
Novel 3,9-Disubstituted Acridines with Strong Inhibition Activity against Topoisomerase I: Synthesis, Biological Evaluation and Molecular Docking Study

 , and
, and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Synthetic Study
2.2. UV-Vis Absorption Spectroscopy
2.3. Thermal Denaturation
2.4. Circular Dichroism
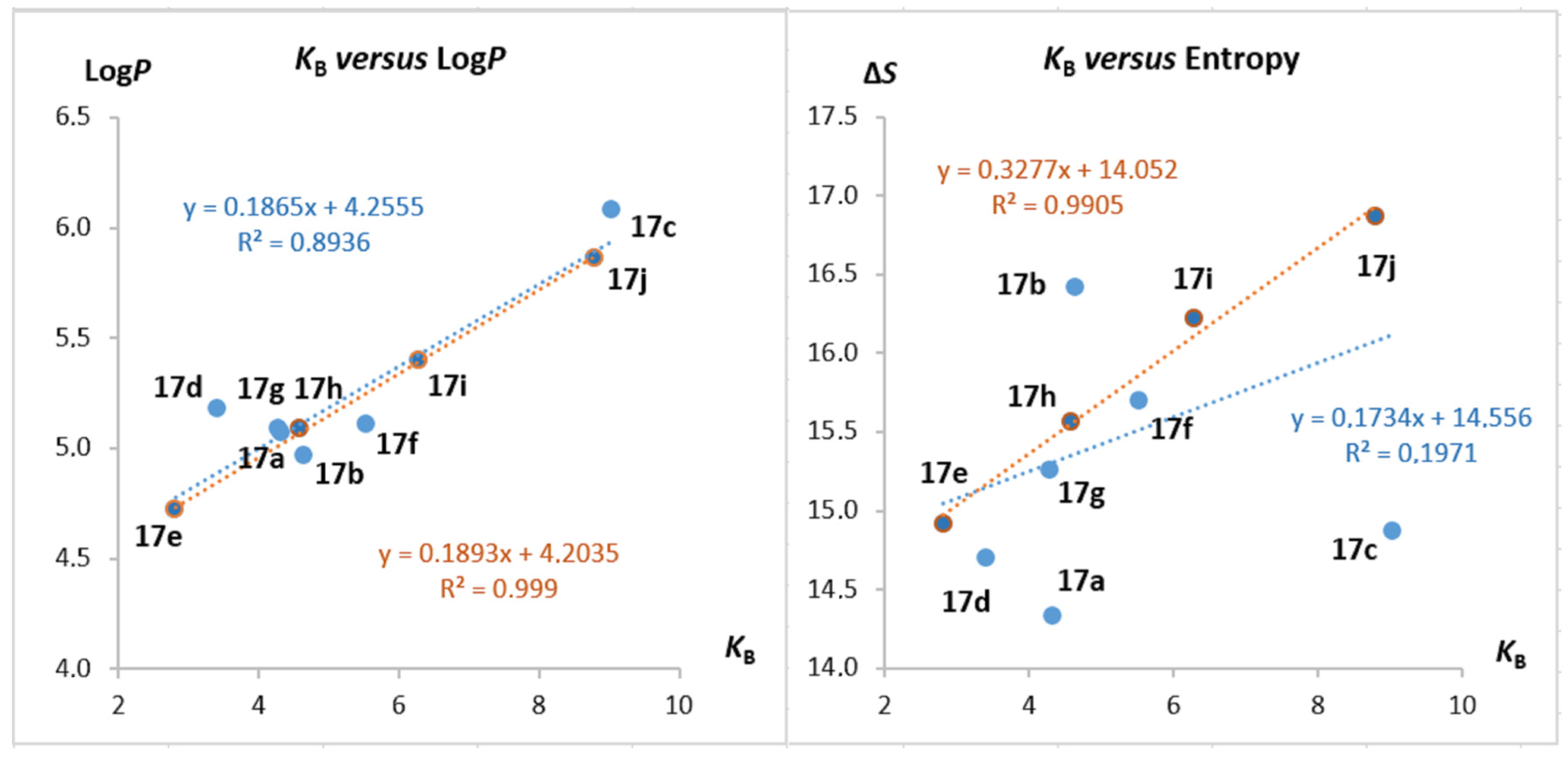
2.5. Structure–Activity Studies
2.6. Inhibition of Human Topoisomerase I and IIα
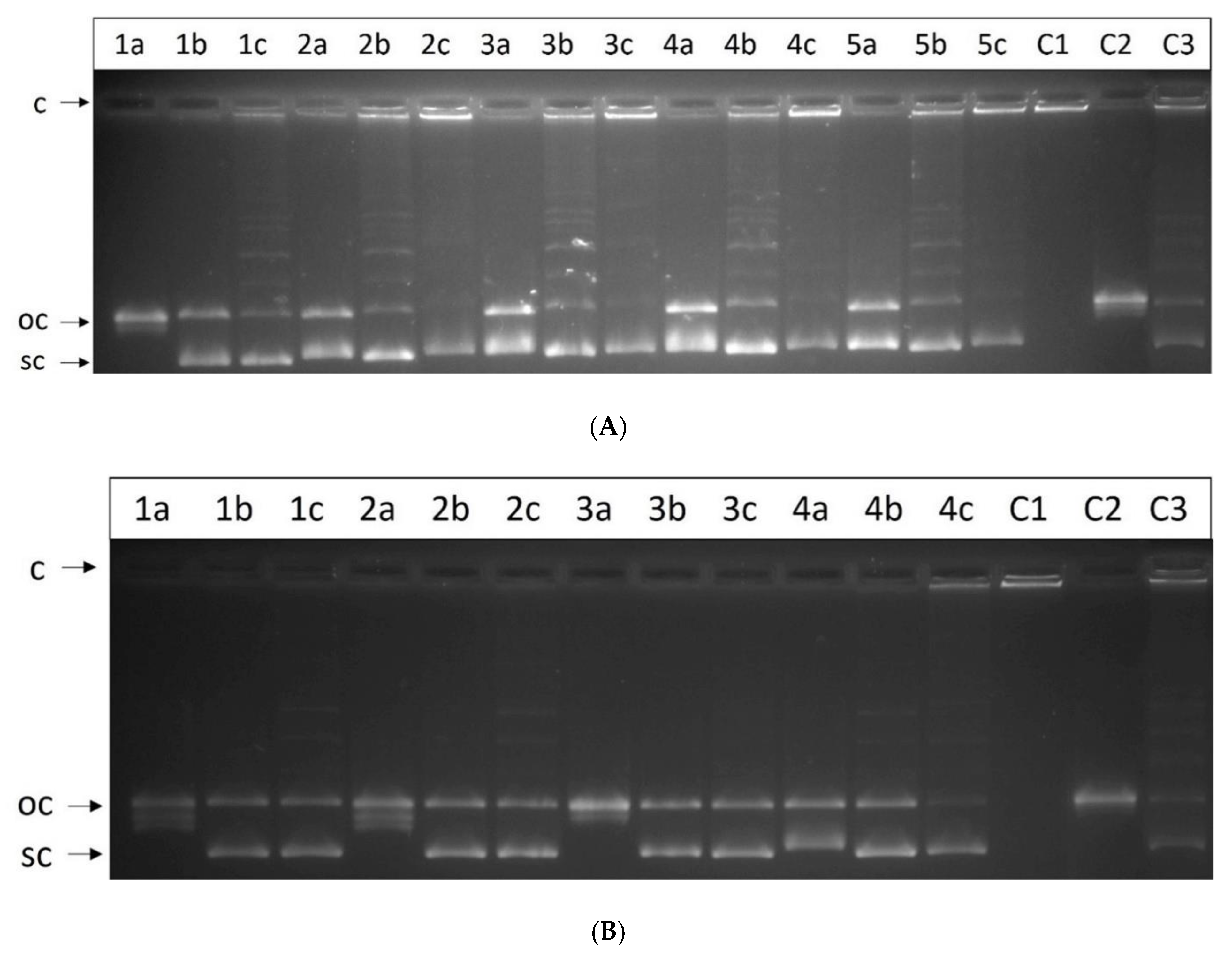
2.7. Relaxation Assay for Human Topoisomerase I
2.8. Unwinding Assay
2.9. Decatenation Assay for Human Topoisomerase IIα
2.10. Cytostatic Activity
2.11. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.2. DNA Binding Experiments
3.3. hTopoisomerase I and IIα Experiments
3.4. Screening of Anticancer Activity-NCI-60 Panels
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Plymale, D.R.; De La Iglesia, F.A. Acridine-induced subcellular and functional changes in isolated human hepatocytes in vitro. J. Appl. Toxicol. 1999, 19, 31. [Google Scholar] [CrossRef]
- Nelson, E.M.; Tewey, K.M.; Liu, L.F. Mechanism of antitumor drug action: Poisoning of mammalian DNA topoisomerase II on DNA by 4’-(9-acridinylamino)-methanesulfon-m-anisidide. Proc. Natl. Acad. Sci. USA 1984, 20, 1361. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell. Biol. 2011, 12, 827. [Google Scholar] [CrossRef] [PubMed]
- Ketron, A.C.; Denny, W.A.; Graves, D.E.; Osheroff, N. Amsacrine as a topoisomerase II poison: Importance of drug-DNA interactions. Biochemistry 2012, 51, 1730. [Google Scholar] [CrossRef] [PubMed]
