
Design, Synthesis, and Antiproliferative Activity of New 5-Chloro-indole-2-carboxylate and Pyrrolo[3,4-b]indol-3-one Derivatives as Potent Inhibitors of EGFRT790M/BRAFV600E Pathways

 ,
,
Abstract
1. Introduction
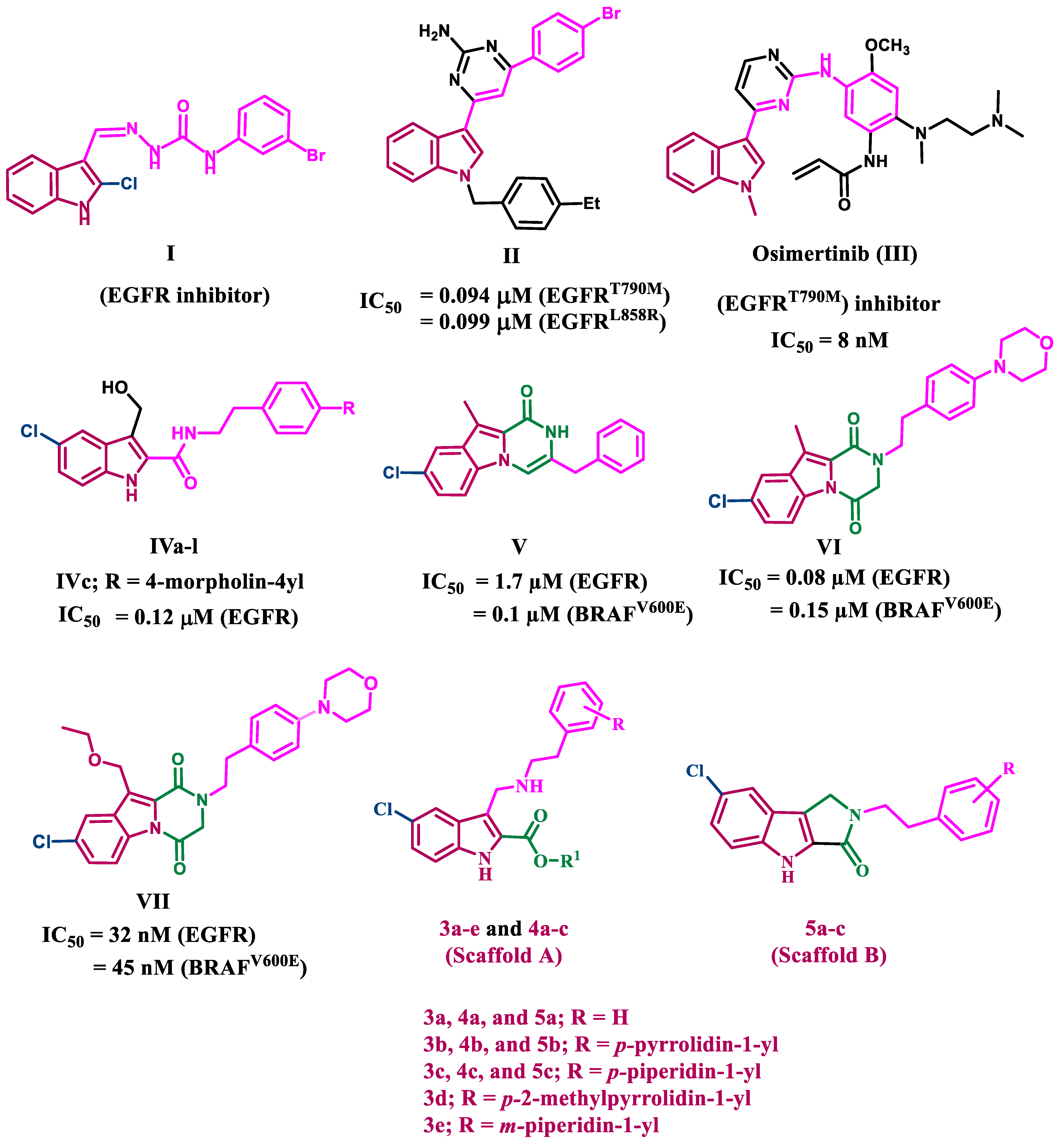
2. Results and Discussion
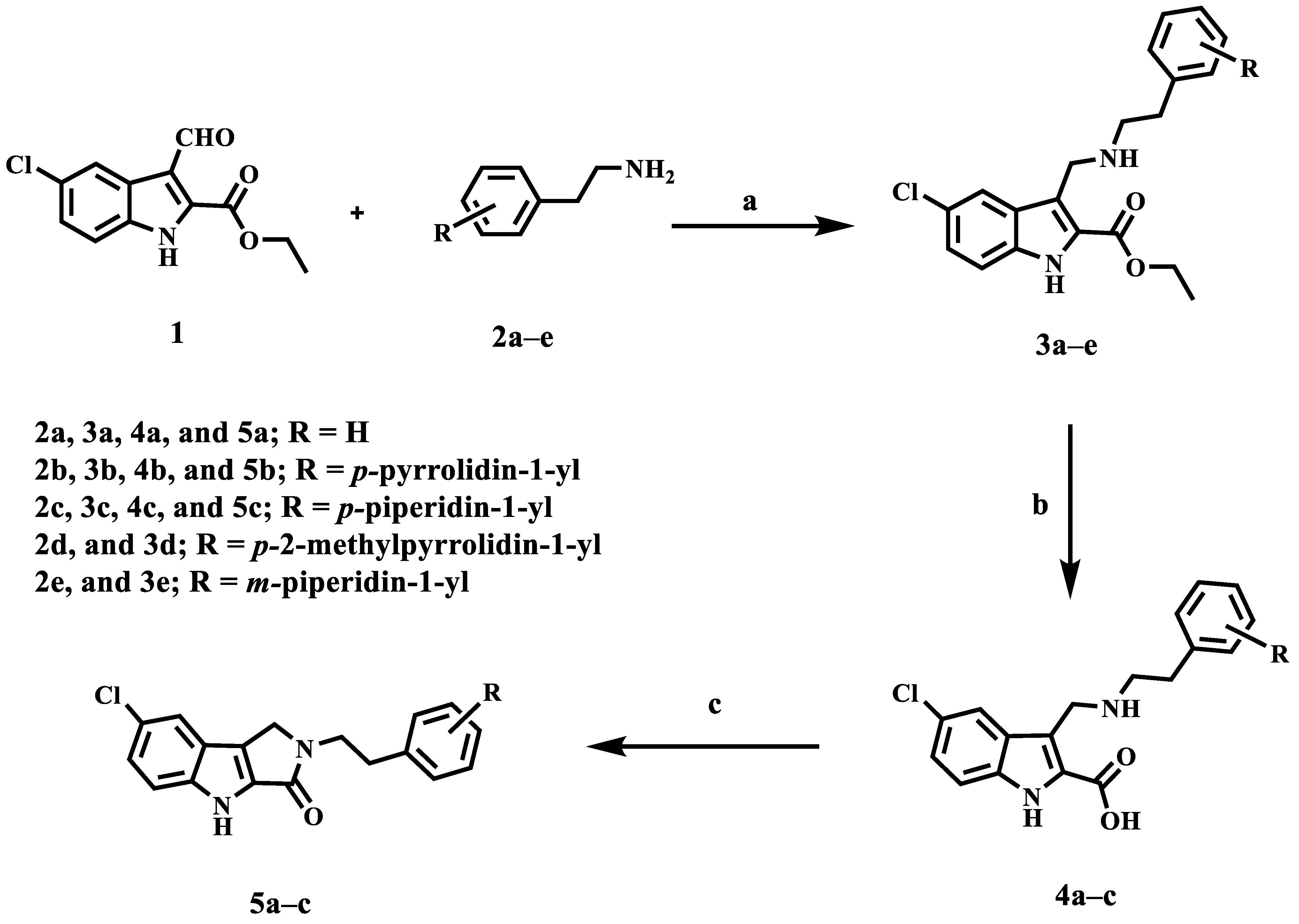
2.1. Chemistry
2.2. Biology
2.2.1. Cell Viability Assay
2.2.2. Antiproliferative Assay
2.2.3. EGFR Inhibitory Assay
2.2.4. BRAFV600E Inhibitory Assay
2.2.5. EGFRT790M Inhibitory Assay
2.2.6. LOX-IMVI Melanoma Cell Line Cytotoxicity Assay
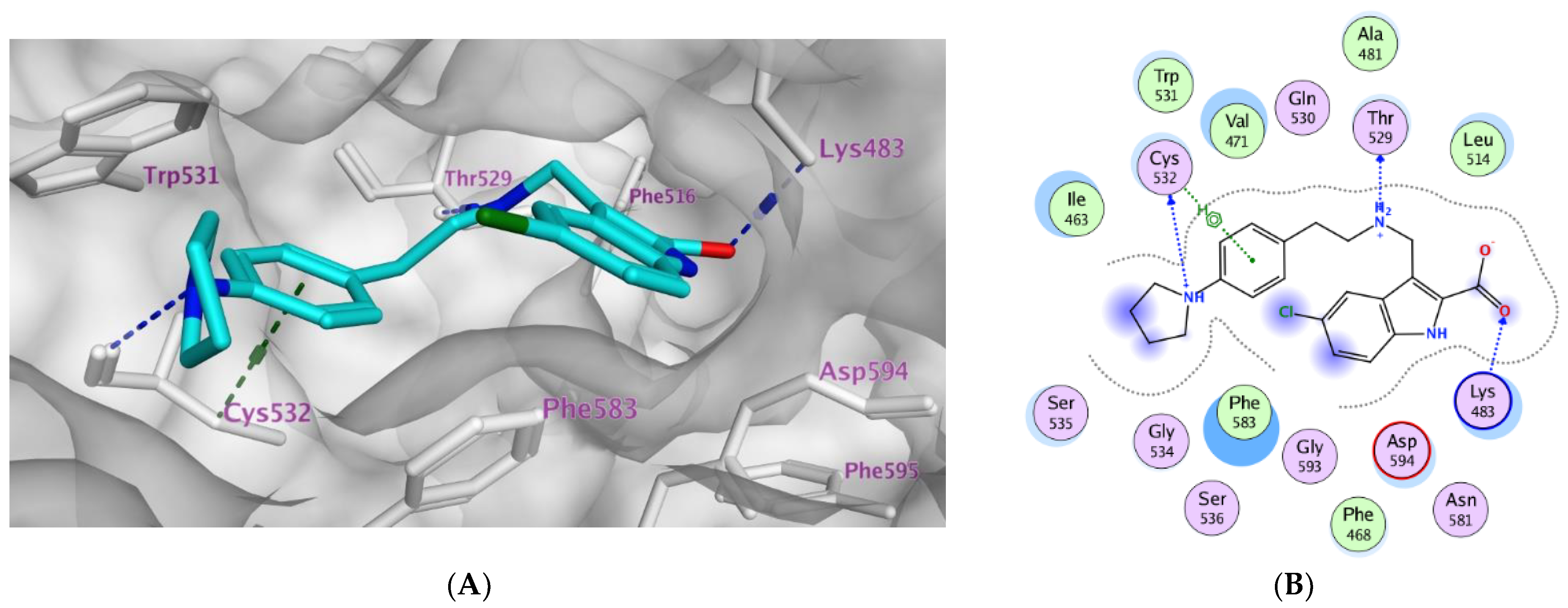
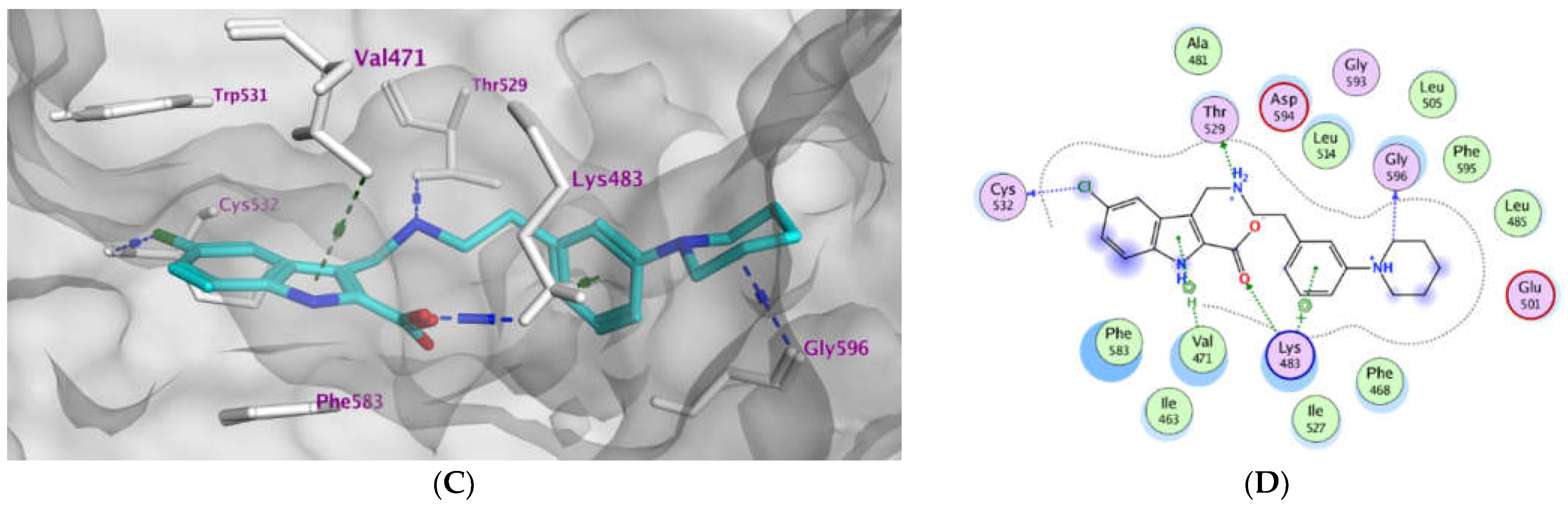
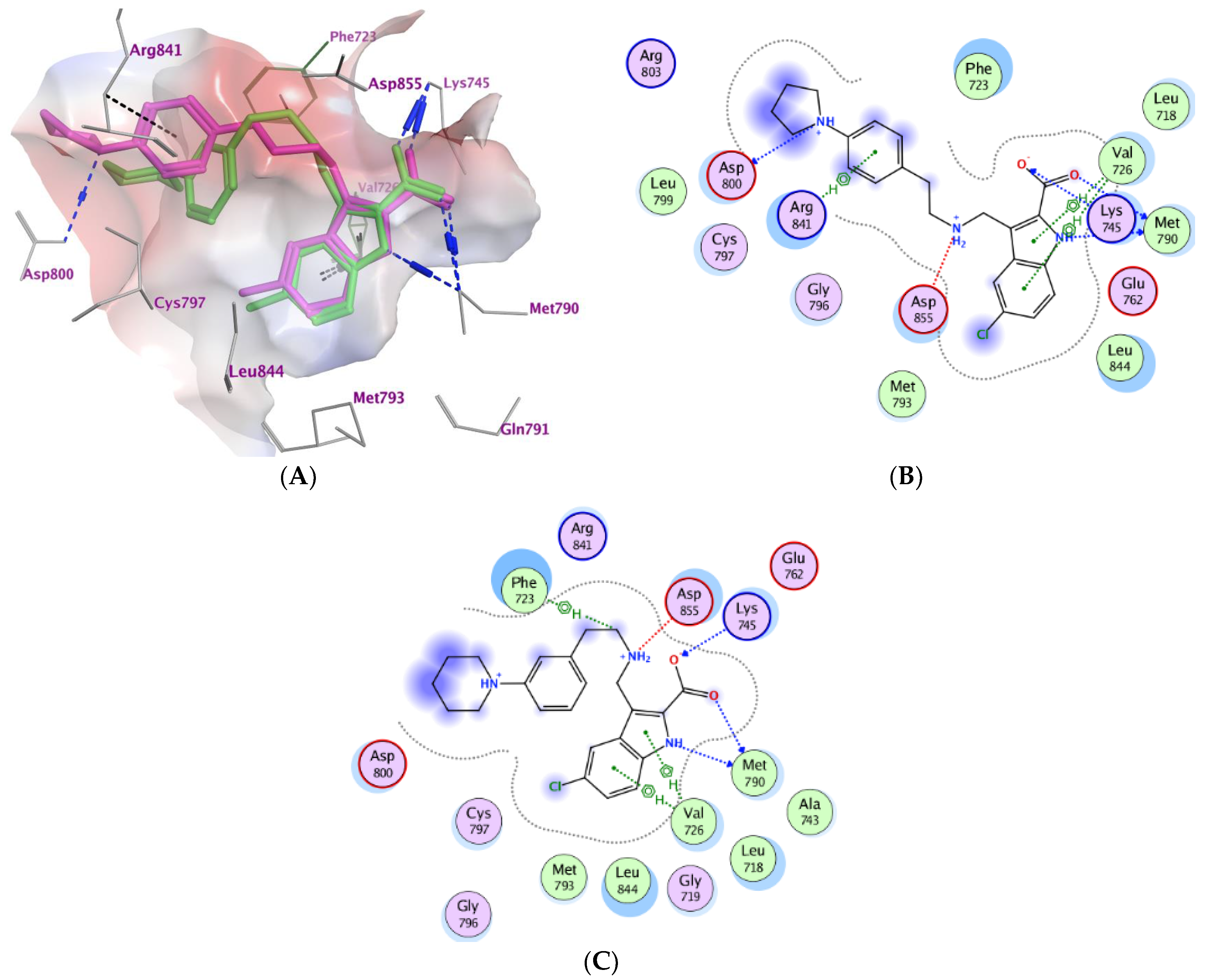
2.3. Molecular Modeling
2.4. In Silico ADME/Pharmacokinetics Studies
3. Experimental
3.1. Chemistry
3.1.1. General Method for the Synthesis of Compounds 3a–e
Ethyl 5-chloro-3-((phenethylamino)methyl)-1H-indole-2-carboxylate (3a)
Ethyl 5-chloro-3-((4-(pyrrolidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylate (3b)
Ethyl 5-chloro-3-((4-(piperidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylate (3c)
Ethyl 5-chloro-3-((4-(2-methylpyrrolidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylate (3d)
Ethyl 5-chloro-3-((3-(piperidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylate (3e)
3.1.2. General Method for the Synthesis of Compounds 4a–c
5-Chloro-3-((phenethylamino)methyl)-1H-indole-2-carboxylic acid (4a)
5-Chloro-3-((4-(pyrrolidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylic acid (4b)
5-Chloro-3-((4-(piperidin-1-yl)phenethylamino)methyl)-1H-indole-2-carboxylic acid (4c)
3.1.3. General Method for the Synthesis of Compounds 5a–c
7-Chloro-2-phenethyl-1,2-dihydropyrrolo [3,4-b]indol-3(4H)-one (5a)
7-Chloro-2-(4-(pyrrolidin-1-yl)phenethyl)-1,2-dihydropyrrolo [3,4-b]indol-3(4H)-one (5b)
7-Chloro-2-(4-(piperidin-1-yl)phenethyl)-1,2-dihydropyrrolo [3,4-b]indol-3(4H)-one (5c)
3.2. Biology
3.2.1. Cell Viability Assay and Evaluation of IC50
MTT Assay
Antiproliferative Test
EGFR Inhibitory Assay
BRAF Kinase Assay
In Vitro Cytotoxicity of LOX-IMVI Melanoma Cell Line
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Al-Sanea, M.M.; Khan, M.S.A.; Abdelazem, A.Z.; Lee, S.H.; Mok, P.L.; Gamal, M.; Shaker, M.E.; Afzal, M.; Youssif, B.G.M.; Omar, N.N. Synthesis, and In Vitro Antiproliferative Activity of New 1-Phenyl-3-(4-(pyridin-3-yl)phenyl)urea Scaffold-Based Compounds. Molecules 2018, 23, 297–309. [Google Scholar] [CrossRef]
