Bergmark, C.; Dewan, A.; Orsoni, A.; Merki, E.; Miller, E.R.; Shin, M.J.; Binder, C.J.; Horkko, S.; Krauss, R.M.; Chapman, M.J.; et al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J. Lipid Res. 2008, 49, 2230–2239. [Google Scholar] [CrossRef] [PubMed]
- Karabina, S.A.; Elisaf, M.C.; Goudevenos, J.; Siamopoulos, K.C.; Sideris, D.; Tselepis, A.D. PAF-acetylhydrolase activity of Lp(a) before and during Cu(2+)-induced oxidative modification in vitro. Atherosclerosis 1996, 125, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Tsironis, L.D.; Tselepis, A.D. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2094–2099. [Google Scholar] [CrossRef]
- Tsimikas, S.; Witztum, J.L. The role of oxidized phospholipids in mediating lipoprotein(a) atherogenicity. Curr. Opin. Lipidol. 2008, 19, 369–377. [Google Scholar] [CrossRef]
- Navab, M.; Hama, S.Y.; Reddy, S.T.; Ng, C.J.; Van Lenten, B.J.; Laks, H.; Fogelman, A.M. Oxidized lipids as mediators of coronary heart disease. Curr. Opin. Lipidol. 2002, 13, 363–372. [Google Scholar] [CrossRef]
- Grainger, D.J.; Kemp, P.R.; Liu, A.C.; Lawn, R.M.; Metcalfe, J.C. Activation of transforming growth factor-beta is inhibited in transgenic apolipoprotein(a) mice. Nature 1994, 370, 460–462. [Google Scholar] [CrossRef]
- Grainger, D.J.; Kirschenlohr, H.L.; Metcalfe, J.C.; Weissberg, P.L.; Wade, D.P.; Lawn, R.M. Proliferation of human smooth muscle cells promoted by lipoprotein(a). Science 1993, 260, 1655–1658. [Google Scholar] [CrossRef]
- Riches, K.; Franklin, L.; Maqbool, A.; Peckham, M.; Adams, M.; Bond, J.; Warburton, P.; Feric, N.T.; Koschinsky, M.L.; O’Regan, D.J.; et al. Apolipoprotein(a) acts as a chemorepellent to human vascular smooth muscle cells via integrin alphaVbeta3 and RhoA/ROCK-mediated mechanisms. Int. J. Biochem. Cell Biol. 2013, 45, 1776–1783. [Google Scholar] [CrossRef]
- Chang, Y.J.; Wang, K.C. Therapeutic perspectives of extracellular vesicles and extracellular microRNAs in atherosclerosis. Curr. Top. Membr. 2021, 87, 255–277. [Google Scholar] [CrossRef]
- Wang, C.; Li, Z.; Liu, Y.; Yuan, L. Exosomes in atherosclerosis: Performers, bystanders, biomarkers, and therapeutic targets. Theranostics 2021, 11, 3996–4010. [Google Scholar] [CrossRef]
- Badimon, L.; Padro, T.; Arderiu, G.; Vilahur, G.; Borrell-Pages, M.; Suades, R. Extracellular vesicles in atherothrombosis: From biomarkers and precision medicine to therapeutic targets. Immunol. Rev. 2022, 312, 6–19. [Google Scholar] [CrossRef]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef]
- Wang, C.; Liu, C.; Shi, J.; Li, H.; Jiang, S.; Zhao, P.; Zhang, M.; Du, G.; Fu, S.; Li, S.; et al. Nicotine exacerbates endothelial dysfunction and drives atherosclerosis via extracellular vesicle-miRNA. Cardiovasc. Res. 2022, cvac140. [Google Scholar] [CrossRef]
- Rogers, M.A.; Atkins, S.K.; Zheng, K.H.; Singh, S.A.; Chelvanambi, S.; Pham, T.H.; Kuraoka, S.; Stroes, E.S.G.; Aikawa, M.; Aikawa, E. Lipoprotein(a) Induces Vesicular Cardiovascular Calcification Revealed With Single-Extracellular Vesicle Analysis. Front. Cardiovasc. Med. 2022, 9, 778919. [Google Scholar] [CrossRef]
- Edelstein, C.; Shapiro, S.D.; Klezovitch, O.; Scanu, A.M. Macrophage metalloelastase, MMP-12, cleaves human apolipoprotein(a) in the linker region between kringles IV-4 and IV-5. Potential relevance to lipoprotein(a) biology. J. Biol. Chem. 1999, 274, 10019–10023. [Google Scholar] [CrossRef]
- Edelstein, C.; Italia, J.A.; Klezovitch, O.; Scanu, A.M. Functional and metabolic differences between elastase-generated fragments of human lipoprotein[a] and apolipoprotein[a]. J. Lipid Res. 1996, 37, 1786–1801. [Google Scholar] [CrossRef]
- Moreau, M.; Brocheriou, I.; Petit, L.; Ninio, E.; Chapman, M.J.; Rouis, M. Interleukin-8 mediates downregulation of tissue inhibitor of metalloproteinase-1 expression in cholesterol-loaded human macrophages: Relevance to stability of atherosclerotic plaque. Circulation 1999, 99, 420–426. [Google Scholar] [CrossRef]
- Ganne, F.; Vasse, M.; Beaudeux, J.L.; Peynet, J.; Francois, A.; Paysant, J.; Lenormand, B.; Collet, J.P.; Vannier, J.P.; Soria, J.; et al. Increased expression of u-PA and u-PAR on monocytes by LDL and Lp(a) lipoproteins—Consequences for plasmin generation and monocyte adhesion. Thromb. Haemost. 1999, 81, 594–600. [Google Scholar] [CrossRef]
- Seimon, T.A.; Nadolski, M.J.; Liao, X.; Magallon, J.; Nguyen, M.; Feric, N.T.; Koschinsky, M.L.; Harkewicz, R.; Witztum, J.L.; Tsimikas, S.; et al. Atherogenic lipids and lipoproteins trigger CD36-TLR2-dependent apoptosis in macrophages undergoing endoplasmic reticulum stress. Cell Metab. 2010, 12, 467–482. [Google Scholar] [CrossRef]
- Oikonomou, E.; Theofilis, P.; Lampsas, S.; Katsarou, O.; Kalogeras, K.; Marinos, G.; Tsatsaragkou, A.; Anastasiou, A.; Lysandrou, A.; Gounaridi, M.I.; et al. Current Concepts and Future Applications of Non-Invasive Functional and Anatomical Evaluation of Coronary Artery Disease. Life 2022, 12, 1803. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ. Res. 2016, 118, 547–563. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Choi, S.H.; Wiesner, P.; Fang, L.; Harkewicz, R.; Hartvigsen, K.; Boullier, A.; Gonen, A.; Diehl, C.J.; Que, X.; et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011, 108, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, P.; Tafelmeier, M.; Chittka, D.; Choi, S.H.; Zhang, L.; Byun, Y.S.; Almazan, F.; Yang, X.; Iqbal, N.; Chowdhury, P.; et al. MCP-1 binds to oxidized LDL and is carried by lipoprotein(a) in human plasma. J. Lipid Res. 2013, 54, 1877–1883. [Google Scholar] [CrossRef] [PubMed]
- Haque, N.S.; Zhang, X.; French, D.L.; Li, J.; Poon, M.; Fallon, J.T.; Gabel, B.R.; Taubman, M.B.; Koschinsky, M.; Harpel, P.C. CC chemokine I-309 is the principal monocyte chemoattractant induced by apolipoprotein(a) in human vascular endothelial cells. Circulation 2000, 102, 786–792. [Google Scholar] [CrossRef]
- Hoover-Plow, J.; Hart, E.; Gong, Y.; Shchurin, A.; Schneeman, T. A physiological function for apolipoprotein(a): A natural regulator of the inflammatory response. Exp. Biol. Med. 2009, 234, 28–34. [Google Scholar] [CrossRef]