- Atwell, G.J.; Rewcastle, G.W.; Baguley, B.C.; Denny, W.A. Potential antitumor agents. 50. In vivo solid-tumor activity of derivatives of N-[2-(dimethylamino)ethyl]acridine-4-carboxamide. J. Med. Chem. 1987, 30, 664. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Lukka, P.B.; Joseph, W.R.; Finlay, G.J.; Paxton, J.W.; McKeage, M.J.; Baguley, B.C. Selective cellular uptake and retention of SN 28049, a new DNA-binding topoisomerase II-directed antitumor agent. Cancer. Chemoth. Pharm. 2014, 74, 25. [Google Scholar] [CrossRef]
- Baguley, B.C.; Drummond, C.J.; Chen, Y.Y.; Finlay, G.J. DNA-binding anticancer drugs: One target, two actions. Molecules 2021, 26, 552. [Google Scholar] [CrossRef]
- McEachern, M.J.; Krauskopf, A.; Blackburn, E.H. Telomeres and their control. Annu. Rev. Genet. 2000, 34, 331. [Google Scholar] [CrossRef]
- Bryan, T.M.; Cech, T.R. Telomerase and the maintenance of chromosome ends. Curr. Opin. Cell. Biol. 1999, 11, 318. [Google Scholar] [CrossRef]
- Read, M.A.; Wood, A.; Harrison, R.; Gowan, S.; Kelland, L.; Dosanjh, H.; Neidle, S. Molecular modelling studies on G-quadruplex complexes of telomerase inhibitors: Structure activity relationships. J. Med. Chem. 1999, 42, 4538. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.A.; Koryudzu, K.; Ishibashi, M. Inubosins A, B, and C are acridine alkaloids isolated from a culture of streptomyces sp. IFM 11440 with Ngn2 promoter activity. J. Nat. Prod. 2015, 78, 311. [Google Scholar] [CrossRef] [PubMed]
- Hamissa, M.F.; Niederhafner, P.; Šafařík, M.; Telus, M.; Kolářová, L.; Koutná, L.; Šestáková, H.; Souček, R.; Šebestík, J. Total synthesis of inubosin B. Tetrahedron Lett. 2020, 61, e152641. [Google Scholar] [CrossRef]
- Goodell, J.R.; Ougolkov, A.V.; Hiasa, H.; Kaur, H.; Remmel, R.; Billadeau, D.D.; Ferguson, D.M. Acridine-based agents with topoisomerase II activity inhibit pancreatic cancer cell proliferation and induce apoptosis. J. Med. Chem. 2008, 51, 179. [Google Scholar] [CrossRef] [PubMed]