- Abdelbaset, M.S.; Abdelrahman, M.H.; Bukhari, S.N.A.; Gouda, A.M.; Youssif, B.G.M.; Abdel-Aziz, M.; Abuo-Rahma, G.E.A. Design, Synthesis, and biological evaluation of new series of pyrrol-2(3H)-one and pyridazin-3(2H)-one derivatives as tubulin polymerization inhibitors. Bioorg. Chem. 2021, 107, 104522. [Google Scholar] [CrossRef]
- Rashid, H.; Xu, Y.; Muhammad, Y.; Wang, L.; Jiang, J. Research advances on anticancer activities of marine and its derivatives: An updated overview. Eur. J. Med. Chem. 2019, 161, 205–238. [Google Scholar] [CrossRef] [PubMed]
- El-Sherief, H.A.M.; Youssif, B.G.M.; Abdelazeem, A.H.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Design, Synthesis and Antiproliferative Evaluation of Novel 1,2,4-Triazole/Schiff Base Hybrids with EGFR and B-RAF Inhibitory Activities. Anti-Cancer Agents Med. Chem. 2019, 19, 697–706. [Google Scholar] [CrossRef]
- Mohassab, A.M.; Hassan, H.A.; Abdelhamid, D.; Gouda, A.M.; Youssif, B.G.M.; Tateishi, H.; Fujita, M.; Otsuka, M.; Abdel-Aziz, M. Design and Synthesis of Novel quinoline/chalcone/1,2,4-triazole hybrids as potent antiproliferative agent targeting EGFR and BRAFV600E kinases. Bioorg. Chem. 2021, 106, 104510. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Youssif, B.G.M.; Taher, E.S.; Abdelazeem, A.H.; Abdelhamid, A.A.; Marzouk, A.A. Design, Synthesis, Biological Evaluation, and Computational Studies of Novel Tri-Aryl Imidazole-Benzene Sulfonamide Hybrids as Promising Selective Carbonic Anhydrase IX and XII Inhibitors. Molecules 2021, 26, 4718. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.-L.; Leng, J.; Youssif, B.G.M.; Amjad, M.W.; Raja, M.A.J.; Hussain, M.A.; Hussaine, Z.; Kazmi, S.N.; Bukhari, S.N.A. Synthesis, and mechanistic studies of curcumin analogs-based oximes as potential anticancer agents. Chem. Biol. Drug Des. 2017, 90, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Zha, G.-F.; Qin, H.-L.; Youssif, B.G.M.; Amjad, M.W.; Raja, M.A.J.; Abdelazeem, A.H.; Bukhari, S.N.A. Discovery of potential anticancer multi-targeted ligustrazine based cyclohexanone and oxime analogs overcoming the cancer multidrug resistance. Eur. J. Med. Chem. 2017, 135, 34–48. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.A.; Gomaa, H.A.M.; Youssif, B.G.M. New 1,3,4-oxadiazoles linked 1,2,3-triazole moiety as antiproliferative agents targeting EGFR-TK. Arch. Pharm. 2022, 355, e2200009. [Google Scholar] [CrossRef]