- Huang, M.; Gong, Y.; Grondolsky, J.; Hoover-Plow, J. Lp(a)/apo(a) modulate MMP-9 activation and neutrophil cytokines in vivo in inflammation to regulate leukocyte recruitment. Am. J. Pathol. 2014, 184, 1503–1517. [Google Scholar] [CrossRef]
- Sabbah, N.; Jaisson, S.; Garnotel, R.; Angles-Cano, E.; Gillery, P. Small size apolipoprotein(a) isoforms enhance inflammatory and proteolytic potential of collagen-primed monocytes. Lipids Health Dis. 2019, 18, 166. [Google Scholar] [CrossRef]
- Schmitz, G.; Orso, E. Lipoprotein(a) hyperlipidemia as cardiovascular risk factor: Pathophysiological aspects. Clin. Res. Cardiol. Suppl. 2015, 10, 21–25. [Google Scholar] [CrossRef]
- Buechler, C.; Ullrich, H.; Aslanidis, C.; Bared, S.M.; Lingenhel, A.; Ritter, M.; Schmitz, G. Lipoprotein (a) downregulates lysosomal acid lipase and induces interleukin-6 in human blood monocytes. Biochim. Biophys. Acta 2003, 1642, 25–31. [Google Scholar] [CrossRef]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Ramharack, R.; Barkalow, D.; Spahr, M.A. Dominant negative effect of TGF-beta1 and TNF-alpha on basal and IL-6-induced lipoprotein(a) and apolipoprotein(a) mRNA expression in primary monkey hepatocyte cultures. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 984–990. [Google Scholar] [CrossRef]
- Brown, S.A.; Morrisett, J.D.; Boerwinkle, E.; Hutchinson, R.; Patsch, W. The relation of lipoprotein[a] concentrations and apolipoprotein[a] phenotypes with asymptomatic atherosclerosis in subjects of the Atherosclerosis Risk in Communities (ARIC) Study. Arterioscler. Thromb. Vasc. Biol. 1993, 13, 1558–1566. [Google Scholar] [CrossRef]
- Maher, V.M.; Brown, B.G.; Marcovina, S.M.; Hillger, L.A.; Zhao, X.Q.; Albers, J.J. Effects of lowering elevated LDL cholesterol on the cardiovascular risk of lipoprotein(a). JAMA 1995, 274, 1771–1774. [Google Scholar] [CrossRef]
- Dangas, G.; Mehran, R.; Harpel, P.C.; Sharma, S.K.; Marcovina, S.M.; Dube, G.; Ambrose, J.A.; Fallon, J.T. Lipoprotein(a) and inflammation in human coronary atheroma: Association with the severity of clinical presentation. J. Am. Coll. Cardiol. 1998, 32, 2035–2042. [Google Scholar] [CrossRef]
- Gazzaruso, C.; Geroldi, D.; Garzaniti, A.; Falcone, C.; Fratino, P.; Finardi, G.; Buscaglia, P. Apolipoprotein(a) phenotypes as genetic markers of coronary atherosclerosis severity. Int. J. Cardiol. 1998, 64, 277–284. [Google Scholar] [CrossRef]
- Kronenberg, F.; Kronenberg, M.F.; Kiechl, S.; Trenkwalder, E.; Santer, P.; Oberhollenzer, F.; Egger, G.; Utermann, G.; Willeit, J. Role of lipoprotein(a) and apolipoprotein(a) phenotype in atherogenesis: Prospective results from the Bruneck study. Circulation 1999, 100, 1154–1160. [Google Scholar] [CrossRef]
- Paultre, F.; Tuck, C.H.; Boden-Albala, B.; Kargman, D.E.; Todd, E.; Jones, J.; Paik, M.C.; Sacco, R.L.; Berglund, L. Relation of Apo(a) size to carotid atherosclerosis in an elderly multiethnic population. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 141–146. [Google Scholar] [CrossRef][Green Version]
- Tsimikas, S.; Kiechl, S.; Willeit, J.; Mayr, M.; Miller, E.R.; Kronenberg, F.; Xu, Q.; Bergmark, C.; Weger, S.; Oberhollenzer, F.; et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: Five-year prospective results from the Bruneck study. J. Am. Coll. Cardiol. 2006, 47, 2219–2228. [Google Scholar] [CrossRef]
- Kiechl, S.; Willeit, J.; Mayr, M.; Viehweider, B.; Oberhollenzer, M.; Kronenberg, F.; Wiedermann, C.J.; Oberthaler, S.; Xu, Q.; Witztum, J.L.; et al. Oxidized phospholipids, lipoprotein(a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: Prospective results from the Bruneck study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, A.; Radwan, J.; Kubicka, A.; Libura, M.; Sacha, T.; Swadzba, J.; Undas, A.; Szczeklik, J.; Jodlowski, J. Plasma fibrinolytic activity in healthy subjects with high and low lipoprotein(a) concentrations. Thromb. Res. 1992, 66, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Testa, R.; Marcovina, S.M. The rate of plasmin formation after in vitro clotting is inversely related to lipoprotein(a) plasma levels. Int. J. Clin. Lab. Res. 1999, 29, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Von Depka, M.; Nowak-Gottl, U.; Eisert, R.; Dieterich, C.; Barthels, M.; Scharrer, I.; Ganser, A.; Ehrenforth, S. Increased lipoprotein (a) levels as an independent risk factor for venous thromboembolism. Blood 2000, 96, 3364–3368. [Google Scholar] [CrossRef]
- Bilgen, D.; Sonmez, H.; Ekmekci, H.; Ulutin, T.; Ozturk, Z.; Kokoglu, E.; Bayram, C.; Soner, A.; Domanic, N. The relationship of TFPI, Lp(a), and oxidized LDL antibody levels in patients with coronary artery disease. Clin. Biochem. 2005, 38, 92–96. [Google Scholar] [CrossRef]
- Di Nisio, M.; ten Wolde, M.; Meijers, J.C.; Buller, H.R. Effects of high plasma lipoprotein (a) levels on tissue factor pathway inhibitor and the protein C pathway. J. Thromb. Haemost. 2005, 3, 2123–2125. [Google Scholar] [CrossRef]
- Undas, A.; Stepien, E.; Tracz, W.; Szczeklik, A. Lipoprotein(a) as a modifier of fibrin clot permeability and susceptibility to lysis. J. Thromb. Haemost. 2006, 4, 973–975. [Google Scholar] [CrossRef]
- Pineda, J.; Marin, F.; Marco, P.; Roldan, V.; Valencia, J.; Ruiz-Nodar, J.M.; Sogorb, F.; Lip, G.Y. Premature coronary artery disease in young (age <45) subjects: Interactions of lipid profile, thrombophilic and haemostatic markers. Int. J. Cardiol. 2009, 136, 222–225. [Google Scholar] [CrossRef]
- Liu, H.; Fu, D.; Luo, Y.; Peng, D. Independent association of Lp(a) with platelet reactivity in subjects without statins or antiplatelet agents. Sci. Rep. 2022, 12, 16609. [Google Scholar] [CrossRef]
- Wu, H.D.; Berglund, L.; Dimayuga, C.; Jones, J.; Sciacca, R.R.; Di Tullio, M.R.; Homma, S. High lipoprotein(a) levels and small apolipoprotein(a) sizes are associated with endothelial dysfunction in a multiethnic cohort. J. Am. Coll. Cardiol. 2004, 43, 1828–1833. [Google Scholar] [CrossRef]
- Anuurad, E.; Rubin, J.; Chiem, A.; Tracy, R.P.; Pearson, T.A.; Berglund, L. High levels of inflammatory biomarkers are associated with increased allele-specific apolipoprotein(a) levels in African-Americans. J. Clin. Endocrinol. Metab. 2008, 93, 1482–1488. [Google Scholar] [CrossRef]