- Ungvarsky, J.; Plsikova, J.; Janovec, L.; Koval, J.; Mikes, J.; Mikesová, L.; Harvanova, D.; Fedorocko, P.; Kristian, P.; Kasparkova, J.; et al. Novel trisubstituted acridines as human telomeric quadruplex binding ligands. Bioorg. Chem. 2014, 57, 13. [Google Scholar] [CrossRef]
- Rehman, S.U.; Sarwar, T.; Husain, M.A.; Ishqi, H.M.; Tabis, M. Studying non-covalent drug-DNA interactions. Arch. Biochem. Biophys. 2015, 576, 49. [Google Scholar] [CrossRef] [PubMed]
- Vardevanyan, P.O.; Élbakyan, V.L.; Shahinyan, M.A.; Minasyants, M.V.; Parsadanyan, M.A.; Sahakyan, N.S. Determination of the isosbestic point in the absorbtion spectra of DNA-ethidium bromide complexes. J. Appl. Spectrosc. 2015, 81, 1060. [Google Scholar] [CrossRef]
- Mukherjee, A.; Sasikala, W.D. Drug-DNA intercalation: From discovery to molecular mechanism. Adv. Protein Chem. Struct. Biol. 2013, 92, 1. [Google Scholar] [PubMed]
- Guédin, A.; Lacroix, L.; Mergny, J.-L. Thermal melting studies of ligand DNA interactions. Methods Mol. Biol. 2010, 613, 25. [Google Scholar]
- Šmidlehrer, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids - tutorial. Beilstein J. Org. Chem. 2018, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Krafčíková, P.; Demkovičová, E.; Víglaský, V. Ebola virus derived G-quadruplexes: Thiazole orange interaction. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1321. [Google Scholar] [CrossRef]
- Xia, K.; Zhang, G.; Gong, D. Deciphering the intercalative binding modes of benzoyl peroxide with calf thymus DNA. Luminescence 2017, 32, 988. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Stewart Computational Chemistry. MOPAC2016, Version: 19.179L. Available online: http://OpenMOPAC.net (accessed on 1 February 2016).