- Youssif, B.G.M.; Gouda, A.M.; Moustafa, A.M.; Abdelhamid, A.A.; Gomaa, H.A.M.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of BRAFV600E/p38α with potential antiproliferative activity. J. Mol. Struct. 2022, 1253, 132218. [Google Scholar] [CrossRef]
- Yu, Z.; Ye, S.; Hu, G.; Lv, M.; Tu, Z.; Zhou, K.; Li, Q. The RAF-MEK-ERK pathway: Targeting ERK to overcome obstacles to effective cancer therapy. Future Med. Chem. 2015, 7, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Hrustanovic, G.; Olivas, V.; Pazarentzos, E.; Tulpule, A.; Asthana, S.; Blakely, C.M.; Okimoto, R.A.; Lin, L.; Neel, D.S.; Sabnis, A.; et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nat. Med. 2015, 21, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Frejat, F.O.A.; Mostafa, Y.A.; Gomaa, H.A.M.; Youssif, B.G.M.; Wu, C. Novel indazole derivatives as potent apoptotic antiproliferative agents by multi-targeted mechanism: Synthesis and biological evaluation. Bioorg. Chem. 2022, 126, 105922. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, J.A.; Gan, P.P.; Erlich, R.B.; Liu, M. TUBB3/betaIII-tubulin acts through the PTEN/AKT signaling axis to promote tumorigenesis and anoikis resistance in nonsmall cell lung cancer. Cancer Res. 2015, 75, 415–425. [Google Scholar] [CrossRef]
- El-Sherief, H.A.; Youssif, B.G.; Bukhari, S.N.A.; Abdel-Aziz, M.; Abdel-Rahman, H.M. Novel 1,2,4-triazole derivatives as potential anticancer agents: Design, synthesis, molecular docking, and mechanistic studies. Bioorg. Chem. 2018, 76, 314–325. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.; Youssif, B.G.M.; Gouda, A.M. Globally approved EGFR inhibitors: Insights into their syntheses, targeted kinases, biological activity, receptor interactions and metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Mishra, R.D.L.A. Anticancer potential of plants and natural products: A review. J. Ethnopharmacol. 2013, 1, 622–628. [Google Scholar]
- Dhimany, A.; Sharmay, R.; Singh, R.K. Target-based anticancer indole derivatives and insight into structure-activity relationship: A mechanistic review update (2018–2021). Acta Pharm. Sin. B 2022, 12, 3006–3027. [Google Scholar] [CrossRef]