- Volpato, S.; Vigna, G.B.; McDermott, M.M.; Cavalieri, M.; Maraldi, C.; Lauretani, F.; Bandinelli, S.; Zuliani, G.; Guralnik, J.M.; Fellin, R.; et al. Lipoprotein(a), inflammation, and peripheral arterial disease in a community-based sample of older men and women (the InCHIANTI study). Am. J. Cardiol. 2010, 105, 1825–1830. [Google Scholar] [CrossRef]
- Nemati, H.; Khodarahmi, R.; Rahmani, A.; Ebrahimi, A.; Amani, M.; Eftekhari, K. Serum lipid profile in psoriatic patients: Correlation between vascular adhesion protein 1 and lipoprotein (a). Cell Biochem. Funct. 2013, 31, 36–40. [Google Scholar] [CrossRef]
- Nishino, M.; Mori, N.; Yoshimura, T.; Nakamura, D.; Lee, Y.; Taniike, M.; Makino, N.; Kato, H.; Egami, Y.; Shutta, R.; et al. Higher serum uric acid and lipoprotein(a) are correlated with coronary spasm. Heart Vessels 2014, 29, 186–190. [Google Scholar] [CrossRef]
- Mu, W.; Chen, M.; Gong, Z.; Zheng, F.; Xing, Q. Expression of vascular cell adhesion molecule-1 in the aortic tissues of atherosclerotic patients and the associated clinical implications. Exp. Ther. Med. 2015, 10, 423–428. [Google Scholar] [CrossRef]
- Topciu-Shufta, V.; Haxhibeqiri, V.; Begolli, L.; Baruti-Gafurri, Z.; Veseli, S.; Haxhibeqiri, S.; Miftari, R.; Kurti, L.; Avdiu, D. Correlation of Inflammation and Lipoprotein (a) with Hypercoagulability in Hemodialysis Patients. Med. Arch. 2015, 69, 232–235. [Google Scholar] [CrossRef]
- Iwabayashi, M.; Taniyama, Y.; Sanada, F.; Azuma, J.; Iekushi, K.; Okayama, K.; Chatterjee, A.; Rakugi, H.; Morishita, R. Inhibition of Lp(a)-induced functional impairment of endothelial cells and endothelial progenitor cells by hepatocyte growth factor. Biochem. Biophys. Res. Commun. 2012, 423, 79–84. [Google Scholar] [CrossRef]
- Wei, D.H.; Zhang, X.L.; Wang, R.; Zeng, J.F.; Zhang, K.; Yang, J.; Li, S.; Lin, X.L.; Jiang, Z.S.; Wang, G.X.; et al. Oxidized lipoprotein(a) increases endothelial cell monolayer permeability via ROS generation. Lipids 2013, 48, 579–586. [Google Scholar] [CrossRef]
- Cho, T.; Jung, Y.; Koschinsky, M.L. Apolipoprotein(a), through its strong lysine-binding site in KIV(10’), mediates increased endothelial cell contraction and permeability via a Rho/Rho kinase/MYPT1-dependent pathway. J. Biol. Chem. 2008, 283, 30503–30512. [Google Scholar] [CrossRef]
- Pellegrino, M.; Furmaniak-Kazmierczak, E.; LeBlanc, J.C.; Cho, T.; Cao, K.; Marcovina, S.M.; Boffa, M.B.; Cote, G.P.; Koschinsky, M.L. The apolipoprotein(a) component of lipoprotein(a) stimulates actin stress fiber formation and loss of cell-cell contact in cultured endothelial cells. J. Biol. Chem. 2004, 279, 6526–6533. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, K.; Li, S.; Tong, Z.; Li, G.; Zhao, Z.; Zhao, Y.; Liu, F.; Lin, X.; Wang, Z.; et al. Apolipoprotein (a) impairs endothelial progenitor cell-mediated angiogenesis. DNA Cell Biol. 2013, 32, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Schulter, V.; Koolwijk, P.; Peters, E.; Frank, S.; Hrzenjak, A.; Graier, W.F.; van Hinsbergh, V.W.; Kostner, G.M. Impact of apolipoprotein(a) on in vitro angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Craig, A.W.; Meldrum, H.D.; Marcovina, S.M.; Elliott, B.E.; Koschinsky, M.L. Apolipoprotein(a) stimulates vascular endothelial cell growth and migration and signals through integrin alphaVbeta3. Biochem. J. 2009, 418, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Muller, G.; Behrens, J.; Nussbaumer, U.; Bohlen, P.; Birchmeier, W. Inhibitory action of transforming growth factor beta on endothelial cells. Proc. Natl. Acad. Sci. USA 1987, 84, 5600–5604. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.; Romagnuolo, R.; Scipione, C.; Boffa, M.B.; Koschinsky, M.L. Apolipoprotein(a) stimulates nuclear translocation of beta-catenin: A novel pathogenic mechanism for lipoprotein(a). Mol. Biol. Cell 2013, 24, 210–221. [Google Scholar] [CrossRef]
- Moeslinger, T.; Friedl, R.; Volf, I.; Brunner, M.; Koller, E.; Spieckermann, P.G. Inhibition of inducible nitric oxide synthesis by oxidized lipoprotein(a) in a murine macrophage cell line. FEBS Lett. 2000, 478, 95–99. [Google Scholar] [CrossRef]
- Rubanyi, G.M.; Freay, A.D.; Lawn, R.M. Endothelium-dependent vasorelaxation in the aorta of transgenic mice expressing human apolipoprotein(a) or lipoprotein(a). Endothelium 2000, 7, 253–264. [Google Scholar] [CrossRef]
- Galle, J.; Winner, B.; Conzelmann, E.; Wanner, C. Impairment of endothelial function induced by glyc-oxidized lipoprotein a [Lp(a)]. Cell. Mol. Biol. 1998, 44, 1035–1045. [Google Scholar]
- Furchgott, R.F. Role of endothelium in responses of vascular smooth muscle. Circ. Res. 1983, 53, 557–573. [Google Scholar] [CrossRef]
- Schlaich, M.P.; John, S.; Langenfeld, M.R.; Lackner, K.J.; Schmitz, G.; Schmieder, R.E. Does lipoprotein(a) impair endothelial function? J. Am. Coll. Cardiol. 1998, 31, 359–365. [Google Scholar] [CrossRef][Green Version]
- Barre, D.E. Lipoprotein (a) reduces platelet aggregation via apo(a)-mediated decreases in thromboxane A(2)production. Platelets 1998, 9, 93–96. [Google Scholar] [CrossRef]
- Pedreno, J.; Fernandez, R.; Cullare, C.; Barcelo, A.; Elorza, M.A.; de Castellarnau, C. Platelet integrin alpha IIb beta 3 (GPIIb-IIIa) is not implicated in the binding of LDL to intact resting platelets. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 156–163. [Google Scholar] [CrossRef]
- Barre, D.E. Arginyl-glycyl-aspartyl (RGD) epitope of human apolipoprotein (a) inhibits platelet aggregation by antagonizing the IIb subunit of the fibrinogen (GPIIb/IIIa) receptor. Thromb. Res. 2007, 119, 601–607. [Google Scholar] [CrossRef]
- Malle, E.; Ibovnik, A.; Stienmetz, A.; Kostner, G.M.; Sattler, W. Identification of glycoprotein IIb as the lipoprotein(a)-binding protein on platelets. Lipoprotein(a) binding is independent of an arginyl-glycyl-aspartate tripeptide located in apolipoprotein(a). Arterioscler. Thromb. Vasc. Biol. 1994, 14, 345–352. [Google Scholar] [CrossRef]
- Hu, D.D.; White, C.A.; Panzer-Knodle, S.; Page, J.D.; Nicholson, N.; Smith, J.W. A new model of dual interacting ligand binding sites on integrin alphaIIbbeta3. J. Biol. Chem 1999, 274, 4633–4639. [Google Scholar] [CrossRef]
- Barre, D.E. Apoprotein (A) antagonises THE GPIIB/IIIA receptor on collagen and adp-stimulated human platelets. Front. Biosci. 2004, 9, 404–410. [Google Scholar] [CrossRef][Green Version]
