



Synthesis of New Azetidine and Oxetane Amino Acid Derivatives through Aza-Michael Addition of NH-Heterocycles with Methyl 2-(Azetidin- or Oxetan-3-Ylidene)Acetates

 ,
,
Abstract
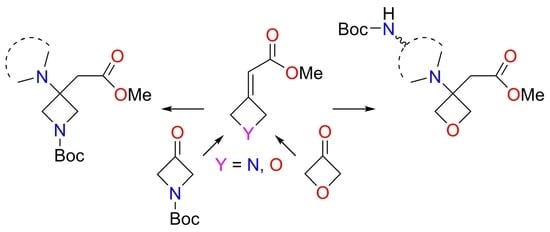
1. Introduction
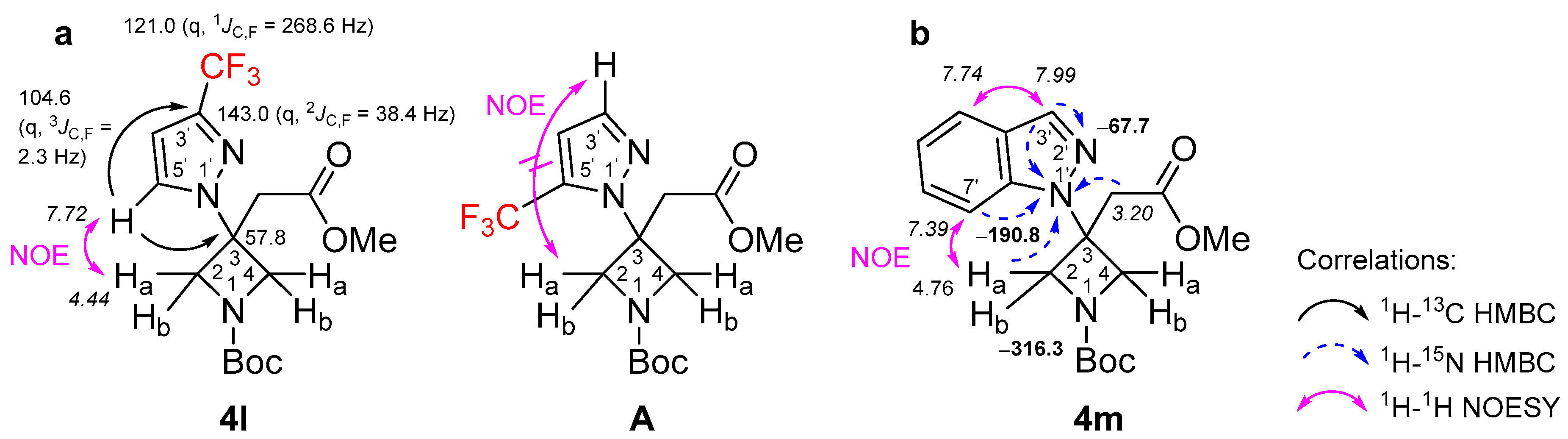
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
3.2.1. tert-Butyl 3-(2-methoxy-2-oxoethylidene)azetidine-1-carboxylate (3)
3.2.2. General Procedure for Compounds 4a–p
tert-Butyl 3′-(2-methoxy-2-oxoethyl)[1,3′-biazetidine]-1′-carboxylate (4a)
tert-Butyl 3-hydroxy-3′-(2-methoxy-2-oxoethyl)[1,3′-biazetidine]-1′-carboxylate (4b)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(pyrrolidin-1-yl)azetidine-1-carboxylate (4c)
tert-Butyl 3-(3,3-difluoropyrrolidin-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4d)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(piperidin-1-yl)azetidine-1-carboxylate (4e)
tert-Butyl 3-(4-hydroxypiperidin-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4f)
tert-Butyl 3-(4-hydroxy-4-phenylpiperidin-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4g)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(morpholin-4-yl)azetidine-1-carboxylate (4h)
tert-Butyl 3-(1,3-dihydro-2H-isoindol-2-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4i)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(1H-pyrazol-1-yl)azetidine-1-carboxylate (4j)
tert-Butyl 3-(4-bromo-1H-pyrazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4k)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[3-(trifluoromethyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (4l)
tert-Butyl 3-(1H-indazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4m)
tert-Butyl 3-(1H-imidazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4n)
tert-Butyl 3-(1H-benzimidazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4o)
tert-Butyl 3-(1H-indol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4p)
3.2.3. General Procedure for Compounds 4q–s
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(1H-1,2,4-triazol-1-yl)azetidine-1-carboxylate (4q)
tert-Butyl 3-(1H-benzotriazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4r)
tert-Butyl 3-(2H-benzotriazol-2-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (4s)
3.2.4. General procedure for compounds 5a–n
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-(4-phenyl-1H-pyrazol-1-yl)azetidine-1-carboxylate (5a)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(4-methylphenyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5b)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(2-methylphenyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5c)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(4-methoxyphenyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5d)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(3-methoxyphenyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5e)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(2-methoxyphenyl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5f)
tert-Butyl 3-[4-(4-fluorophenyl)-1H-pyrazol-1-yl]-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (5g)
tert-Butyl 3-[4-(4-chlorophenyl)-1H-pyrazol-1-yl]-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (5h)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(pyridin-4-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5i)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(pyridin-3-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5j)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(6-methoxypyridin-3-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5k)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(thiophen-3-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5l)
tert-Butyl 3-(2-methoxy-2-oxoethyl)-3-[4-(4-methylthiophen-3-yl)-1H-pyrazol-1-yl]azetidine-1-carboxylate (5m)
tert-Butyl 3-(4-cyclopropyl-1H-pyrazol-1-yl)-3-(2-methoxy-2-oxoethyl)azetidine-1-carboxylate (5n)
3.2.5. Methyl(oxetan-3-ylidene)acetate (7)
3.2.6. General Procedure for Compounds 8a–g
Methyl(3-{3-[(tert-butoxycarbonyl)amino]azetidin-1-yl}oxetan-3-yl)acetate (8a)