- Pajaniradje, S.; Kumar, M.K.; Radhakrishanan, R.; Sufi, S.A.; Subramaniam, S.; Anaikutti, P. Indole curcumin reverses multidrug resistance by reducing the expression of ABCD1 and COX2 in induced multidrug resistant human lung cancer cells. Lett. Drug Des. Discov. 2020, 17, 1146–1154. [Google Scholar] [CrossRef]
- Song, J.; Yoo, J.; Kwon, A.; Kim, D.; Nguyen, H.K.; Lee, B.-Y.; Suh, W.; Min, K.H. Structure-Activity Relationship of Indole-Tethered Pyrimidine Derivatives that Concurrently Inhibit Epidermal Growth Factor Receptor and Other Angiokinases. PLoS ONE 2015, 10, e0138823. [Google Scholar] [CrossRef]
- Li, W.; Qi, Y.-Y.; Wang, Y.-Y.; Gan, Y.-Y.; Shao, L.-H.; Zhang, L.-Q.; Tang, Z.-H.; Zhu, M.; Tang, S.-Y.; Wang, Z.-C.; et al. Design, synthesis, and biological evaluation of sorafenib derivatives containing indole (ketone) semicarbazide analogs as antitumor agents. J. Heterocycl. Chem. 2020, 57, 2548–2560. [Google Scholar] [CrossRef]
- Singh, P.K.; Silakari, O. Molecular dynamics guided development of indole based dual inhibitors of EGFR (T790M) and c-MET. Bioorg. Chem. 2018, 79, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H. Three generations of epidermal growth factor receptor tyrosine kinase inhibitors developed to revolutionize the therapy of lung cancer. Drug Des. Dev. Ther. 2016, 10, 3867–3872. [Google Scholar] [CrossRef]
- Mohamed, F.A.M.; Gomaa, H.A.M.; Hendawy, O.M.; Ali, A.T.; Farghaly, H.S.; Gouda, A.M.; Abdelazeem, A.H.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Design, synthesis, and biological evaluation of novel EGFR inhibitors containing 5-chloro-3-hydroxymethyl-indole-2-carboxamide scaffold with apoptotic antiproliferative activity. Bioorg. Chem. 2021, 112, 104960. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.M.; Abdelrahman, M.H.; Abdelazeem, A.H.; Abdelgawad, M.A.; Ibrahim, H.M.; Salem, O.I.A.; Mohamed, M.F.A.; Treamblu, L.; Bukhari, S.N.A. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino[1,2-a]indol-1(2H)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem. 2018, 146, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.A.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.M.; Youssif, B.G.M. Design, and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef]