- Barre, D.E. Apolipoprotein (a) mediates the lipoprotein (a)-induced biphasic shift in human platelet cyclic AMP. Thromb. Res. 2003, 112, 321–324. [Google Scholar] [CrossRef]
- Martinez, C.; Rivera, J.; Loyau, S.; Corral, J.; Gonzalez-Conejero, R.; Lozano, M.L.; Vicente, V.; Angles-Cano, E. Binding of recombinant apolipoprotein(a) to human platelets and effect on platelet aggregation. Thromb. Haemost. 2001, 85, 686–693. [Google Scholar] [CrossRef]
- Barre, D. Human lipoprotein (a)-induced reduction of platelet aggregation is not mediated by apolipoprotein A’s lysine-binding regions. Front. Biosci. 2003, 8, s1226–s1228. [Google Scholar] [CrossRef][Green Version]
- Malle, E.; Ibovnik, A.; Leis, H.J.; Kostner, G.M.; Verhallen, P.F.; Sattler, W. Lysine modification of LDL or lipoprotein(a) by 4-hydroxynonenal or malondialdehyde decreases platelet serotonin secretion without affecting platelet aggregability and eicosanoid formation. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 377–384. [Google Scholar] [CrossRef]
- Rand, M.L.; Sangrar, W.; Hancock, M.A.; Taylor, D.M.; Marcovina, S.M.; Packham, M.A.; Koschinsky, M.L. Apolipoprotein(a) enhances platelet responses to the thrombin receptor-activating peptide SFLLRN. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Tsironis, L.D.; Mitsios, J.V.; Milionis, H.J.; Elisaf, M.; Tselepis, A.D. Effect of lipoprotein (a) on platelet activation induced by platelet-activating factor: Role of apolipoprotein (a) and endogenous PAF-acetylhydrolase. Cardiovasc. Res. 2004, 63, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Bacchetti, T.; Johnston, T.P.; Banach, M.; Pirro, M.; Sahebkar, A. Lipoprotein(a): A missing culprit in the management of athero-thrombosis? J. Cell Physiol. 2018, 233, 2966–2981. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.S.; Kuppuswamy, M.N.; Saito, H.; Spitzer, S.G.; Bajaj, S.P. Cultured normal human hepatocytes do not synthesize lipoprotein-associated coagulation inhibitor: Evidence that endothelium is the principal site of its synthesis. Proc. Natl. Acad. Sci. USA 1990, 87, 8869–8873. [Google Scholar] [CrossRef]
- Caplice, N.M.; Panetta, C.; Peterson, T.E.; Kleppe, L.S.; Mueske, C.S.; Kostner, G.M.; Broze, G.J., Jr.; Simari, R.D. Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: A novel link between lipoproteins and thrombosis. Blood 2001, 98, 2980–2987. [Google Scholar] [CrossRef]
- Broze, G.J., Jr.; Girard, T.J. Tissue factor pathway inhibitor: Structure-function. Front. Biosci. Landmark 2012, 17, 262–280. [Google Scholar] [CrossRef]
- Beisiegel, U.; Niendorf, A.; Wolf, K.; Reblin, T.; Rath, M. Lipoprotein(a) in the arterial wall. Eur. Heart J. 1990, 11 (Suppl. E), 174–183. [Google Scholar] [CrossRef]
- Castellino, F.J. Biochemistry of human plasminogen. Semin. Thromb. Hemost. 1984, 10, 18–23. [Google Scholar] [CrossRef]
- Hervio, L.; Chapman, M.J.; Thillet, J.; Loyau, S.; Angles-Cano, E. Does apolipoprotein(a) heterogeneity influence lipoprotein(a) effects on fibrinolysis? Blood 1993, 82, 392–397. [Google Scholar] [CrossRef]
- Angles-Cano, E.; de la Pena Diaz, A.; Loyau, S. Inhibition of fibrinolysis by lipoprotein(a). Ann. N. Y. Acad. Sci. 2001, 936, 261–275. [Google Scholar] [CrossRef]
- Harpel, P.C.; Gordon, B.R.; Parker, T.S. Plasmin catalyzes binding of lipoprotein (a) to immobilized fibrinogen and fibrin. Proc. Natl. Acad. Sci. USA 1989, 86, 3847–3851. [Google Scholar] [CrossRef]
- Miles, L.A.; Fless, G.M.; Levin, E.G.; Scanu, A.M.; Plow, E.F. A potential basis for the thrombotic risks associated with lipoprotein(a). Nature 1989, 339, 301–303. [Google Scholar] [CrossRef]
- Hajjar, K.A.; Gavish, D.; Breslow, J.L.; Nachman, R.L. Lipoprotein(a) modulation of endothelial cell surface fibrinolysis and its potential role in atherosclerosis. Nature 1989, 339, 303–305. [Google Scholar] [CrossRef]
- Ezratty, A.; Simon, D.I.; Loscalzo, J. Lipoprotein(a) binds to human platelets and attenuates plasminogen binding and activation. Biochemistry 1993, 32, 4628–4633. [Google Scholar] [CrossRef]
- Romanic, A.M.; Arleth, A.J.; Willette, R.N.; Ohlstein, E.H. Factor XIIIa cross-links lipoprotein(a) with fibrinogen and is present in human atherosclerotic lesions. Circ. Res. 1998, 83, 264–269. [Google Scholar] [CrossRef]
- Sangrar, W.; Bajzar, L.; Nesheim, M.E.; Koschinsky, M.L. Antifibrinolytic effect of recombinant apolipoprotein(a) in vitro is primarily due to attenuation of tPA-mediated Glu-plasminogen activation. Biochemistry 1995, 34, 5151–5157. [Google Scholar] [CrossRef]
- Hoylaerts, M.; Rijken, D.C.; Lijnen, H.R.; Collen, D. Kinetics of the activation of plasminogen by human tissue plasminogen activator. Role of fibrin. J. Biol. Chem. 1982, 257, 2912–2919. [Google Scholar] [CrossRef]
- Horrevoets, A.J.; Pannekoek, H.; Nesheim, M.E. A steady-state template model that describes the kinetics of fibrin-stimulated [Glu1]- and [Lys78]plasminogen activation by native tissue-type plasminogen activator and variants that lack either the finger or kringle-2 domain. J. Biol. Chem. 1997, 272, 2183–2191. [Google Scholar] [CrossRef]
- Hancock, M.A.; Boffa, M.B.; Marcovina, S.M.; Nesheim, M.E.; Koschinsky, M.L. Inhibition of plasminogen activation by lipoprotein(a): Critical domains in apolipoprotein(a) and mechanism of inhibition on fibrin and degraded fibrin surfaces. J. Biol. Chem. 2003, 278, 23260–23269. [Google Scholar] [CrossRef]
- Simon, D.I.; Fless, G.M.; Scanu, A.M.; Loscalzo, J. Tissue-type plasminogen activator binds to and is inhibited by surface-bound lipoprotein(a) and low-density lipoprotein. Biochemistry 1991, 30, 6671–6677. [Google Scholar] [CrossRef]
- Loscalzo, J.; Weinfeld, M.; Fless, G.M.; Scanu, A.M. Lipoprotein(a), fibrin binding, and plasminogen activation. Arteriosclerosis 1990, 10, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Hajjar, K.A.; Hamel, N.M.; Harpel, P.C.; Nachman, R.L. Binding of tissue plasminogen activator to cultured human endothelial cells. J. Clin. Investig. 1987, 80, 1712–1719. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.N.; Harpel, P.C.; Pannell, R.; Gurewich, V. Lipoprotein(a): A kinetic study of its influence on fibrin-dependent plasminogen activation by prourokinase or tissue plasminogen activator. Biochemistry 1993, 32, 9694–9700. [Google Scholar] [CrossRef] [PubMed]
