
Fundamentals of Rhenium-188 Radiopharmaceutical Chemistry




Abstract
:1. Introduction
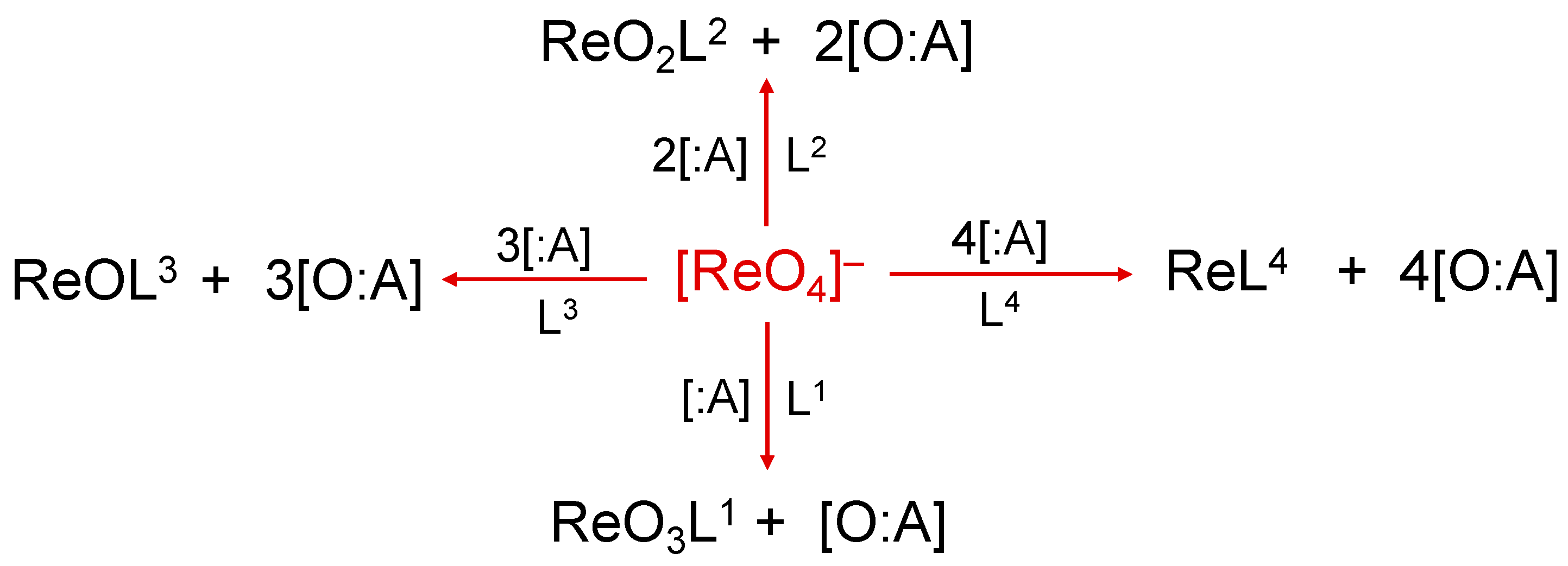
2. The Reduction of the Perrhenate Anion [188Re][ReO4]−
- (i)
- generator-eluted [188Re][ReO4]− in high-specific activity,
- (ii)
- an appropriate oxygen atom acceptor ‘:A’ (historically called ‘reducing agent’),
- (iii)
- appropriate ligands for stabilization of the metallic complex.
3. The Expansion of the Coordination Sphere
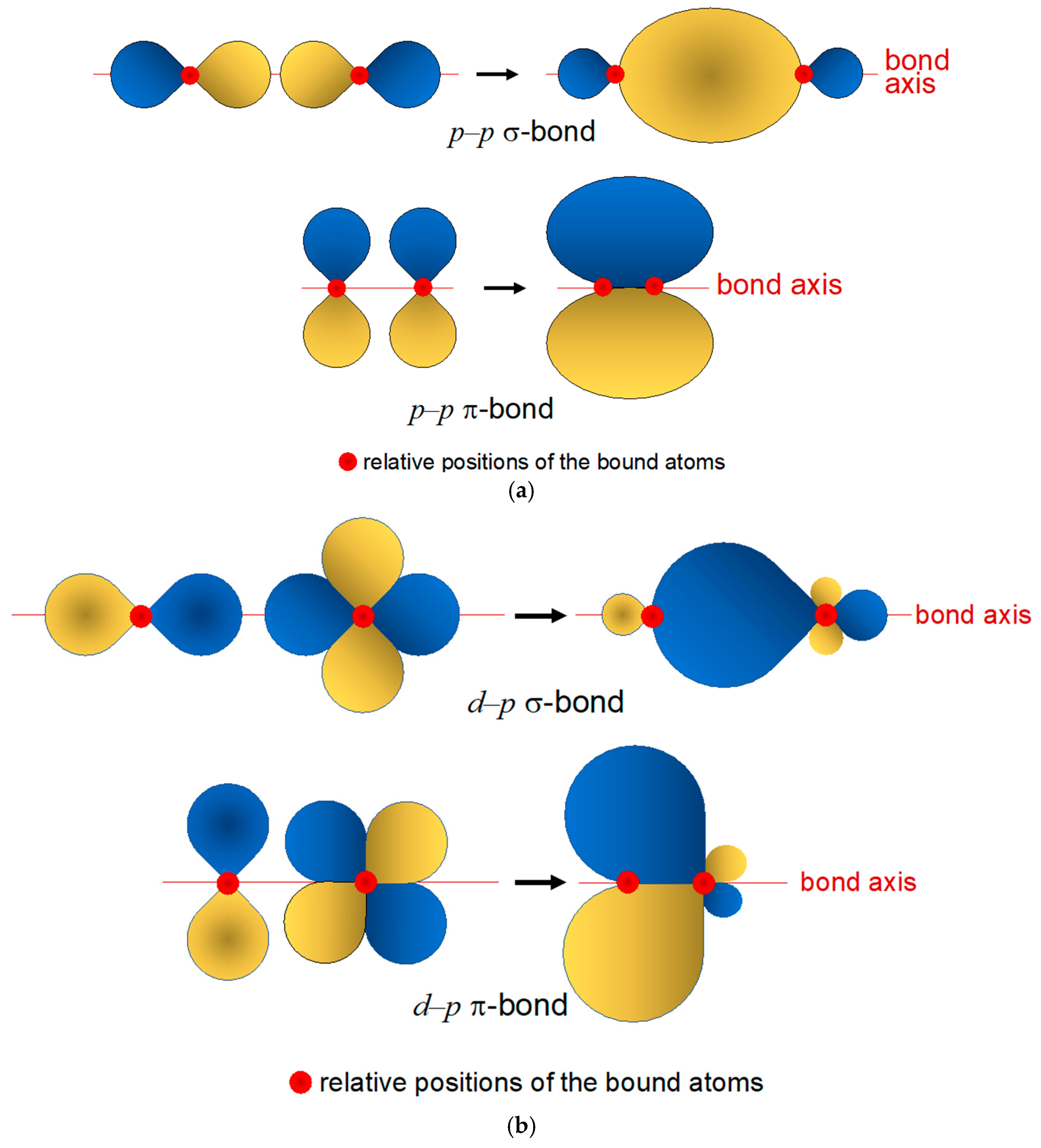
4. Basic Rhenium Coordination Chemistry
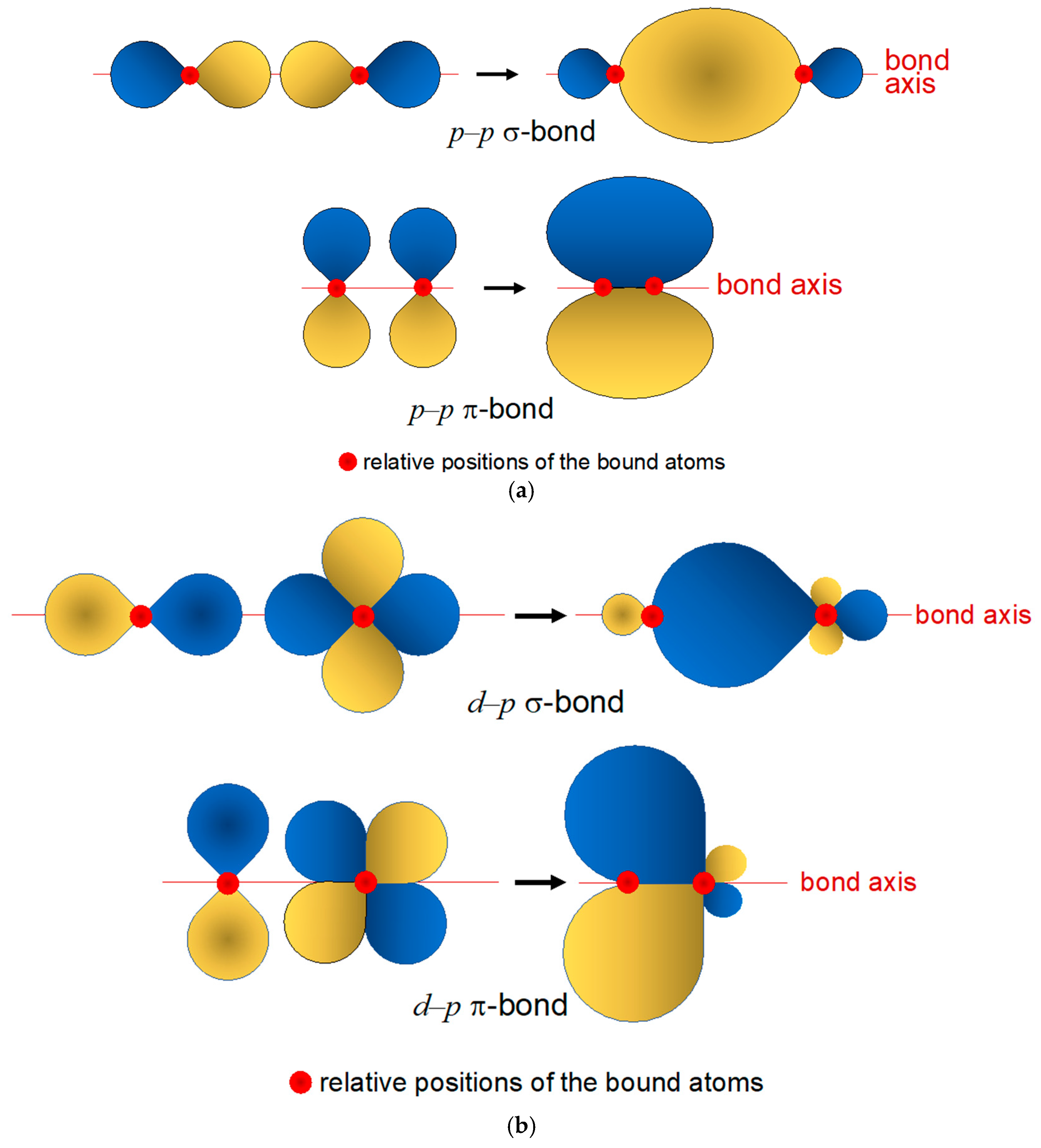
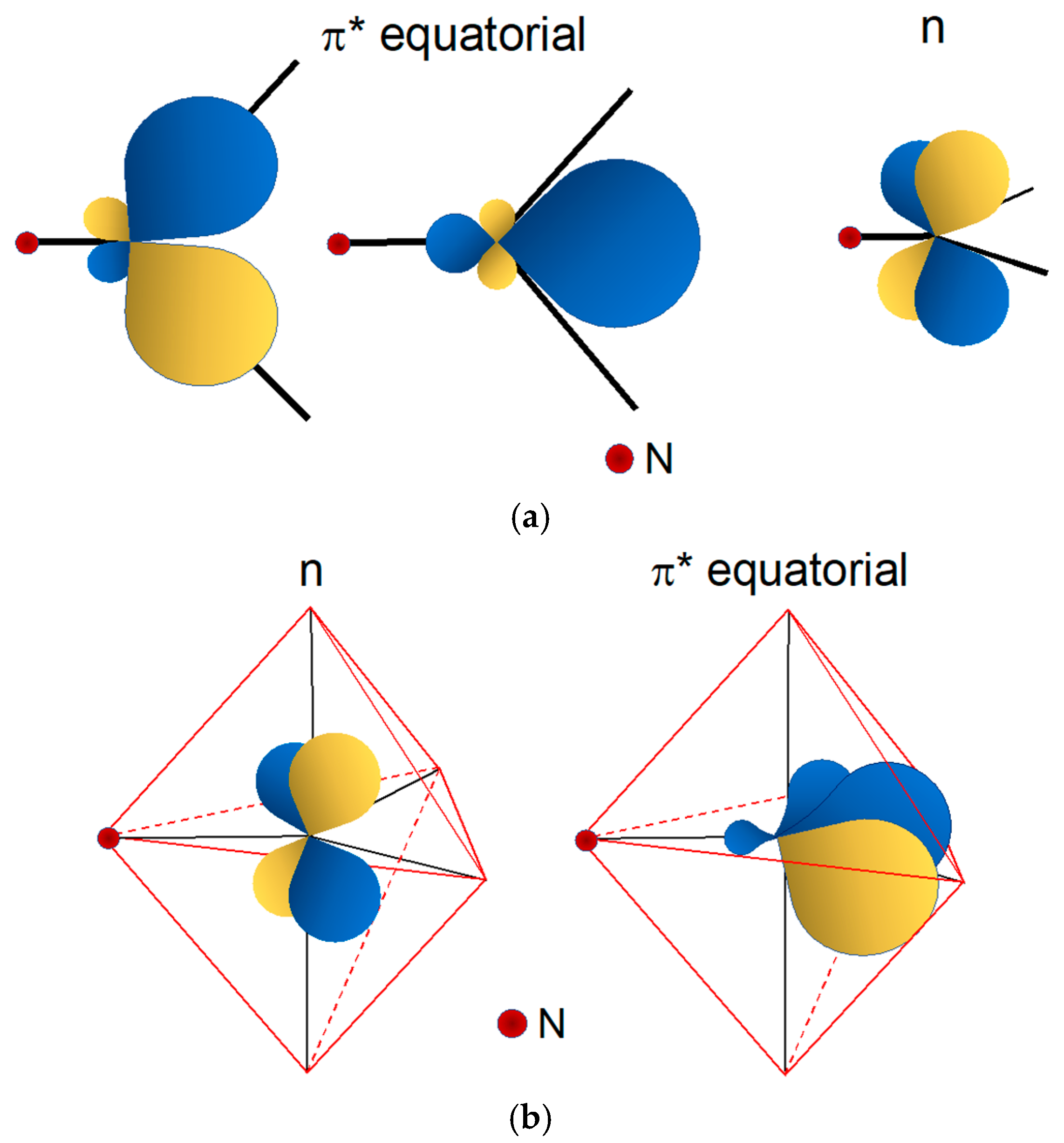
Bonding in Rhenium Complexes
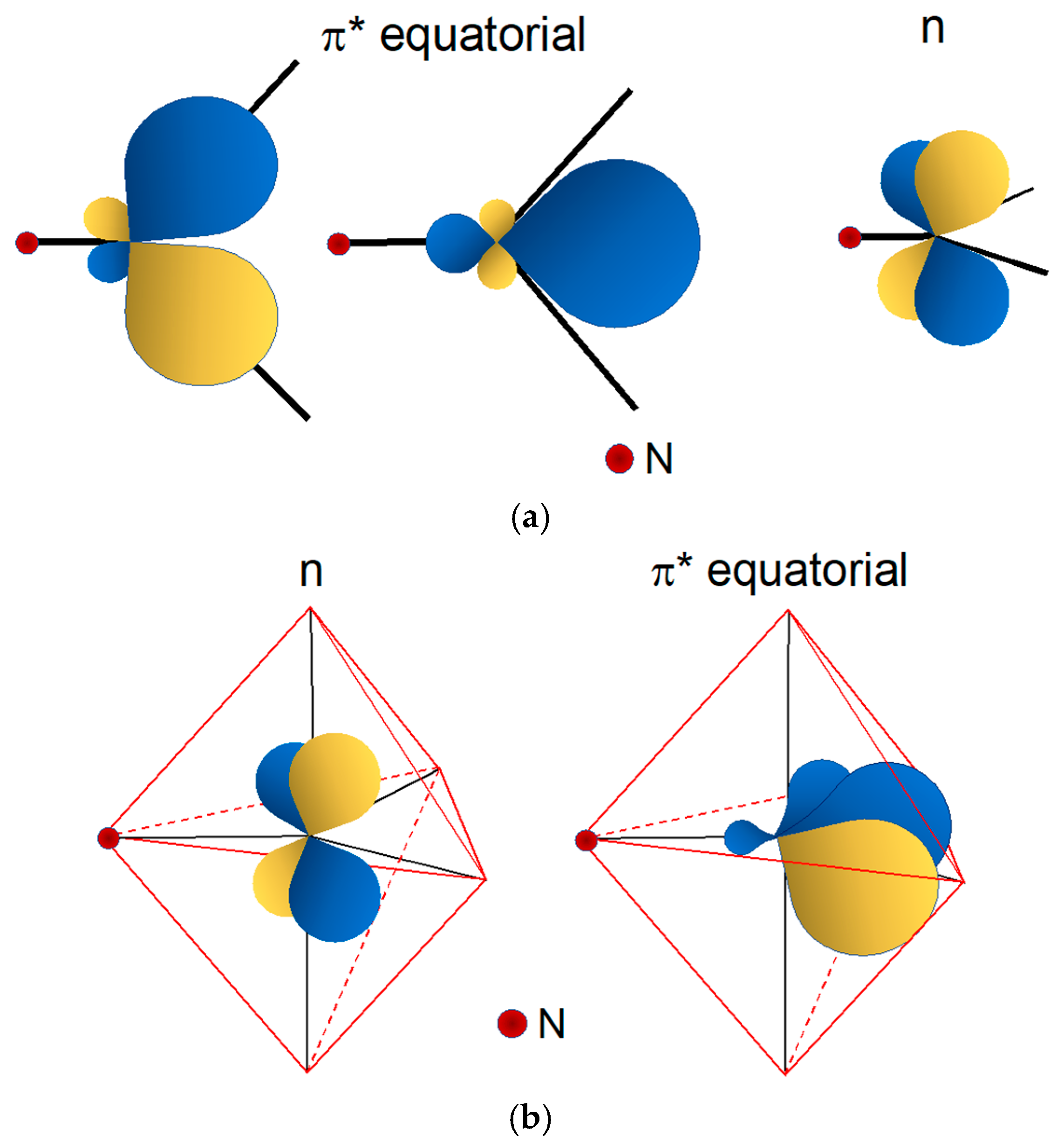
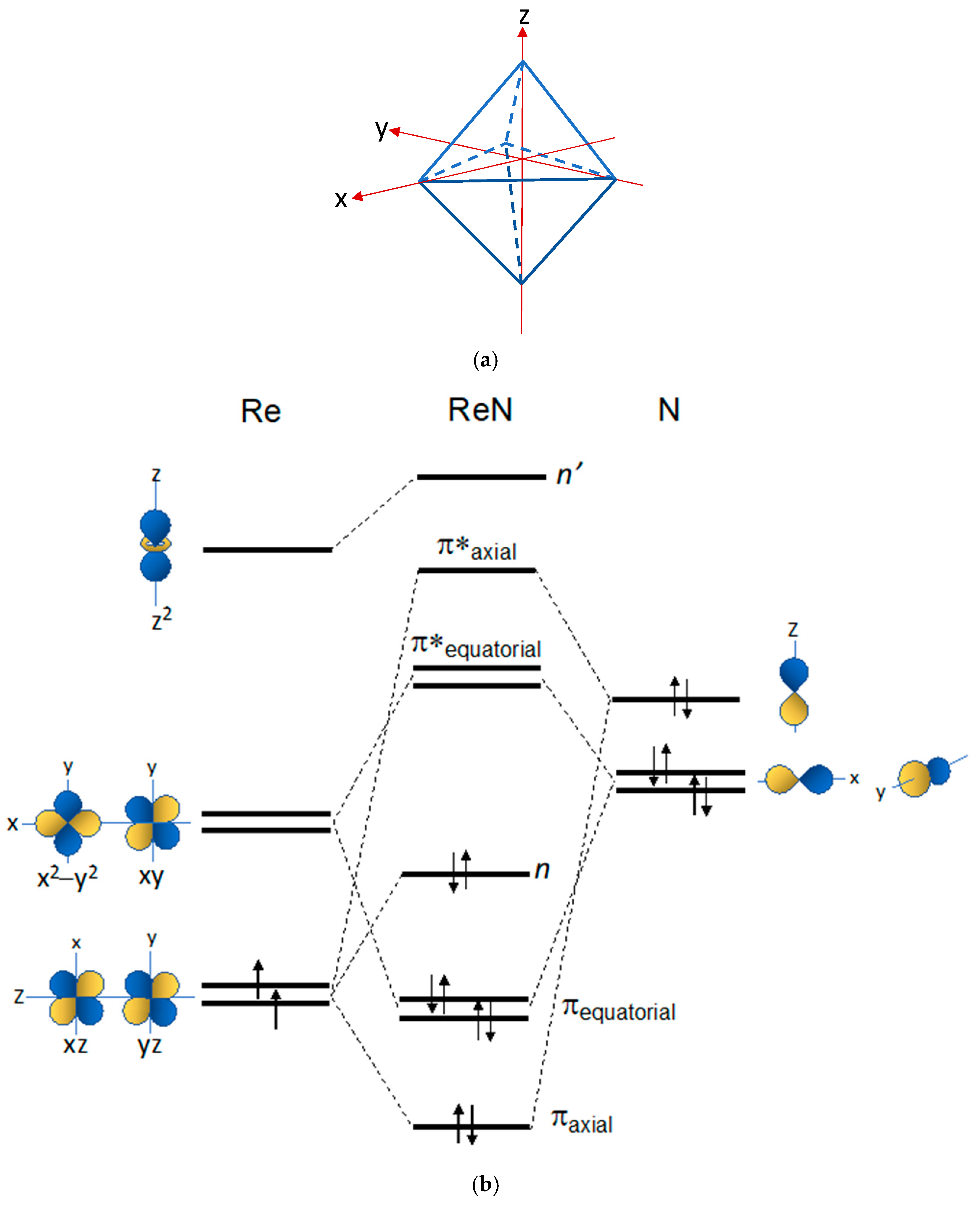
5. Chemical Differences between the Re(N) and Re(O) Cores
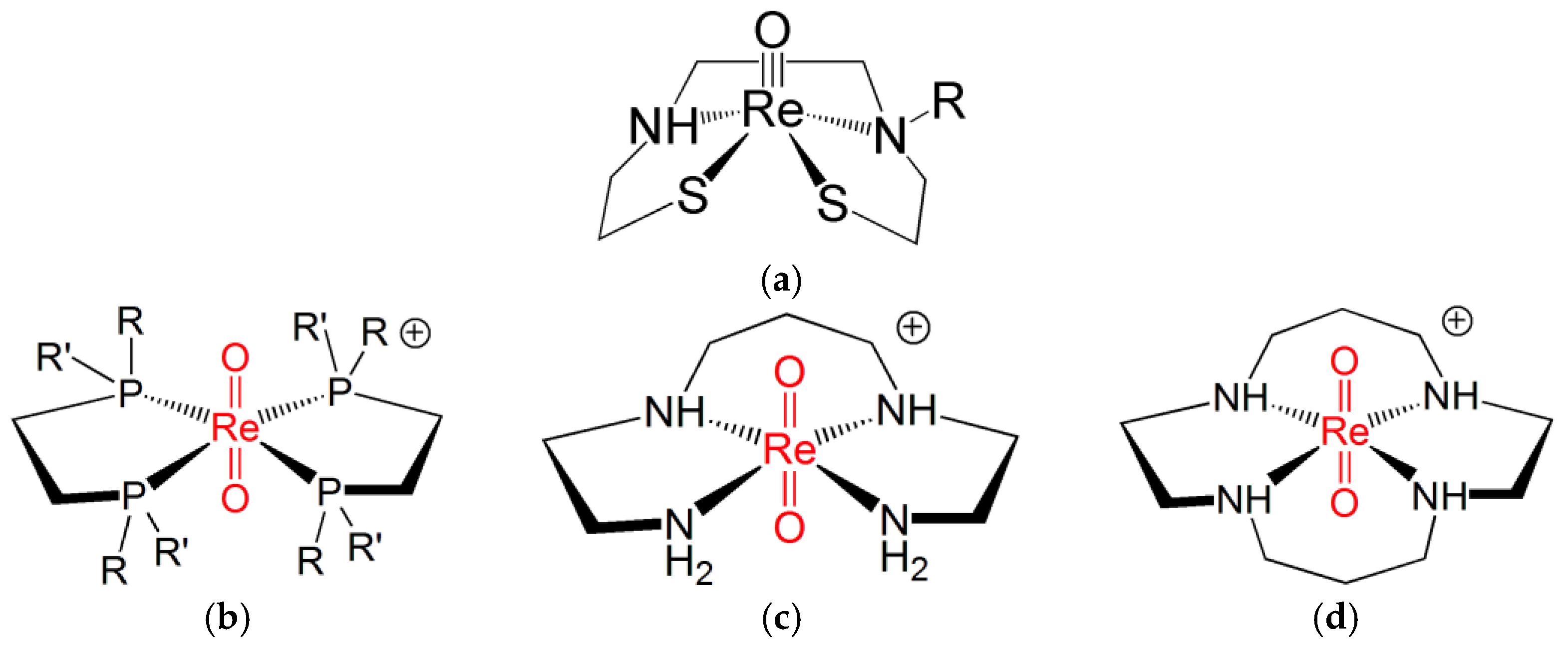
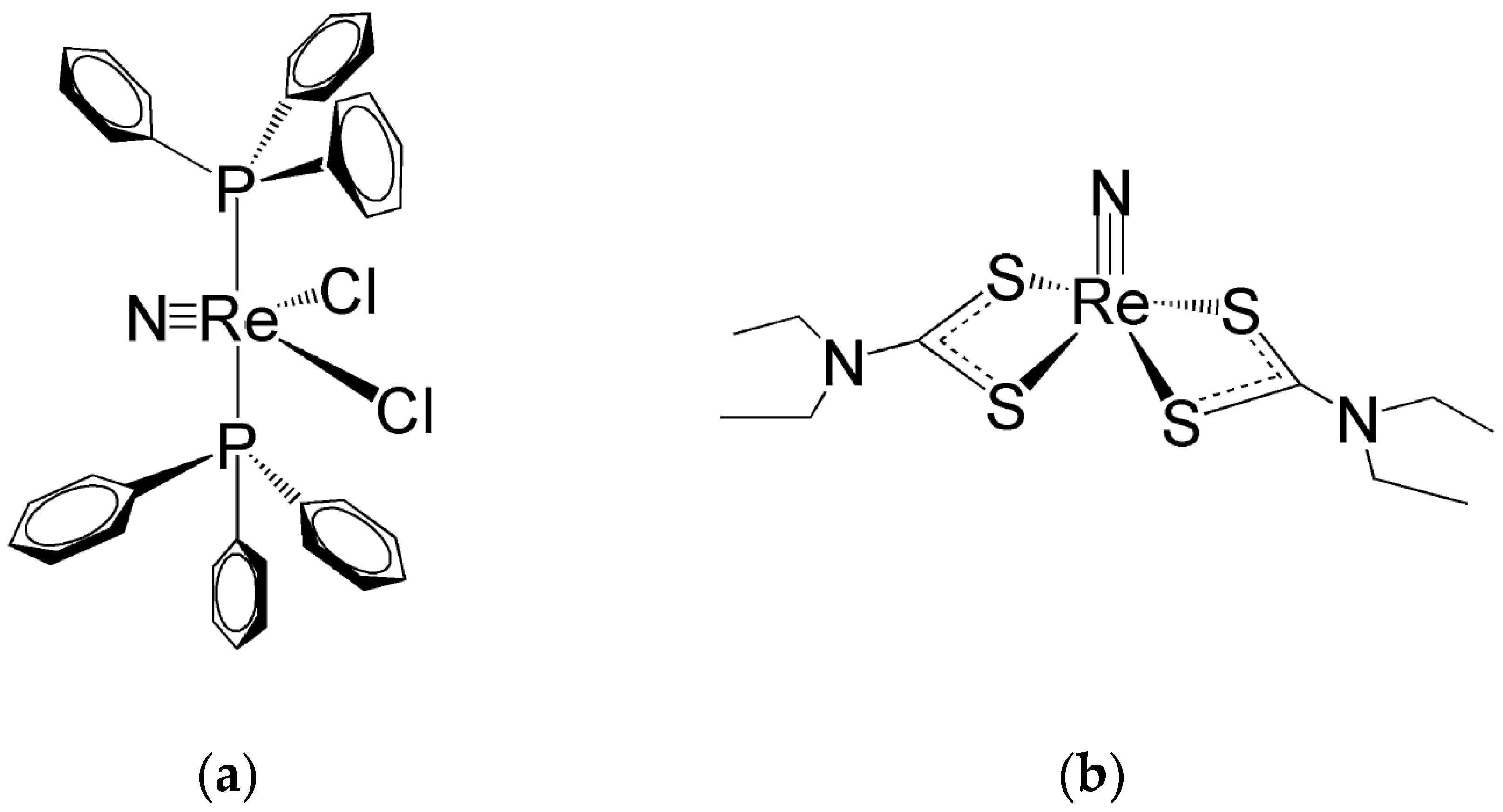
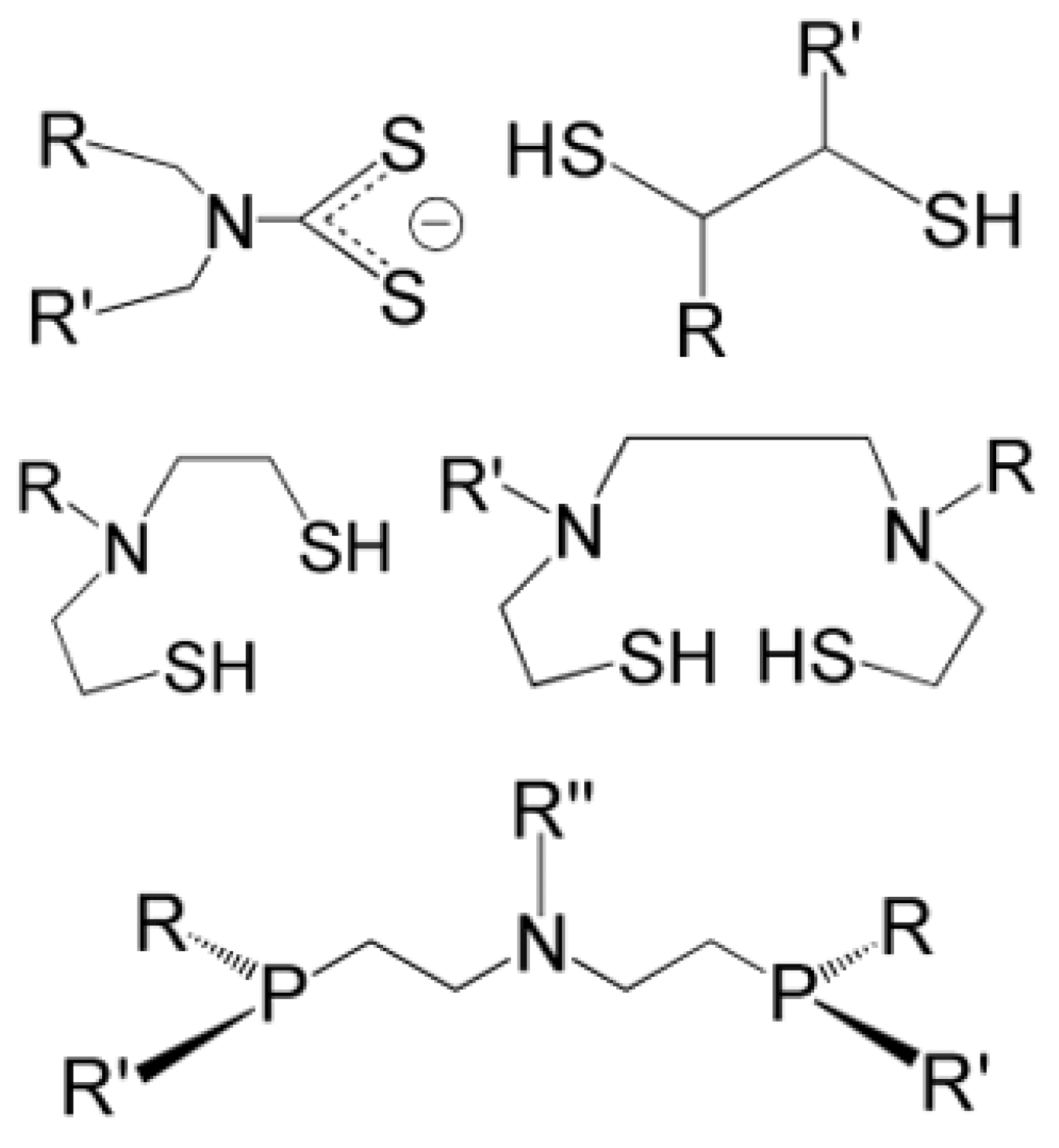
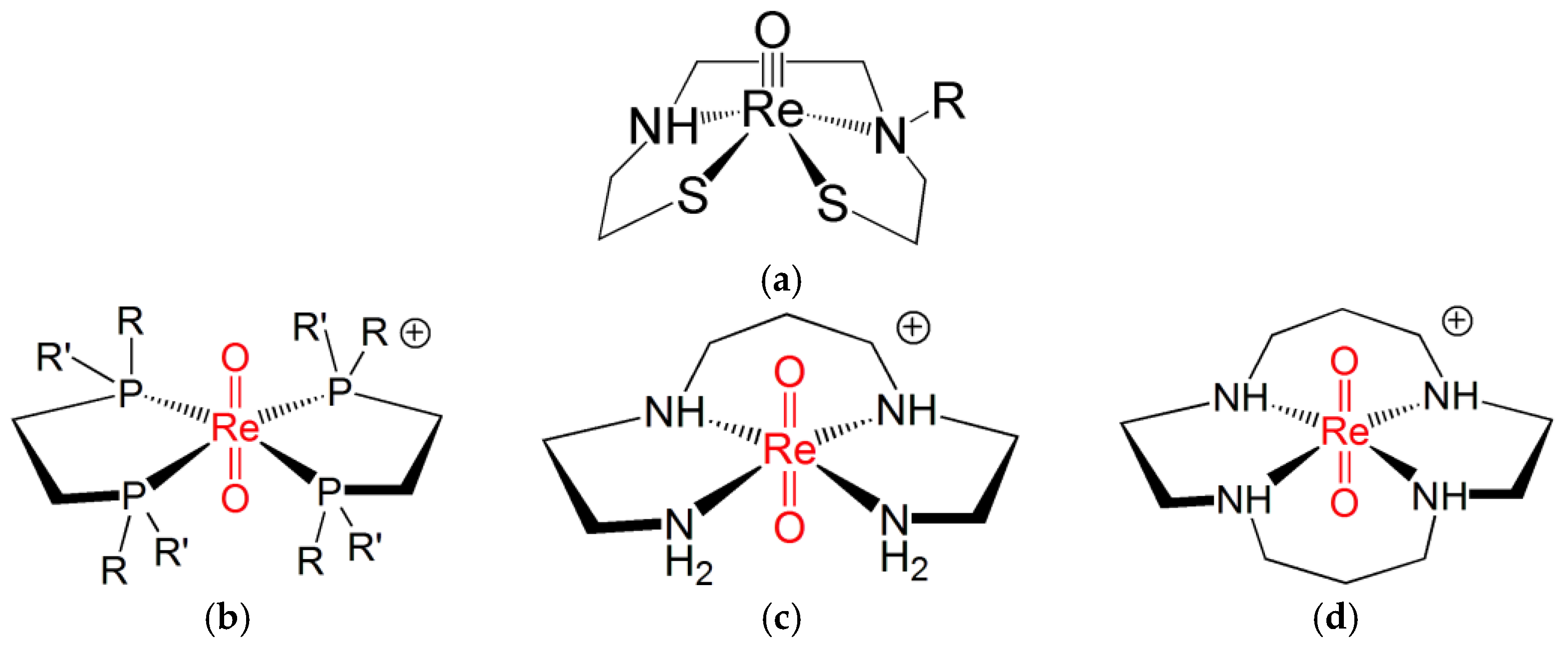
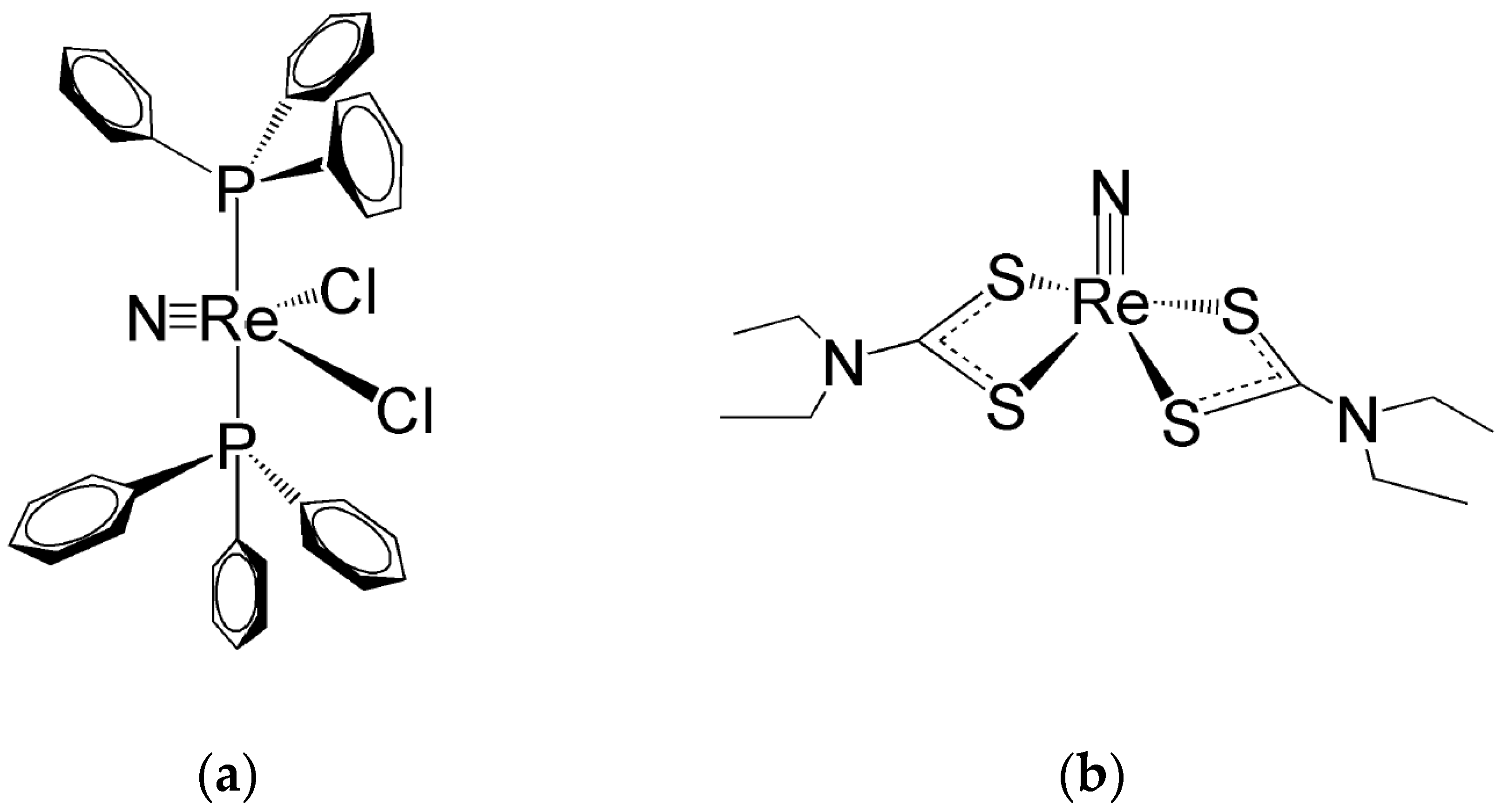
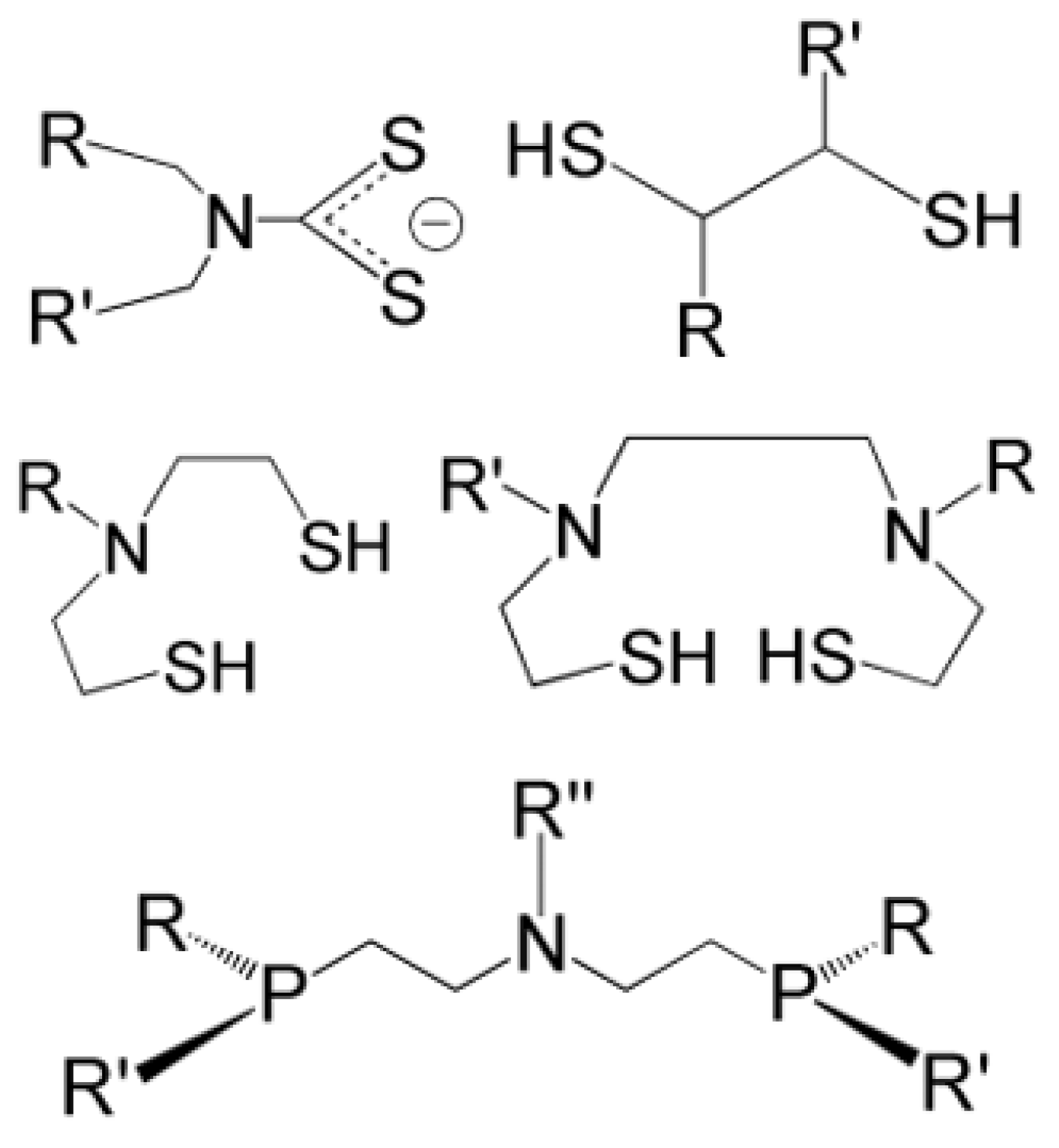
Ligands for the Re≡N and Re≡O Functional Groups
6. Matching the Nuclear and Biological Properties of 188Re Radiopharmaceuticals
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dilworth, J.R.; Parrott, S.J. The biomedical chemistry of technetium and rhenium. Chem. Soc. Rev. 1998, 27, 43–55. [Google Scholar] [CrossRef]
- Esquinas, P.L.; Rodríguez-Rodríguez, C.; Carlos De La Vega, J.; Bokharaei, M.; Saatchi, K.; Shirmohammad, M.; Häfeli, U.O.; Sossi, V.; Celler, A. 188Re image performance assessment using small animal multi-pinhole SPECT/PET/CT system. Phys. Med. 2017, 33, 26–37. [Google Scholar] [CrossRef]
- Lepareur, N.; Lacœuille, F.; Bouvry, C.; Hindré, F.; Garcion, E.; Chérel, M.; Noiret, N.; Etienne Garin, E.; Knapp, F.F.R., Jr. Rhenium-188 labeled radiopharmaceuticals: Current clinical applications in oncology and promising perspectives. Front. Med. 2019, 6, 132. [Google Scholar] [CrossRef]
- Uccelli, L.; Martini, P.; Urso, L.; Ghirardi, T.; Marvelli, L.; Cittanti, C.; Carnevale, A.; Giganti, M.; Bartolomei, M.; Boschi, A. Rhenium radioisotopes for medicine, a focus on production and applications. Molecules 2022, 27, 5283. [Google Scholar] [CrossRef]
- Deutsch, E.; Libson, K.; Vanderheyden, J.L.; Ketring, A.R.; Maxon, H.R. The chemistry of rhenium and technetium as related to the use of isotopes of these elements in therapeutic and diagnostic nuclear medicine. Int. J. Rad. Appl. Instrum. B. 1986, 13, 465–477. [Google Scholar] [CrossRef]
- Duatti, A. Fundamentals of technetium and rhenium chemistry. In Sampson’s Textbook of Radiopharmacy, 4th ed.; Theobald, T., Ed.; Pharmaceutical Press: London, UK, 2011; ISBN 10 0853697892/13 978-0853697893. [Google Scholar]