- Gomaa, H.A.M.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.M.; Mohamed, F.A.M.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Mahmoud, M.A.; Mostafa, Y.A.; Raslan, A.E.; Youssif, B.G.M. Novel piperine-carboximidamide hybrids: Design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzyme Inhib. Med. Chem. 2023, 38, 376–386. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Beshr, E.A.M.; Gomaa, H.A.M.; Mostafa, Y.A.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Discovery of new cyanopyridine/chalcone hybrids as dual inhibitors of EGFR/BRAFV600E with promising antiproliferative properties. Arch. Pharm. 2023; in press. [Google Scholar]
- Tawfeek, H.N.; Hassan, A.A.; Bräse, S.; Nieger, M.; Mostafa, Y.A.; Gomaa, H.A.M.; Youssif, B.G.M.; El-Shreef, E.M. Design, Synthesis, Crystal Structures and Biological Evaluation of Some 1,3-Thiazolidin-4-ones as Dual CDK2/EGFR Potent Inhibitors with Potential Apoptotic Antiproliferative Effects. Arab. J. Chem. 2022, 15, 104280. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Aboraia, A.S.; Youssif, B.G.M.; Elsadek, B.E.M. Design, Synthesis and Pharmacophoric Model Building of New 3-Alkoxymethyl / 3-Phenyl indole-2-carboxamides with Potential Antiproliferative Activity. Chem. Biol. Drug Des. 2017, 90, 64–82. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Youssif, B.G.M.; Abdelgawad, M.A.; Abdelazeem, A.H.; Ibrahim, H.M.; Moustafa, A.A.; Treamblu, T.; Bukhari, S.N.A. Synthesis, Biological Evaluation, Docking Study and Ulcerogenicity Profiling of Some Novel Quinoline-2-Carboxamides as Dual COXs/LOX Inhibitors Endowed with Anti-Inflammatory Activity. Eur. J. Med. Chem. 2017, 127, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.M.; El-Sherief, H.A.M.; Hussein, S.; Gouda, A.M.; Salem, O.I.A.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G.M. Novel 1,2,4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020, 105, 104369. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.M.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A.M. 5-chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]
- Marzouk, A.A.; Abdel-Aziz, S.A.; Abdelrahman, K.S.; Wanas, A.S.; Gouda, A.M.; Youssif, B.G.M.; Abdel-Aziz, M. Design, and synthesis of new 1,6-dihydropyrimidin-2-thio derivatives targeting VEGFR-2: Molecular docking and antiproliferative evaluation. Bioorg. Chem. 2020, 102, 104090. [Google Scholar] [CrossRef]