- ACD/ChemSketch package 2020.2.0. Available online: www.acdlabs.com (accessed on 1 February 2016). (freeware).
- Allouche, A.-R. A graphical user interface for computational chemistry softwares. J. Comput. Chem 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Mod. 2013, 19, 1. [Google Scholar] [CrossRef] [PubMed]
- Sushko, I.; Novotarskyi, S.; Körner, R.; Pandey, A.K.; Rupp, M.; Teetz, W.; Brandmaier, S.; Abdelaziz, A.; Prokopenko, V.V.; Tanchuk, V.Y.; et al. Online chemical modeling environment (OCHEM): Web platform for data storage, model development and publishing of chemical information. J. Comput. Aided Mol. Des. 2011, 25, 533. [Google Scholar] [CrossRef]
- Sushko, I.; Salmina, E.; Potemkin, V.A.; Poda, G.; Tetko, I.V. ToxAlerts: A web server of structural alerts for toxic chemicals and compounds with potential adverse reactions. J. Chem. Inf. Model. 2012, 52, 2310. [Google Scholar] [CrossRef] [PubMed]
- Oprisiu, I.; Novotarskyi, S.; Tetko, I.V. Modeling of non-additive mixture properties using the online chemical database and modeling environment (OCHEM). J. Cheminform. 2013, 5, e4. [Google Scholar] [CrossRef]
- Tetko, I.V.; Novotarskyi, S.; Sushko, I.; Ivanov, V.; Petrenko, A.E.; Dieden, R.; Lebon, F.; Mathieu, B. Development of dimethyl sulfoxide solubility models using 163,000 molecules: Using a domain applicability metric to select more reliable predictions. J. Chem. Inf. Model. 2013, 53, 1990. [Google Scholar] [CrossRef]
- Hargrove, A.E.; Zhong, Z.; Sessier, J.L.; Anslyn, E.V. Algorithms for the determination of binding constants and enantiomeric excess in complex host: Guest equilibria using optical measurements. New J. Chem. 2010, 34, 348. [Google Scholar] [CrossRef]
- Janovec, L.; Kovacova, E.; Semelakova, M.; Kvakova, M.; Kupka, D.; Jager, D.; Kozurkova, M. Synthesis of novel biologically active proflavine ureas designed on the basis of predicted entropy changes. Molecules 2021, 26, 4860. [Google Scholar] [CrossRef] [PubMed]
- Record, M.T., Jr.; Ha, J.-H.; Fisher, M.A. Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol. 1991, 208, 291. [Google Scholar] [PubMed]
- Ren, J.; Jenkins, T.C.; Chaires, J.B. Energetics of DNA intercalation reactions. Biochemistry 2000, 39, 8439. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhang, P.; Yu, B.; Jin, C.; Ji, L.; Chao, H. Synthesis, characterization and biological evaluation of mixed-ligand ruthenium(II) complexes for photodynamic therapy. Dalton Trans. 2015, 44, 17335. [Google Scholar] [CrossRef] [PubMed]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Human DNA topoisomerase I (70 Kda) in complex with the poison camptothecin and covalent complex with a 22 base pair DNA duplex. J. Med. Chem. 2005, 48, 2336. [Google Scholar] [CrossRef] [PubMed]
- Michel, F.; Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph Model. 1999, 17, 57. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theor. Comput. 2015, 11, 3696. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639. [Google Scholar] [CrossRef]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Krogh, B.O.; Shuman, S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell 2000, 5, 1035. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Champoux, J.J.; Hol, W.G.J. Novel insights into catalytic mechanism from a crystal structure of human topoisomerase I in complex with DNA. Biochemistry 2000, 39, 6832. [Google Scholar] [CrossRef]