- Dilworth, J.R. Rhenium chemistry—Then and now. Coord. Chem. Rev. 2021, 436, 213822. [Google Scholar] [CrossRef]
- Duatti, A. Review on 99mTc radiopharmaceuticals with emphasis on new advancements. Nucl. Med. Biol. 2021, 92, 202–216. [Google Scholar] [CrossRef]
- Ferro-Flores, G.; de Murphy, C.A. Pharmacokinetics and dosimetry of 188Re-pharmaceuticals. Adv. Drug Deliv. Rev. 2008, 60, 1389–1401. [Google Scholar] [CrossRef]
- Blower, P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015, 44, 4819–4844. [Google Scholar] [CrossRef]
- Bodei, L.; Herrmann, K.; Schöder, H.; Scott, A.M.; Lewis, J.S. Radiotheranostics in oncology: Current challenges and emerging opportunities. Nat. Rev. Clin. Oncol. 2022, 19, 534–550. [Google Scholar] [CrossRef]
- Knapp, F.F., Jr. Rhenium-188--a generator-derived radioisotope for cancer therapy. Cancer Biother. Radiopharm. 1998, 13, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Pillai, M.R.; Dash, A.; Knapp, F.F., Jr. Rhenium-188: Availability from the 188W/188Re generator and status of current applications. Curr. Radiopharm. 2012, 5, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, G.; Hartmann, H.; Andreeff, M.; Kotzerke, J. A semi-automated system for concentration of rhenium-188 for radiopharmaceutical applications. Appl. Radiat. Isot. 2008, 66, 1876–1880. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, S.C.; Ginsberg, A.P.; Knox, K. Transition Metal-Hydrogen Compounds. II. The Crystal and Molecular Structure of Potassium Rhenium Hydride, K2ReH9. Inorg. Chem. 1964, 3, 558–567. [Google Scholar] [CrossRef]
- Ginsberg, A.P.; Sprinkle, C.R. Nonahydridorhenate Salts. Inorg. Synth. 1972, 13, 219–225. [Google Scholar] [CrossRef]
- Vajo, J.J.; Aikens, D.A.; Ashley, L.; Poeltl, D.E.; Bailey, R.A.; Clark, H.M.; Bunce, S.C. Facile electroreduction of perrhenate in weakly acidic citrate and oxalate media. Inorg. Chem. 1981, 20, 3328–3333. [Google Scholar] [CrossRef]
- Boschi, A.; Bolzati, C.; Uccelli, L.; Duatti, A. High-yield synthesis of the terminal 188Re≡N multiple bond from generator-produced [188ReO4]−. Nucl. Med. Biol. 2003, 30, 381–387. [Google Scholar] [CrossRef]
- Bolzati, C.; Boschi, A.; Uccelli, L.; Duatti, A.; Franceschini, R.; Piffanelli, A. An alternative approach to the preparation of 188Re radiopharmaceuticals from generator-produced [188ReO4]−: Efficient synthesis of 188Re(V)-meso-2,3-dimercaptosuccinic acid. Nucl. Med. Biol. 2000, 27, 309–314. [Google Scholar] [CrossRef]