- Hisham, M.; Hassan, H.A.; Gomaa, H.A.M.; Youssif, B.G.M.; Hayallah, A.M.; Abdel-Aziz, M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as EGFR inhibitors. J. Mol. Struct. 2022, 1254, 132422. [Google Scholar] [CrossRef]
- Alshammari, M.B.; Aly, A.A.; Youssif, B.G.M.; Bräse, S.; Ahmad, A.; Brown, A.B.; Ibrahim, M.A.A.; Mohamed, A.A. Design and synthesis of new thiazolidinone/uracil derivatives as antiproliferative agents targeting EGFR and/or BRAFV600E. Front. Chem. 2022, 10, 1076383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, Z.; Xia, S.; Liu, Q.; Gou, S. Design, synthesis, and biological evaluation of sulfamoyl phenyl quinazoline derivatives as potential EGFR/CAIX dual inhibitors. Eur. J. Med. Chem. 2021, 216, 113300. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Gray, D.C.; Eby, M.T.; Tien, J.Y.; Wong, L.; Bower, J.; Gogineni, A.; Zha, J.; Cole, M.J.; Stern, H.M.; et al. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006, 66, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, T.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Frejat, F.O.A.; Cao, Y.; Zhai, H.; Abdel-Aziz, S.A.; Gomaa, H.A.M.; Youssif, B.G.M.; Wu1, C. Novel 1,2,4-oxadiazole/pyrrolidine hybrids as DNA gyrase and Topoisomerase IV inhibitors with potential antibacterial activity. Arab. J. Chem. 2022, 15, 103538. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.; Becker, C.; Lategahn, J.; Keul, M.; Ketzer, J.; Mühlenberg, T.; Kollipara, L.; Schultz-Fademrecht, C.; Zahedi, R.P.; Bauer, S.; et al. Insight into the Inhibition of Drug-Resistant Mutants of the Receptor Tyrosine Kinase EGFR. Angew. Chem. Int. Ed. 2016, 55, 10909–10912. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness, and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Dong, J.; Wang, N.-N.; Yao, Z.-J.; Zhang, L.; Cheng, Y.; Ouyang, D.; Lu, A.-P.; Cao, D.-S. ADMETlab: A platform for systematic ADMET evaluation based on a comprehensively collected ADMET database. J. Cheminform. 2018, 10, 29. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average (GI50) | ||
| 3a | 87 | 33 ± 3 | 35 ± 3 | 36 ± 3 | 36 ± 3 | 35 |
| 3b | 91 | 30 ± 3 | 32 ± 3 | 30 ± 3 | 32 ± 3 | 31 |
| 3c | 89 | 41 ± 4 | 45 ± 4 | 42 ± 4 | 44 ± 4 | 42 |
| 3d | 90 | 37 ± 3 | 40 ± 4 | 38 ± 3 | 38 ± 3 | 38 |