- Palabrica, T.M.; Liu, A.C.; Aronovitz, M.J.; Furie, B.; Lawn, R.M.; Furie, B.C. Antifibrinolytic activity of apolipoprotein(a) in vivo: Human apolipoprotein(a) transgenic mice are resistant to tissue plasminogen activator-mediated thrombolysis. Nat. Med. 1995, 1, 256–259. [Google Scholar] [CrossRef]
- Edelberg, J.M.; Gonzalez-Gronow, M.; Pizzo, S.V. Lipoprotein a inhibits streptokinase-mediated activation of human plasminogen. Biochemistry 1989, 28, 2370–2374. [Google Scholar] [CrossRef]
- Etingin, O.R.; Hajjar, D.P.; Hajjar, K.A.; Harpel, P.C.; Nachman, R.L. Lipoprotein (a) regulates plasminogen activator inhibitor-1 expression in endothelial cells. A potential mechanism in thrombogenesis. J. Biol. Chem. 1991, 266, 2459–2465. [Google Scholar] [CrossRef]
- Buechler, C.; Ullrich, H.; Ritter, M.; Porsch-Oezcueruemez, M.; Lackner, K.J.; Barlage, S.; Friedrich, S.O.; Kostner, G.M.; Schmitz, G. Lipoprotein (a) up-regulates the expression of the plasminogen activator inhibitor 2 in human blood monocytes. Blood 2001, 97, 981–986. [Google Scholar] [CrossRef]
- Hoff, H.F.; Beck, G.J.; Skibinski, C.I.; Jurgens, G.; O’Neil, J.; Kramer, J.; Lytle, B. Serum Lp(a) level as a predictor of vein graft stenosis after coronary artery bypass surgery in patients. Circulation 1988, 77, 1238–1244. [Google Scholar] [CrossRef]
- Hearn, J.A.; Donohue, B.C.; Ba’albaki, H.; Douglas, J.S.; King, S.B., 3rd; Lembo, N.J.; Roubin, G.S.; Sgoutas, D.S. Usefulness of serum lipoprotein (a) as a predictor of restenosis after percutaneous transluminal coronary angioplasty. Am. J. Cardiol. 1992, 69, 736–739. [Google Scholar] [CrossRef]
- Desmarais, R.L.; Sarembock, I.J.; Ayers, C.R.; Vernon, S.M.; Powers, E.R.; Gimple, L.W. Elevated serum lipoprotein(a) is a risk factor for clinical recurrence after coronary balloon angioplasty. Circulation 1995, 91, 1403–1409. [Google Scholar] [CrossRef]
- Kamitani, T.; Taniguchi, T.; Miyai, N.; Kawasaki, T.; Kawasaki, S.; Sugihara, H. Association between plasma lipoprotein(a) concentration and restenosis after stent implantation. Circ. J. 2005, 69, 644–649. [Google Scholar] [CrossRef]
- Tenda, K.; Saikawa, T.; Maeda, T.; Sato, Y.; Niwa, H.; Inoue, T.; Yonemochi, H.; Maruyama, T.; Shimoyama, N.; Aragaki, S.; et al. The relationship between serum lipoprotein(a) and restenosis after initial elective percutaneous transluminal coronary angioplasty. Jpn. Circ. J. 1993, 57, 789–795. [Google Scholar] [CrossRef][Green Version]
- Shah, P.K.; Amin, J. Low high density lipoprotein level is associated with increased restenosis rate after coronary angioplasty. Circulation 1992, 85, 1279–1285. [Google Scholar] [CrossRef]
- Cooke, T.; Sheahan, R.; Foley, D.; Reilly, M.; D’Arcy, G.; Jauch, W.; Gibney, M.; Gearty, G.; Crean, P.; Walsh, M. Lipoprotein(a) in restenosis after percutaneous transluminal coronary angioplasty and coronary artery disease. Circulation 1994, 89, 1593–1598. [Google Scholar] [CrossRef][Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Ray, K.; Boren, J.; Andreotti, F.; Watts, G.F.; Ginsberg, H.; Amarenco, P.; Catapano, A.; Descamps, O.S.; et al. Lipoprotein(a) as a cardiovascular risk factor: Current status. Eur. Heart J. 2010, 31, 2844–2853. [Google Scholar] [CrossRef]
- Rogers, C.; Edelman, E.R.; Simon, D.I. A mAb to the beta2-leukocyte integrin Mac-1 (CD11b/CD18) reduces intimal thickening after angioplasty or stent implantation in rabbits. Proc. Natl. Acad. Sci. USA 1998, 95, 10134–10139. [Google Scholar] [CrossRef]
- Zioncheck, T.F.; Powell, L.M.; Rice, G.C.; Eaton, D.L.; Lawn, R.M. Interaction of recombinant apolipoprotein(a) and lipoprotein(a) with macrophages. J. Clin. Investig. 1991, 87, 767–771. [Google Scholar] [CrossRef]
- Araki, T.; Nakamura, M.; Sugi, K. Characterization of in-stent neointimal tissue components following drug-eluting stent implantation according to the phase of restenosis using a 40-MHz intravascular ultrasound imaging system. J. Cardiol. 2014, 64, 423–429. [Google Scholar] [CrossRef]
- Takano, M.; Yamamoto, M.; Inami, S.; Murakami, D.; Ohba, T.; Seino, Y.; Mizuno, K. Appearance of lipid-laden intima and neovascularization after implantation of bare-metal stents extended late-phase observation by intracoronary optical coherence tomography. J. Am. Coll. Cardiol. 2009, 55, 26–32. [Google Scholar] [CrossRef]
- Hasegawa, K.; Tamai, H.; Kyo, E.; Kosuga, K.; Ikeguchi, S.; Hata, T.; Okada, M.; Fujita, S.; Tsuji, T.; Takeda, S.; et al. Histopathological findings of new in-stent lesions developed beyond five years. Catheter Cardiovasc. Interv. 2006, 68, 554–558. [Google Scholar] [CrossRef]
- Ross, R.; Glomset, J.; Kariya, B.; Harker, L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro. Proc. Natl. Acad. Sci. USA 1974, 71, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Braga, S.F.; Neves, J.R.; Ferreira, J.; Carrilho, C.; Simoes, J.C.; Mesquita, A. Neointimal Hyperplasia. Rev. Port. Cir. Cardiotorac. Vasc. 2019, 26, 213–217. [Google Scholar] [PubMed]
- Schwartz, R.S.; Holmes, D.R., Jr.; Topol, E.J. The restenosis paradigm revisited: An alternative proposal for cellular mechanisms. J. Am. Coll. Cardiol. 1992, 20, 1284–1293. [Google Scholar] [CrossRef]
- McNamara, C.A.; Sarembock, I.J.; Gimple, L.W.; Fenton, J.W., 2nd; Coughlin, S.R.; Owens, G.K. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J. Clin. Investig. 1993, 91, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Gasic, G.P.; Arenas, C.P.; Gasic, T.B.; Gasic, G.J. Coagulation factors X, Xa, and protein S as potent mitogens of cultured aortic smooth muscle cells. Proc. Natl. Acad. Sci. USA 1992, 89, 2317–2320. [Google Scholar] [CrossRef]
- Oltrona, L.; Speidel, C.M.; Recchia, D.; Wickline, S.A.; Eisenberg, P.R.; Abendschein, D.R. Inhibition of tissue factor-mediated coagulation markedly attenuates stenosis after balloon-induced arterial injury in minipigs. Circulation 1997, 96, 646–652. [Google Scholar] [CrossRef]
- Jang, Y.; Guzman, L.A.; Lincoff, A.M.; Gottsauner-Wolf, M.; Forudi, F.; Hart, C.E.; Courtman, D.W.; Ezban, M.; Ellis, S.G.; Topol, E.J. Influence of blockade at specific levels of the coagulation cascade on restenosis in a rabbit atherosclerotic femoral artery injury model. Circulation 1995, 92, 3041–3050. [Google Scholar] [CrossRef]