- Chiozzone, R.; Gonzales, R.; Kremer, C.; De Munno, G.; Faus, J. Oxalato complexes of Re(V). Synthesis and structural characterization of [ReO(OCH3)(ox)(L)] (L = bipy, dppe and dppee). Inorg. Chim. Acta 2001, 325, 203–207. [Google Scholar] [CrossRef]
- Martínez-Lillo, J.; Mastropietro, T.F.; Lhotel, E.; Paulsen, C.; Cano, J.; De Munno, G.; Faus, J.; Lloret, F.; Julve, M.; Nellutla, S.; et al. Highly anisotropic rhenium(IV) complexes: New examples of mononuclear single-molecule magnets. J. Am. Chem. Soc. 2013, 135, 13737–13748. [Google Scholar] [CrossRef]
- Rouschias, G. Recent advances in the chemistry of rhenium. Chem. Rev. 1974, 74, 531–566. [Google Scholar] [CrossRef]
- Mallia, M.B.; Chirayil, V.; Dash, A. Improved freeze-dried kit for the preparation of 188ReN-DEDC/lipiodol for the therapy of unresectable hepatocellular carcinoma. Appl. Radiat. Isot. 2018, 137, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, E.R.; Chirayil, V.; Pandiyan, A.; Subramanian, S.; Mallia, M.B.; Kamaleshwaran, K.K.; Shinto, A. Preparation of rhenium-188-lipiodol using freeze-dried kits for transarterial radioembolization: An overview and experience in a hospital radiopharmacy. Cancer Biother. Radiopharm. 2022, 37, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Melis, D.R.; Burgoyne, A.R.; Ooms, M.; Gasser, G. Bifunctional chelators for radiorhenium: Past, present and future outlook. RSC Med. Chem. 2022, 13, 217–245. [Google Scholar] [CrossRef]
- Bolzati, C.; Carta, D.; Salvarese, N.; Refosco, F. Chelating Systems for 99mTc/188Re in the development of radiolabeled peptide pharmaceuticals. Anti Cancer Agents Med. Chem. 2012, 12, 428–464. [Google Scholar] [CrossRef]
- Shinto, A.; Knapp, F.F. Important clinical applications of 188Rhenium for radionuclide therapy. Int. J. Nucl. Medi. Res. Spec. Issue 2017, 16–38. [Google Scholar] [CrossRef]
- Lepareur, N.; Ardisson, V.; Noiret, N.; Garin, E. (188)Re-SSS/Lipiodol: Development of a potential treatment for HCC from bench to bedside. Int. J. Mol. Imaging 2012, 2012, 278306. [Google Scholar] [CrossRef]
- Ter Heine, R.; Lange, R.; Breukels, O.B.; Bloemendal, H.J.; Rummenie, R.G.; Wakker, A.M.; de Graaf, H.; Beekman, F.J.; van der Westerlaken, M.M.; Malingré, M.M.; et al. Bench to bedside development of GMP grade Rhenium-188-HEDP, a radiopharmaceutical for targeted treatment of painful bone metastases. Int. J. Pharm. 2014, 465, 317–324. [Google Scholar] [CrossRef]
- Liepe, K.; Kropp, J.; Runge, R.; Kotzerke, J. Therapeutic efficiency of rhenium-188-HEDP in human prostate cancer skeletal metastases. Br. J. Cancer 2003, 89, 625–629. [Google Scholar] [CrossRef]
- Elian, M.; Hoffmann, R. Bonding capabilities of transition metal complexes. Inorg. Chem. 1975, 14, 1058–1076. [Google Scholar] [CrossRef]