| 3e | 92 | 27 ± 2 | 30 ± 3 | 29 ± 3 | 30 ± 3 | 29 |
| 4a | 91 | 75 ± 7 | 79 ± 7 | 78 ± 7 | 78 ± 7 | 78 |
| 4b | 89 | 65 ± 6 | 69 ± 6 | 68 ± 6 | 68 ± 6 | 68 |
| 4c | 87 | 69 ± 7 | 73 ± 7 | 72 ± 7 | 75 ± 7 | 72 |
| 5a | 89 | 46 ± 4 | 48 ± 5 | 50 ± 5 | 49 ± 5 | 48 |
| 5b | 89 | 58 ± 5 | 61 ± 6 | 64 ± 6 | 66 ± 6 | 62 |
| 5c | 91 | 51± 5 | 53 ± 5 | 55 ± 5 | 57 ± 5 | 54 |
| Erlotinib | - | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Compound | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) | EGFRT790M Inhibition IC50 ± SEM (nM) |
|---|---|---|---|
| 3a | 85 ± 6 | 43 ± 4 | -- |
| 3b | 74 ± 5 | 39 ± 3 | 9.2 ± 2 |
| 3c | 89 ± 6 | 67 ± 6 | -- |
| 3d | 82 ± 7 | 54 ± 5 | -- |
| 3e | 68 ± 5 | 35 ± 3 | 8.6 ± 2 |
| Erlotinib | 80 ± 5 | 60 ± 5 | -- |
| Vemurafenib | ND | 30 ± 3 | -- |
| Osimertinib | -- | -- | 8 ± 2 |
| Compound | LOX-IMVI Melanoma IC50 ± SEM (µM) |
|---|---|
| 3b | 1.12 ± 0.01 |
| 3e | 0.96 ± 0.01 |
| Staurosporine | 7.10 ± 0.05 |
| Compd. | MOE Score kcal/mol | Hydrogen Bond Interactions | Hydrophobic Interactions | Other Interactions |
|---|---|---|---|---|
| Vemurafenib | −11.78 | Thr529 Gln530 Cys532 Asp594 Gly596 | Trp531, Phe583, Cys532, Ile463, Thr592, val471, Lys483, Leu514 | Lys483 (ionic) |
| 3a | −8.08 | Lys483 | Phe583, Cys532, Thr592, val471, Lys483, Leu514 | Lys483 (Pi-cation) |
| 3b | −10.12 | Thr529 Cys532 Lys483 | Trp531, Phe583, Cys532, Ile463, Thr592, val471, Lys483, Leu514 | Lys483 (ionic) Cys532 (Pi-H) |
| 3c | −9.27 | Thr529 Asp594 | Trp531, Phe583, Cys532, Ile463, Thr592, val471, Lys483, Leu514 | Lys483 (ionic) |
| 3d | −9.14 | Thr529 | Trp531, Phe583, Cys532, Ile463, Thr592, val471, Lys483, Leu514 | Lys483 (ionic) |
| 3e | −10.40 | Thr529 Gly596 Cys532 Lys483 | Trp531, Phe583, Cys532, Ile463, Thr592, val471, Lys483, Leu514, Gly596 | Val471 (Pi-H) Lys483 (ionic) Lys483 Pi-cation) |
| Compd. | MOE Score kcal/mol | Hydrogen Bond Interactions | Hydrophobic Interactions | Other Interactions |
|---|---|---|---|---|
| Co-crystalized ligand (6HJ) a | −10.42 | Met790 Gln791 Met793 | Leu844, Cys797, Leu718, Val726, Met790 | Val726 (Pi-H) Cys797 (covalent) |
| 3b | −6.71 | Met790 Asp800 Lys745 | Asp800, Phe723, Leu844, Cys797, Leu718, Val726, Met790, Lys745. | Lys745 (Ionic) Asp855 (Ionic) Asp800 (Ionic) Val726 (pi-H) Arg841 (pi-H) |