- Brown, D.M.; Kania, N.M.; Choi, E.T.; Lantieri, L.A.; Pasia, E.N.; Wun, T.C.; Khouri, R.K. Local irrigation with tissue factor pathway inhibitor inhibits intimal hyperplasia induced by arterial interventions. Arch. Surg. 1996, 131, 1086–1090. [Google Scholar] [CrossRef]
- Kojima, S.; Harpel, P.C.; Rifkin, D.B. Lipoprotein (a) inhibits the generation of transforming growth factor beta: An endogenous inhibitor of smooth muscle cell migration. J. Cell Biol. 1991, 113, 1439–1445. [Google Scholar] [CrossRef]
- Miyata, M.; Biro, S.; Kaieda, H.; Tanaka, H. Lipoprotein(a) stimulates the proliferation of cultured human arterial smooth muscle cells through two pathways. FEBS Lett. 1995, 377, 493–496. [Google Scholar] [CrossRef]
- Strandkjaer, N.; Hansen, M.K.; Nielsen, S.T.; Frikke-Schmidt, R.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Tabor, A.; Bundgaard, H.; Iversen, K.; Kamstrup, P.R. Lipoprotein(a) Levels at Birth and in Early Childhood: The COMPARE Study. J. Clin. Endocrinol. Metab. 2022, 107, 324–335. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Ceska, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Frohlich, J.; Dobiasova, M.; Adler, L.; Francis, M. Gender differences in plasma levels of lipoprotein (a) in patients with angiographically proven coronary artery disease. Physiol. Res. 2004, 53, 481–486. [Google Scholar]
- Nago, N.; Kayaba, K.; Hiraoka, J.; Matsuo, H.; Goto, T.; Kario, K.; Tsutsumi, A.; Nakamura, Y.; Igarashi, M. Lipoprotein(a) levels in the Japanese population: Influence of age and sex, and relation to atherosclerotic risk factors. The Jichi Medical School Cohort Study. Am. J. Epidemiol. 1995, 141, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Simony, S.B.; Mortensen, M.B.; Langsted, A.; Afzal, S.; Kamstrup, P.R.; Nordestgaard, B.G. Sex differences of lipoprotein(a) levels and associated risk of morbidity and mortality by age: The Copenhagen General Population Study. Atherosclerosis 2022, 355, 76–82. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein (a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef]
- Corsetti, J.P.; Sterry, J.A.; Sparks, J.D.; Sparks, C.E.; Weintraub, M. Effect of weight loss on serum lipoprotein(a) concentrations in an obese population. Clin. Chem. 1991, 37, 1191–1195. [Google Scholar] [CrossRef]
- Jenner, J.L.; Ordovas, J.M.; Lamon-Fava, S.; Schaefer, M.M.; Wilson, P.W.; Castelli, W.P.; Schaefer, E.J. Effects of age, sex, and menopausal status on plasma lipoprotein(a) levels. The Framingham Offspring Study. Circulation 1993, 87, 1135–1141. [Google Scholar] [CrossRef]
- Catapano, A.L.; Graham, I.; De Backer, G.; Wiklund, O.; Chapman, M.J.; Drexel, H.; Hoes, A.W.; Jennings, C.S.; Landmesser, U.; Pedersen, T.R.; et al. 2016 ESC/EAS Guidelines for the Management of Dyslipidaemias. Eur. Heart J. 2016, 37, 2999–3058. [Google Scholar] [CrossRef]
- Conroy, R.M.; Pyorala, K.; Fitzgerald, A.P.; Sans, S.; Menotti, A.; De Backer, G.; De Bacquer, D.; Ducimetiere, P.; Jousilahti, P.; Keil, U.; et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: The SCORE project. Eur. Heart J. 2003, 24, 987–1003. [Google Scholar] [CrossRef]
- Davidson, M.H.; Ballantyne, C.M.; Jacobson, T.A.; Bittner, V.A.; Braun, L.T.; Brown, A.S.; Brown, W.V.; Cromwell, W.C.; Goldberg, R.B.; McKenney, J.M.; et al. Clinical utility of inflammatory markers and advanced lipoprotein testing: Advice from an expert panel of lipid specialists. J. Clin. Lipidol. 2011, 5, 338–367. [Google Scholar] [CrossRef]
- Anderson, T.J.; Gregoire, J.; Hegele, R.A.; Couture, P.; Mancini, G.B.; McPherson, R.; Francis, G.A.; Poirier, P.; Lau, D.C.; Grover, S.; et al. 2012 update of the Canadian Cardiovascular Society guidelines for the diagnosis and treatment of dyslipidemia for the prevention of cardiovascular disease in the adult. Can. J. Cardiol. 2013, 29, 151–167. [Google Scholar] [CrossRef]
- Cegla, J.; Neely, R.D.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J.; Datta, D.; Capps, N.; Shoulders, C.; Qureshi, N.; et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70. [Google Scholar] [CrossRef]
- Cegla, J.; France, M.; Marcovina, S.M.; Neely, R.D.G. Lp(a): When and how to measure it. Ann. Clin. Biochem. 2021, 58, 16–21. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Cho, L.; Nicholls, S.J.; Kastelein, J.; Leitersdorf, E.; Landmesser, U.; Blaha, M.; Lincoff, A.M.; Morishita, R.; et al. Lipoprotein(a) levels in a global population with established atherosclerotic cardiovascular disease. Open Heart 2022, 9, e002060. [Google Scholar] [CrossRef]
- Farnier, M.; Chague, F.; Maza, M.; Bichat, F.; Masson, D.; Cottin, Y.; Zeller, M. High lipoprotein(a) levels predict severity of coronary artery disease in patients hospitalized for acute myocardial infarction. Data from the French RICO survey. J. Clin. Lipidol. 2022, 16, P685–P693. [Google Scholar] [CrossRef]
- Clarke, R.; Hammami, I.; Sherliker, P.; Valdes-Marquez, E.; Watkins, H.; Hill, M.; Yang, X.; Tsimikas, S.; Hopewell, J.C.; Consortium, P. Oxidized phospholipids on apolipoprotein B-100 versus plasminogen and risk of coronary heart disease in the PROCARDIS study. Atherosclerosis 2022, 354, 15–22. [Google Scholar] [CrossRef]
- Nurmohamed, N.S.; Kaiser, Y.; Schuitema, P.C.E.; Ibrahim, S.; Nierman, M.; Fischer, J.C.; Chamuleau, S.A.J.; Knaapen, P.; Stroes, E.S.G. Finding very high lipoprotein(a): The need for routine assessment. Eur. J. Prev. Cardiol. 2022, 29, 769–776. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-Lowering by 50 mg/dL (105 nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention: A Population-Based Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 255–266. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Arsenault, B.J.; Petrides, F.; Tabet, F.; Bao, W.; Hovingh, G.K.; Boekholdt, S.M.; Ramin-Mangata, S.; Meilhac, O.; DeMicco, D.; Rye, K.A.; et al. Effect of atorvastatin, cholesterol ester transfer protein inhibition, and diabetes mellitus on circulating proprotein subtilisin kexin type 9 and lipoprotein(a) levels in patients at high cardiovascular risk. J. Clin. Lipidol. 2018, 12, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, J.; Ju, J.; Fan, Y.; Xu, H. Effect of different types and dosages of statins on plasma lipoprotein(a) levels: A network meta-analysis. Pharmacol. Res. 2021, 163, 105275. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration Group. Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef]