- Albright, T.A.; Burdett, J.K.; Whangbo, M.-H. Orbital Interactions in Chemistry, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Corfield, P.W.R.; Doedens, R.J.; Ibers, A.J. Studies of metal-nitrogen multiple bonds. II. Crystal and molecular structure of nitridodichlorobistriphenylphosphine)rhenium(V), ReNCl2(P(C6H5)3)2. Inorg. Chem. 1967, 6, 204–210. [Google Scholar] [CrossRef]
- Gernert, M.B.; Hiller, W.; Dilworth, J.R.; Parrott, S.J. Crystal structure of dichlorobis(triphenylphosphine)nitridotechnetium(V), TcNCl2((C6H5)3P)2. Z. Für Krist. 1995, 210, 961–962. [Google Scholar] [CrossRef]
- Bolzati, C.; Boschi, A.; Duatti, A.; Prakash, S.; Uccelli, L.; Refosco, F.; Tisato, F.; Bandoli, G. Geometrically controlled selective formation of nitrido technetium(V) asymmetrical heterocomplexes with bidentate ligands. J. Am. Chem. Soc. 2000, 122, 4510–4511. [Google Scholar] [CrossRef]
- Boschi, A.; Bolzati, C.; Uccelli, L.; Duatti, A.; Benini, E.; Refosco, F.; Tisato, F.; Piffanelli, A. A class of asymmetrical nitrido 99mTc heterocomplexes as heart imaging agents with improved biological properties. Nucl. Med. Commun. 2002, 23, 689–693. [Google Scholar] [CrossRef] [PubMed]
- Bolzati, C.; Boschi, A.; Uccelli, L.; Tisato, F.; Refosco, F.; Cagnolini, A.; Duatti, A.; Prakash, S.; Bandoli, G.; Vittadini, A. Chemistry of the strong electrophilic metal fragment [99Tc(N)(PXP)]2+ (PXP = diphosphine ligand). A novel tool for the selective labeling of small molecules. J. Am. Chem. Soc. 2002, 124, 11468–11479. [Google Scholar] [CrossRef]
- Bolzati, C.; Cavazza-Ceccato, M.; Agostini, S.; Tisato, F.; Bandoli, G. Technetium and rhenium in five-coordinate symmetrical and dissymmetrical nitrido complexes with alkyl phosphino-thiol ligands. Synthesis and structural characterization. Inorg. Chem. 2008, 47, 11972–11983. [Google Scholar] [CrossRef] [PubMed]
- Tisato, F.; Refosco, F.; Porchia, M.; Bolzati, C.; Bandoli, G.; Dolmella, A.; Duatti, A.; Boschi, A.; Jung, C.M.; Pietzsch, H.J.; et al. The crucial role of the diphosphine heteroatom X in the stereochemistry and stabilization of the substitution-inert [M(N)(PXP)]2+ metal fragments (M = Tc, Re; PXP = diphosphine ligand). Inorg. Chem. 2004, 43, 8617–8625. [Google Scholar] [CrossRef]
- Bolzati, C.; Cavazza-Ceccato, M.; Agostini, S.; Refosco, F.; Yamamichi, Y.; Tokunaga, S.; Carta, D.; Salvarese, N.; Bernardini, D.; Bandoli, G. Biological in vitro and in vivo studies of a series of new asymmetrical cationic [99mTc(N)(DTC-Ln)(PNP)]+ complex (DTC-Ln = alicyclic dithiocarbamate and PNP = diphosphinoamine). Bioconjug. Chem. 2010, 21, 928–939. [Google Scholar] [CrossRef]