| 3e | −6.64 | Met790 Lys745 | Asp800, Phe723, Leu844, Cys797, Leu718, Val726, Met790 | Lys745 (Ionic) Asp855 (Ionic) Phe723 (Pi-H) Val726 (pi-H) |
| Compd. | MW | ROTB | HBA | HBD | Violations | MR | TPSA | Log P |
|---|---|---|---|---|---|---|---|---|
| 3a | 357 | 8 | 3 | 2 | 0 | 101 | 54.12 | 4.23 |
| 3b | 426 | 9 | 3 | 2 | 0 | 126 | 57.36 | 4.59 |
| 3c | 440 | 9 | 3 | 2 | 0 | 131 | 57.36 | 4.82 |
| 3d | 440 | 9 | 3 | 2 | 0 | 131 | 57.36 | 4.79 |
| 3e | 440 | 9 | 3 | 2 | 0 | 131 | 57.36 | 4.76 |
| Compd. | GI Abs. | BBB | P-gp Substrate | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | CYP2D6 Inhibitor | CYP3A4 Inhibitor |
|---|---|---|---|---|---|---|---|---|
| 3a | 88.904 | 0.312 | −−− | +++ | ++ | ++ | ++ | ++ |
| 3b | 90.505 | 0.239 | −−− | ++ | ++ | + | + | ++ |
| 3c | 90.116 | 0.252 | −−− | ++ | ++ | + | + | ++ |
| 3d | 90.12 | 0.252 | −−− | + | ++ | + | + | ++ |
| 3e | 89.884 | 0.223 | −−− | ++ | ++ | + | + | ++ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Mohammed, A.F.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G.M. Design, Synthesis, and Antiproliferative Activity of New 5-Chloro-indole-2-carboxylate and Pyrrolo[3,4-b]indol-3-one Derivatives as Potent Inhibitors of EGFRT790M/BRAFV600E Pathways. Molecules 2023, 28, 1269. https://doi.org/10.3390/molecules28031269
Al-Wahaibi LH, Mohammed AF, Abdelrahman MH, Trembleau L, Youssif BGM. Design, Synthesis, and Antiproliferative Activity of New 5-Chloro-indole-2-carboxylate and Pyrrolo[3,4-b]indol-3-one Derivatives as Potent Inhibitors of EGFRT790M/BRAFV600E Pathways. Molecules. 2023; 28(3):1269. https://doi.org/10.3390/molecules28031269
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Anber F. Mohammed, Mostafa H. Abdelrahman, Laurent Trembleau, and Bahaa G. M. Youssif. 2023. "Design, Synthesis, and Antiproliferative Activity of New 5-Chloro-indole-2-carboxylate and Pyrrolo[3,4-b]indol-3-one Derivatives as Potent Inhibitors of EGFRT790M/BRAFV600E Pathways" Molecules 28, no. 3: 1269. https://doi.org/10.3390/molecules28031269
APA StyleAl-Wahaibi, L. H., Mohammed, A. F., Abdelrahman, M. H., Trembleau, L., & Youssif, B. G. M. (2023). Design, Synthesis, and Antiproliferative Activity of New 5-Chloro-indole-2-carboxylate and Pyrrolo[3,4-b]indol-3-one Derivatives as Potent Inhibitors of EGFRT790M/BRAFV600E Pathways. Molecules, 28(3), 1269. https://doi.org/10.3390/molecules28031269