- Sahebkar, A.; Simental-Mendia, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirtori, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef]
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of nicotinic acid on serum cholesterol in man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Guyton, J.R.; Blazing, M.A.; Hagar, J.; Kashyap, M.L.; Knopp, R.H.; McKenney, J.M.; Nash, D.T.; Nash, S.D. Extended-release niacin vs gemfibrozil for the treatment of low levels of high-density lipoprotein cholesterol. Niaspan-Gemfibrozil Study Group. Arch. Intern. Med. 2000, 160, 1177–1184. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Sahebkar, A.; Reiner, Z.; Simental-Mendia, L.E.; Ferretti, G.; Cicero, A.F. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Julius, U.; Fischer, S. Nicotinic acid as a lipid-modifying drug—A review. Atheroscler. Suppl. 2013, 14, 7–13. [Google Scholar] [CrossRef]
- Reyes-Soffer, G.; Pavlyha, M.; Ngai, C.; Thomas, T.; Holleran, S.; Ramakrishnan, R.; Karmally, W.; Nandakumar, R.; Fontanez, N.; Obunike, J.; et al. Effects of PCSK9 Inhibition With Alirocumab on Lipoprotein Metabolism in Healthy Humans. Circulation 2017, 135, 352–362. [Google Scholar] [CrossRef]
- Tavori, H.; Christian, D.; Minnier, J.; Plubell, D.; Shapiro, M.D.; Yeang, C.; Giunzioni, I.; Croyal, M.; Duell, P.B.; Lambert, G.; et al. PCSK9 Association With Lipoprotein(a). Circ. Res. 2016, 119, 29–35. [Google Scholar] [CrossRef]
- Stein, E.A.; Raal, F.J. Insights into PCSK9, low-density lipoprotein receptor, and low-density lipoprotein cholesterol metabolism: Of mice and man. Circulation 2013, 127, 2372–2374. [Google Scholar] [CrossRef][Green Version]
- Sun, H.; Samarghandi, A.; Zhang, N.; Yao, Z.; Xiong, M.; Teng, B.B. Proprotein convertase subtilisin/kexin type 9 interacts with apolipoprotein B and prevents its intracellular degradation, irrespective of the low-density lipoprotein receptor. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1585–1595. [Google Scholar] [CrossRef]
- Farmakis, I.; Doundoulakis, I.; Pagiantza, A.; Zafeiropoulos, S.; Antza, C.; Karvounis, H.; Giannakoulas, G. Lipoprotein(a) Reduction With Proprotein Convertase Subtilisin/Kexin Type 9 Inhibitors: A Systematic Review and Meta-analysis. J. Cardiovasc. Pharmacol. 2021, 77, 397–407. [Google Scholar] [CrossRef]
- Romagnuolo, R.; Scipione, C.A.; Boffa, M.B.; Marcovina, S.M.; Seidah, N.G.; Koschinsky, M.L. Lipoprotein(a) catabolism is regulated by proprotein convertase subtilisin/kexin type 9 through the low density lipoprotein receptor. J. Biol. Chem. 2015, 290, 11649–11662. [Google Scholar] [CrossRef]
- Villard, E.F.; Thedrez, A.; Blankenstein, J.; Croyal, M.; Tran, T.T.; Poirier, B.; Le Bail, J.C.; Illiano, S.; Nobecourt, E.; Krempf, M.; et al. PCSK9 Modulates the Secretion But Not the Cellular Uptake of Lipoprotein(a) Ex Vivo: An Effect Blunted by Alirocumab. JACC Basic Transl. Sci. 2016, 1, 419–427. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Greco, M.F.; Rizzuto, A.S.; Zara, M.; Cafora, M.; Favero, C.; Solazzo, G.; Giusti, I.; Adorni, M.P.; Zimetti, F.; Dolo, V.; et al. PCSK9 Confers Inflammatory Properties to Extracellular Vesicles Released by Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2022, 23, 13065. [Google Scholar] [CrossRef]
- Moriarty, P.M.; Parhofer, K.G.; Babirak, S.P.; Cornier, M.A.; Duell, P.B.; Hohenstein, B.; Leebmann, J.; Ramlow, W.; Schettler, V.; Simha, V.; et al. Alirocumab in patients with heterozygous familial hypercholesterolaemia undergoing lipoprotein apheresis: The ODYSSEY ESCAPE trial. Eur. Heart J. 2016, 37, 3588–3595. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Gipe, D.; Bergeron, J.; Gaudet, D.; Weiss, R.; Dufour, R.; Wu, R.; Pordy, R. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: A phase 2 randomised controlled trial. Lancet 2012, 380, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Rader, D.J.; Raal, F.J.; Guyton, J.R.; Baccara-Dinet, M.T.; Lorenzato, C.; Pordy, R.; Stroes, E. Efficacy and Safety of Alirocumab in Patients with Heterozygous Familial Hypercholesterolemia and LDL-C of 160 mg/dl or Higher. Cardiovasc. Drugs Ther. 2016, 30, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Stein, E.A.; Dufour, R.; Turner, T.; Civeira, F.; Burgess, L.; Langslet, G.; Scott, R.; Olsson, A.G.; Sullivan, D.; et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 331–340. [Google Scholar] [CrossRef]
- Raal, F.; Scott, R.; Somaratne, R.; Bridges, I.; Li, G.; Wasserman, S.M.; Stein, E.A. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: The Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012, 126, 2408–2417. [Google Scholar] [CrossRef]
- Raal, F.J.; Honarpour, N.; Blom, D.J.; Hovingh, G.K.; Xu, F.; Scott, R.; Wasserman, S.M.; Stein, E.A.; Investigators, T. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): A randomised, double-blind, placebo-controlled trial. Lancet 2015, 385, 341–350. [Google Scholar] [CrossRef]
- Ge, X.; Zhu, T.; Zeng, H.; Yu, X.; Li, J.; Xie, S.; Wan, J.; Yang, H.; Huang, K.; Zhang, W. A Systematic Review and Meta-Analysis of Therapeutic Efficacy and Safety of Alirocumab and Evolocumab on Familial Hypercholesterolemia. Biomed Res. Int. 2021, 2021, 8032978. [Google Scholar] [CrossRef]
- Borrelli, M.J.; Youssef, A.; Boffa, M.B.; Koschinsky, M.L. New Frontiers in Lp(a)-Targeted Therapies. Trends Pharmacol. Sci. 2019, 40, 212–225. [Google Scholar] [CrossRef]
- Graham, M.J.; Viney, N.; Crooke, R.M.; Tsimikas, S. Antisense inhibition of apolipoprotein (a) to lower plasma lipoprotein (a) levels in humans. J. Lipid Res. 2016, 57, 340–351. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Study | Study Design | Population Characteristics | Key Findings |
|---|---|---|---|
| Atherosclerosis | |||
| Brown et al., 1993 [126] | Case-control | 171 cases with preclinical extracranial carotid atherosclerosis and 274 control subjects free of carotid atherosclerosis. |