- Salvarese, N.; Carta, D.; Marzano, C.; Gerardi, G.; Melendez-Alafort, L.; Bolzati, C. [99mTc][Tc(N)(DASD)(PNPn)]+ (DASD = 1,4-dioxa-8-azaspiro [4,5]decandithiocarbamate, PNPn = bisphosphinoamine) for myocardial imaging: Synthesis, pharmacological and pharmacokinetic studies. J. Med. Chem. 2018, 61, 11114–11126. [Google Scholar] [CrossRef]
- Bolzati, C.; Salvarese, N.; Carpanese, D.; Seraglia, R.; Meléndez-Alafort, L.; Rosato, A.; Capasso, D.; Saviano, M.; Del Gatto, A.; Comegna, D.; et al. [99mTc][Tc(N)PNP43]-labeled RGD peptides as new probes for a selective detection of αvβ3 integrin: Synthesis, structure–activity and pharmacokinetic studies. J. Med. Chem. 2018, 61, 9596–9610. [Google Scholar] [CrossRef]
- Bolzati, C.; Salvarese, N.; Spolaore, B.; Vittadini, A.; Forrer, D.; Brunello, S.; Ghiani, S.; Maiocchi, A. Water-soluble [Tc(N)(PNP)] moiety for room-temperature 99mTc labeling of sensitive target vectors. Mol. Pharm. 2022, 19, 876–894. [Google Scholar] [CrossRef] [PubMed]
- Salvarese, N.; Carpanese, D.; Meléndez-Alafort, L.; De Nardo, L.; Calderan, A.; Biondi, B.; Ruzza, P.; Rosato, A.; Bolzati, C. Impact of different [Tc(N)PNP]-scaffolds on the biological properties of the small CRGDfK peptide: Synthesis, in vitro and in vivo evaluations. Molecules 2022, 27, 2548. [Google Scholar] [CrossRef] [PubMed]
- Lebuis, A.-M.; Beauchamp, A.L. Stereoisomerism of oxotrichloro(triphenylphosphine)rhenium(V), ReOCl3(PPh3)2: A reinvestigation. Can. J. Chem. 1993, 71, 441–449. [Google Scholar] [CrossRef]
- Kremer, C.; Rivero, M.; Kremer, E.; Suescun, L.; Mombrú, A.W.; Mariezcurrena, R.; Domínguez, S.; Mederos, A.; Midollini, S.; Castiñeiras, A. Synthesis, characterization and crystal structures of rhenium(V) complexes with diphosphines. Inorg. Chim. Acta 1999, 294, 47–55. [Google Scholar] [CrossRef]
- Blake, A.J.; Greig, J.A.; Schröder, M. Rhenium complexes of tetra-aza macrocycles: The synthesis and single-crystal X-ray structure of trans-[Re(O)2(cyclam)]Cl·2(BPh3·H2O). J. Chem. Soc. Dalton Trans. 1988, 2645–2647. [Google Scholar] [CrossRef]
- Abram, U.; Braun, M.; Abram, S.; Kirmse, R.; Voigt, A. [NBu4][ReNCl4]: Facile synthesis, structure, electron paramagnetic resonance spectroscopy and reactions. J. Chem. Soc. Dalton Trans. 1998, 231–238. [Google Scholar] [CrossRef]
- Thieme, S.; Agostini, S.; Bergmann, R.; Pietzsch, J.; Pietzsch, H.-J.; Carta, D.; Salvarese, N.; Refosco, F.; Bolzati, C. Synthesis, Characterization and Biological Evaluation of [188Re(N)(Cys∼)(PNP)]+/0 mixed-ligand complexes as prototypes for the development of 188Re(N)-based target-specific radiopharmaceuticals. Nucl. Med. Biol. 2011, 38, 399–415. [Google Scholar] [CrossRef]