- Wang, Y.R.; Chen, S.F.; Wu, C.C.; Liao, Y.W.; Lin, T.S.; Liu, K.T.; Chen, Y.S.; Li, T.K.; Chien, T.C.; Chan, N.L. Producing irreversible topoisomerase II-mediated DNA breaks by site-specific Pt(ii)-methionine coordination chemistry. Nucleic Acids Res. 2017, 45, 10861. [Google Scholar] [CrossRef]
- Wu, C.C.; Li, Y.C.; Wang, Y.R.; Li, T.K.; Chan, N.L. Human topoisomerase IIbeta in complex with DNA and amsacrine. Nucleic Acids Res. 2013, 41, 10630. [Google Scholar] [CrossRef]
- Drwal, M.N.; Marinello, J.; Manzo, S.G.; Wakelin, L.P.G.; Capranico, G.; Griffith, R. Novel DNA topoisomerase IIa inhibitors from combined ligand- and structure-based virtual screening. PLoS ONE 2014, 9, e114904. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605. [Google Scholar] [CrossRef] [PubMed]
- Couch, G.S.; Hendrix, D.K.; Ferrin, T.E. Designed divergent evolution of enzyme function. Nucleic Acids Res. 2006, 34, e29. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | LogP [a] | Charge [δ] [b] | ΔS° [cal/K.mol] [c] | KB [×104 M−1] [d] |
|---|---|---|---|---|
| 17a | 5.07 | 0.270 | 14.3 | 4.32 |
| 17b | 4.97 | 0.313 | 16.4 | 4.65 |
| 17c | 6.08 | 0.254 | 14.9 | 9.03 |
| 17d | 5.18 | 0.264 | 14.7 | 3.41 |
| 17e | 4.72 | 0.323 | 14.9 | 2.81 |
| 17f | 5.11 | 0.330 | 15.7 | 5.53 |
| 17g | 5.09 | 0.327 | 15.3 | 4.28 |
| 17h | 5.09 | 0.344 | 15.6 | 4.59 |
| 17i | 5.40 | 0.350 | 16.2 | 6.28 |
| 17j | 5.86 | 0.349 | 16.9 | 8.80 [e] |
| Cell Line [a] | Ams. [b] | Dox. [c] | Pac.[d] | 17a | 17b | 17e | 17f | 17g | 17h | R2 |
|---|---|---|---|---|---|---|---|---|---|---|
| GI50 [µM] [d] | σ [e] | |||||||||
| MOLT-4 | 0.016 | 0.019 | 0.007 | 0.046 | 0.045 | 0.38 | 0.31 | 0.38 | 0.32 | 0.62 |
| SR | 0.016 | 0.006 | 0.043 | 0.046 | 0.038 | 0.26 | 0.30 | 0.35 | 0.18 | 0.49 |
| A549/ATCC | 0.030 | 0.029 | 0.010 | 0.141 | 0.065 | 0.37 | 0.47 | 0.38 | 0.33 | 0.74 |
| NCI-H460 | 0.013 | 0.006 | 0.007 | 0.537 | 0.043 | 0.31 | 0.36 | 0.33 | 0.30 | 0.69 |
| DU-145 | nd | 0.068 | 0.021 | 0.214 | 0.138 | 0.39 | 0.46 | 0.40 | 0.38 | 0.81 |
| MCF7 | nd | 0.009 | 0.007 | 0.019 | 0.776 | 0.30 | 0.47 | 0.32 | 0.31 | 0.52 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krochtová, K.; Halečková, A.; Janovec, L.; Blizniaková, M.; Kušnírová, K.; Kožurková, M. Novel 3,9-Disubstituted Acridines with Strong Inhibition Activity against Topoisomerase I: Synthesis, Biological Evaluation and Molecular Docking Study. Molecules 2023, 28, 1308. https://doi.org/10.3390/molecules28031308
Krochtová K, Halečková A, Janovec L, Blizniaková M, Kušnírová K, Kožurková M. Novel 3,9-Disubstituted Acridines with Strong Inhibition Activity against Topoisomerase I: Synthesis, Biological Evaluation and Molecular Docking Study. Molecules. 2023; 28(3):1308. https://doi.org/10.3390/molecules28031308
Chicago/Turabian StyleKrochtová, Kristína, Annamária Halečková, Ladislav Janovec, Michaela Blizniaková, Katarína Kušnírová, and Mária Kožurková. 2023. "Novel 3,9-Disubstituted Acridines with Strong Inhibition Activity against Topoisomerase I: Synthesis, Biological Evaluation and Molecular Docking Study" Molecules 28, no. 3: 1308. https://doi.org/10.3390/molecules28031308
APA StyleKrochtová, K., Halečková, A., Janovec, L., Blizniaková, M., Kušnírová, K., & Kožurková, M. (2023). Novel 3,9-Disubstituted Acridines with Strong Inhibition Activity against Topoisomerase I: Synthesis, Biological Evaluation and Molecular Docking Study. Molecules, 28(3), 1308. https://doi.org/10.3390/molecules28031308