|
| Maher et al., 1995 [127] | Randomized controlled trial | 146 men aged 62 years or younger with CAD and apo B-100 levels ≥125 mg/dL. Step II Diet and lovastatin (40 mg daily) plus colestipol (30 g daily), niacin (4 g daily) plus colestipol or placebo (plus colestipol if LDL > 90th percentile) for 2.5 years. |
|
| Dangas et al., 1997 [128] | Cross-sectional | Coronary atheroma removed from 72 patients with stable or unstable angina. |
|
| Gazzaruso et al., 1998 [129] | Cross-sectional | 267 patients with CAD. |
|
| Kronenberg et al., 1999 [130] | Prospective cohort | 826 individuals (500 without carotid atherosclerosis and 326 with preexisting carotid artery disease). |
|
| Paultre et al., 2002 [131] | Cross-sectional | Randomly selected elderly multiethnic population (173 men and 253 women, consisting of 135 African Americans, 146 Hispanics and 145 whites; mean age 70.5 ± 11.4 years). |
|
| Tsimikas et al., 2006 [132] | Prospective cohort | Sample from the Bruneck study (recruitment in 1990 and follow-up in 1995 and 2000 and 2005). 826 subjects had ultrasonographic follow-up of the carotid and femoral arteries in 1995 and 684 in 2000, respectively. Assessment of OxPL/apo B-100 was achieved in 765/826 subjects in 1995 and 671/684 subjects in 2000. |
|
| Kiechl et al., 2007 [133] | Prospective cohort | Sample from the Bruneck study: 765 subjects with assessment of oxPL/apoB in 1995. |
|
| Van der Valk et al., 2016 [42] | Cross-sectional | 30 subjects with elevated Lp(a) levels (50–195 mg/dL) and 30 subjects with normal Lp(a) (2–28 mg/dL) matched for age, sex and body mass index. |
|
| Atherothrombosis | |||
| Szczeklik et al., 1992 [134] | Cross-sectional | 50 healthy men aged 22–55 years divided into 2 groups: (a) with Lp(a) level > 30 mg/dL and (b) with Lp(a) < 5 mg/dL. |
|
| Testa et al., 1999 [135] | Cross-sectional | 45 healthy subjects, 27 males and 18 females (mean age 37.54 ± 11.02 years). Intervention: different amounts of purified Lp(a) were added to a plasma with very low Lp(a) concentration (<1 mg/dL). Equal amounts of LDL were added to the plasma sample. |
|
| Von Depka et al., 2000 [136] | Case-control | A total of 685 consecutive patients with at least one episode of VTE and 266 sex- and age-matched healthy controls. |
|
| Bilgen et al., 2005 [137] | Case-control | 167 individuals: 136 CAD cases (37 with no vascular disease, 36 subjects with single vessel, 29 with double vessel and 34 with triple vessel disease) and 31 controls. |
|
| Di Nisio et al., 2005 [138] | Cross-sectional | 62 outpatients (51 males, 11 females) of a clinic were included in the study with CAD history, VTE history, family history of CAD and CAD risk factors comparable among 2 groups: patients with Lp(a) above or below the median (300 mg/L). |
|
| Undas et al., 2006 [139] | Cross-sectional | 52 apparently healthy men, aged 38–63 years and 24 male survivors of MI, aged 42–65 years. |
|
| Pineda et al., 2009 [140] | Case-control | 142 subjects presenting with MI at a young age (≤45 years) and 95 controls. |
|
| Liu et al., 2022 [141] | Cross-sectional | 92 subjects without statins or antiplatelet agents. |
|
| Inflammation and Endothelial Dysfunction | |||
| Wu et al., 2004 [142] | Cross-sectional | Multiethnic study of 89 healthy subjects (age 42 ± 9 years; 50 men, 39 women) free of other cardiac risk factors. |
|
| Anuurad et al., 2008 [143] | Cross-sectional | 167 African Americans and 259 Caucasians. |
|
| Volpato et al., 2010 [144] | Prospective cohort | Sample from InCHIANTI study (1002 Italian subjects, 60 to 96 years of age over a 6-year follow-up with an ankle–brachial index (ABI) < 1.5. Longitudinal analysis was limited to 686 participants. |
|
| Nemati et al., 2013 [145] | Case-control | 90 patients with psoriasis and 90 age-matched controls. |
|
| Nishino et al., 2014 [146] | Cross-sectional | 441 patients with suspected vasospastic angina (VSA). |
|
| Mu et al., 2015 [147] | Cross-sectional | 42 patients undergoing CABG (26 males and 8 females aged 48–76 years) and 12 renal artery specimens from kidney transplant donors served as controls. |
|
| Topçiu-Shufta et al., 2016 [148] | Cross-sectional | 78 patients undergoing hemodialysis treatment for a period longer than 6 months with no evidence of CVD history. |
|
| Lp(a) Should be Measured Once Over a Lifespan in Individuals with: |
|---|
| (1) premature CVD |
| (2) familial hypercholesterolemia, or other genetic forms of dyslipidemia |
| (3) recurrent CVD despite optimal lipid-lowering treatment |
| (4) ≥5% 10-year risk of fatal CVD according to ESC SCORE Guidelines |
| (5) family history of premature CVD and/or elevated Lp(a) (≥50 mg/dL) |
| (6) ≥10% 10-year risk of fetal CVD according to US Guidelines |
| (7) calcific aortic valve stenosis |
| (8) 10–19% Framingham risk according to the 2012 Canadian Cardiovascular Society recommendations |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lampsas, S.; Xenou, M.; Oikonomou, E.; Pantelidis, P.; Lysandrou, A.; Sarantos, S.; Goliopoulou, A.; Kalogeras, K.; Tsigkou, V.; Kalpis, A.; et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules 2023, 28, 969. https://doi.org/10.3390/molecules28030969
Lampsas S, Xenou M, Oikonomou E, Pantelidis P, Lysandrou A, Sarantos S, Goliopoulou A, Kalogeras K, Tsigkou V, Kalpis A, et al. Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules. 2023; 28(3):969. https://doi.org/10.3390/molecules28030969
Chicago/Turabian StyleLampsas, Stamatios, Maria Xenou, Evangelos Oikonomou, Panteleimon Pantelidis, Antonios Lysandrou, Savvas Sarantos, Athina Goliopoulou, Konstantinos Kalogeras, Vasiliki Tsigkou, Athanasios Kalpis, and et al. 2023. "Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment" Molecules 28, no. 3: 969. https://doi.org/10.3390/molecules28030969
APA StyleLampsas, S., Xenou, M., Oikonomou, E., Pantelidis, P., Lysandrou, A., Sarantos, S., Goliopoulou, A., Kalogeras, K., Tsigkou, V., Kalpis, A., Paschou, S. A., Theofilis, P., Vavuranakis, M., Tousoulis, D., & Siasos, G. (2023). Lipoprotein(a) in Atherosclerotic Diseases: From Pathophysiology to Diagnosis and Treatment. Molecules, 28(3), 969. https://doi.org/10.3390/molecules28030969