- Carta, D.; Jentschel, C.; Thieme, S.; Salvarese, N.; Morellato, N.; Refosco, F.; Ruzza, P.; Bergmann, R.; Pietzsch, H.-J.; Bolzati, C. Assessment of the best N3– donors in preparation of [M(N)(PNP)]-Based (M = 99mTc-; 188Re) target-specific radiopharmaceuticals: Comparison among succinic dihydrazide (SDH), N-methyl-S-methyl dithiocarbazate (HDTCZ) and PEGylated N-methyl-S-methyl dithiocarbazate (HO2C-PEG600-DTCZ). Nucl. Med. Biol. 2014, 41, 570–581. [Google Scholar] [CrossRef]
- Fletcher, S.R.; Rowbottom, J.F.; Skapski, A.C.; Wilkinson, G. X-Ray crystal structure of diethyldithiocarbamate complexes of rhenium(V) with oxo- and nitrido-ligands. Chem. Commun. 1970, 22, 1572–1573. [Google Scholar] [CrossRef]
- Libson, K.; Deutsch, E.; Barnett, B.L. Structural characterization of a technetium-99-diphosphonate complex. Implications for the chemistry of technetium-99m skeletal imaging agents. J. Am. Chem. Soc. 1980, 102, 2476–2478. [Google Scholar] [CrossRef]
- Ramakurup, R.E.K.; Chirayil, V.; Pandiyan, A.; Mallia, M.B.; Kameswaran, M.; Shinto, A.; Dash, A. Rhenium-188 hydroxyethane 1,1-diphosphonic acid (HEDP) for bone pain palliation using BARC-HEDP kits versus pars-HEDP kits: A comparison on preparation and performance aspects at hospital radiopharmacy. Indian J. Nucl. Med. 2018, 33, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Lange, R.; ter Heine, R.; Van der Gronde, T.; Selles, S.; De Klerk, J.; Bloemendal, H.; Hendrikse, H. Applying quality by design principles to the small-scale preparation of bone-targeting therapeutic radiopharmaceutical rhenium-188-HEDP. Eur. J. Pharm. Sci. 2016, 90, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Mallia, M.B.; Shitno, A.S.; Kameswarana, M.; Kameleshwaran, K.K.; Kalarikal, R.; Aswathy, K.K.; Banerjee, S. A freeze-dried kit for the preparation of 188Re-HEDP for bone pain palliation: Preparation and preliminary clinical evaluation. Cancer Biother. Radiopharm. 2016, 31, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Savio, E.; Gaudiano, J.; Robles, A.M.; Balter, H.; Paolino, A.; López, A.; Hermida, J.C.; De Marco, E.; Martinez, G.; Osinaga, E.; et al. Re-HEDP: Pharmacokinetic characterization, clinical and dosimetric evaluation in osseous metastatic patients with two levels of radiopharmaceutical dose. BMC Nucl. Med. 2001, 1, 2. [Google Scholar] [CrossRef]
- Palmedo, H.; Manka-Waluch, A.; Albers, P.; Schmidt-Wolf, I.G.; Reinhardt, M.; Ezziddin, S.; Joe, A.; Roedel, R.; Fimmers, R.; Knapp, F.F., Jr.; et al. Repeated bone-targeted therapy for hormone-refractory prostate carcinoma: Tandomized phase II trial with the new, high-energy radiopharmaceutical rhenium-188 hydroxyethylidenediphosphonate. J. Clin. Oncol. 2003, 21, 2869–2875. [Google Scholar] [CrossRef]
- Biersack, H.J.; Palmedo, H.; Andris, A.; Rogenhofer, S.; Knapp, F.F.; Guhlke, S.; Ezziddin, S.; Bucerius, J.; von Mallek, D. Palliation and survival after repeated 188Re-HEDP therapy of hormone-refractory bone metastases of prostate cancer: A retrospective analysis. J. Nucl. Med. 2011, 52, 1721–1726. [Google Scholar] [CrossRef]
- Manafi-Farid, R.; Masoumi, F.; Divband, G.; Saidi, B.; Ataeninia, B.; Hertel, F.; Schweighofer-Zwink, G.; Morgenroth, A.; Beheshti, M. Targeted palliative radionuclide therapy for metastatic bone pain. J. Clin. Med. 2020, 9, 2622. [Google Scholar] [CrossRef]
- Boschi, A.; Uccelli, L.; Duatti, A.; Colamussi, P.; Cittanti, C.; Filice, A.; Rose, A.H.; Martindale, A.A.; Claringbold, P.G.; Kearney, D.; et al. A kit formulation for the preparation of 188Re-lipiodol: Preclinical studies and preliminary therapeutic evaluation in patients with unresectable hepatocellular carcinoma. Nucl. Med. Commun. 2004, 25, 691–699, Erratum in Nucl. Med. Commun. 2004, 25, 983. [Google Scholar] [CrossRef]
- Uccelli, L.; Pasquali, M.; Boschi, A.; Giganti, M.; Duatti, A. Automated preparation of Re-188 lipiodol for the treatment of hepatocellular carcinoma. Nucl. Med. Biol. 2011, 38, 207–213. [Google Scholar] [CrossRef]
- Cipriani, C.; Desantis, M.; Dahlhoff, G.; Brown, S.D., III; Wendler, T.; Olmeda, M.; Pietsch, G.; Eberlein, B. Personalized irradiation therapy for NMSC by rhenium-188 skin cancer therapy: A long-term retrospective study. J. Dermatolog. Treat. 2022, 33, 969–975. [Google Scholar] [CrossRef]
- Castellucci, P.; Savoia, F.; Farina, A.; Lima, G.M.; Patrizi, A.; Baraldi, C.; Zagni, F.; Vichi, S.; Pettinato, C.; Morganti, A.G.; et al. High dose brachytherapy with non sealed 188Re (rhenium) resin in patients with non-melanoma skin cancers (NMSCs): Single center preliminary results. Eur. J. Nucl. Med. Mol. Imaging. 2021, 48, 1511–1521, Erratum in Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1706. [Google Scholar] [CrossRef] [PubMed]
- Dadachova, E.; Bouzahzah, B.; Zuckier, L.S.; Pestell, R.G. Rhenium-188 as an alternative for iodine-131 for treatment of breast tumours expressing the sodium/iodide symporter (NIS). Nucl. Med. Biol. 2002, 29, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Zang, M.; Shi, S.; Guo, R.; Miao, Y.; Li, B. Use of rhenium-188 for in vivo imaging and treatment of human cervical cancer cells transfected with lentivirus expressing sodium iodide symporter. Oncol. Rep. 2016, 36, 2289–2297. [Google Scholar] [CrossRef] [PubMed]
- Spitzweg, C.; Nelson, P.J.; Wagner, E.; Bartenstein, P.; Weber, W.A.; Schwaiger, M.; Morris, J.C. The sodium iodide symporter (NIS): Novel applications for radionuclide imaging and treatment. Endocr. Relat. Cancer. 2021, 28, T193–T213. [Google Scholar] [CrossRef] [PubMed]
- Lindner, T.; Altmann, A.; Krämer, S.; Kleist, C.; Loktev, A.; Kratochwil, C.; Giesel, F.; Mier, W.; Marme, F.; Debus, J.; et al. Design and development of 99mTc-Labeled FAPI tracers for SPECT imaging and 188Re therapy. J. Nucl. Med. 2020, 61, 1507–1513. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.I.; Wu, F.S.; Menda, Y.; Kim, H. Radiopharmaceuticals for neuroendocrine tumors. Semin. Radiat. Oncol. 2021, 31, 60–70. [Google Scholar] [CrossRef]
- Chandran, E.; Figg, W.D.; Madan, R. Lutetium-177-PSMA-617: A vision of the future. Cancer Biol. Ther. 2022, 23, 186–190. [Google Scholar] [CrossRef]
- Pollet, R.J.; Levey, G.S. Principles of membrane receptor physiology and their application to clinical medicine. Ann. Intern. Med. 1980, 92, 663–680. [Google Scholar] [CrossRef] [PubMed]
- Pich, O.; Bailey, C.; Watkins, T.B.K.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. The translational challenges of precision oncology. Cancer Cell. 2022, 40, 458–478. [Google Scholar] [CrossRef] [PubMed]
- Lepareur, N.; Ardisson, V.; Noiret, N.; Boucher, E.; Raoul, J.L.; Clément, B.; Garin, E. Automation of labelling of Lipiodol with high-activity generator-produced 188Re. Appl. Radiat. Isot. 2011, 6, 426–430. [Google Scholar] [CrossRef]
- Thakral, P.; Jyotsna Tandon, P.; Dureja, S.; Pant, V.; Sen, I. Radiation dose to the occupational worker during the synthesis of 188Re-labeled radiopharmaceuticals in the nuclear medicine department. Indian J. Nucl. Med. 2018, 33, 1–5. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kleynhans, J.; Duatti, A.; Bolzati, C. Fundamentals of Rhenium-188 Radiopharmaceutical Chemistry. Molecules 2023, 28, 1487. https://doi.org/10.3390/molecules28031487
Kleynhans J, Duatti A, Bolzati C. Fundamentals of Rhenium-188 Radiopharmaceutical Chemistry. Molecules. 2023; 28(3):1487. https://doi.org/10.3390/molecules28031487
Chicago/Turabian StyleKleynhans, Janke, Adriano Duatti, and Cristina Bolzati. 2023. "Fundamentals of Rhenium-188 Radiopharmaceutical Chemistry" Molecules 28, no. 3: 1487. https://doi.org/10.3390/molecules28031487
APA StyleKleynhans, J., Duatti, A., & Bolzati, C. (2023). Fundamentals of Rhenium-188 Radiopharmaceutical Chemistry. Molecules, 28(3), 1487. https://doi.org/10.3390/molecules28031487