Methyl(3-{(3S)-3-[(tert-butoxycarbonyl)amino]pyrrolidin-1-yl}oxetan-3-yl)acetate (8b)
Methyl(3-{(3R)-3-[(tert-butoxycarbonyl)amino]pyrrolidin-1-yl}oxetan-3-yl)acetate (8c)
Methyl(3-{(3S)-3-[(tert-butoxycarbonyl)amino]piperidin-1-yl}oxetan-3-yl)acetate (8d)
Methyl(3-{(3R)-3-[(tert-butoxycarbonyl)amino]piperidin-1-yl}oxetan-3-yl)acetate (8e)
Methyl(3-{4-[(tert-butoxycarbonyl)amino]piperidin-1-yl}oxetan-3-yl)acetate (8f)
Methyl[3-(4-{[(tert-butoxycarbonyl)amino]methyl}piperidin-1-yl)oxetan-3-yl]acetate (8g)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gupta, R.R.; Kumar, M.; Gupta, V. Four-membered heterocycles. In Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 1998; pp. 357–410. [Google Scholar] [CrossRef]
- Singh, G.S.; D’hooghe, M.; De Kimpe, N. Azetidines, azetines and azetes: Monocyclic. In Comprehensive Heterocyclic Chemistry III; Stevens, C.V., Ed.; Elsevier: Oxford, UK, 2008; Volume 2, Chapter 2.01; pp. 1–110. [Google Scholar]
- Mehra, V.; Lumb, I.; Anand, A.; Kumar, V. Recent advances in synthetic facets of immensely reactive azetidines. RSC Adv. 2017, 7, 45763–45783. [Google Scholar] [CrossRef]
- Brandi, A.; Cicchi, S.; Cordero, F.M. Novel syntheses of azetidines and azetidinones. Chem. Rev. 2008, 108, 3988–4035. [Google Scholar] [CrossRef] [PubMed]
- Ohshita, K.; Ishiyama, H.; Takahashi, Y.; Ito, J.; Mikami, Y.; Kobayashi, J. Synthesis of penaresidin derivatives and its biological activity. Bioorg. Med. Chem. 2007, 15, 4910–4916. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.; Cheng, J.-F.; Ishibashi, M.; Wälchli, M.R.; Yamamur, S.; Ohizumi, Y. Penaresidin A and B, two novel azetidine alkaloids with potent actomyosin ATPase-activating activity from the Okinawan marine sponge Penares sp. J. Chem. Soc. Perkin Trans. 1991, 5, 1135–1137. [Google Scholar] [CrossRef]
- Sata, T.; Saito, H. Sustained calcium antagonists azelnidipine (Calblock pharmacological properties of) the clinical effects. Jpn. J. Pharmacol. 2003, 122, 539–547. [Google Scholar] [CrossRef]
- Watanabe, M.; Hirano, T.; Okamoto, S.; Shiraishi, S.; Tomiguchi, S.; Uchino, M. Azelnidipine, a long-acting calcium channel blocker, could control hypertension without decreasing cerebral blood flow in post-ischemic stroke patients. A 123I-IMP SPECT follow-up study. Hypertens. Res. 2010, 33, 43–48. [Google Scholar] [CrossRef]
- Faust, M.; Höfner, G.; Pabel, J.; Wanner, K. Azetidine derivatives as novel γ-aminobutyric acid uptake inhibitors: Synthesis, biological evaluation, and structure–activity relationship. Eur. J. Med. Chem. 2010, 45, 2453–2466. [Google Scholar] [CrossRef]
- Žukauskaitė, A.; Mangelinckx, S.; Buinauskaitė, V.; Šačkus, A.; De Kimpe, N. Synthesis of new functionalized aziridine-2- and azetidine-3-carboxylic acid derivatives of potential interest for biological and foldameric applications. Amino Acids 2011, 41, 541–558. [Google Scholar] [CrossRef]
- Žukauskaitė, A.; Moretto, A.; Peggion, C.; De Zotti, M.; Šačkus, A.; Formaggio, F.; De Kimpe, N.; Mangelinckx, S. Synthesis and conformational study of model peptides containing N-substituted 3-aminoazetidine-3-carboxylic Acids. Eur. J. Org. Chem. 2014, 2014, 2312–2321. [Google Scholar] [CrossRef]
- Rubenstein, E.; Zhou, H.; Krasinska, K.M.; Chien, A.; Becker, C.H. Azetidine-2-carboxylic acid in garden beets (Beta vulgaris). Phytochemistry 2006, 67, 898–903. [Google Scholar] [CrossRef]
- Ashino, H.; Shimamura, M.; Nakajima, H.; Dombou, M.; Kawanaka, S.; Oikawa, T.; Iwaguchi, T.; Kawashima, S. Novel function of ascorbic acid as an angiostatic factor. Angiogenesis 2003, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Tsai, F.H.; Overberger, C.G.R.; Zand, R. Synthesis and peptide bond orientation in tetrapeptides containing L-azetidine-2-carboxylic acid and L-proline. Biopolymers 1990, 30, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Reiners, F.; Joseph, E.; Nißl, B.; Didier, D. Stereoselective access to azetidine-based α-amino acids and applications to small peptide synthesis. Org. Lett. 2020, 22, 8533–8537. [Google Scholar] [CrossRef] [PubMed]
- Torino, D.; Mollica, A.; Pinnen, F.; Lucente, G.; Feliciani, F.; Davis, P.; Lai, J.; Ma, S.-W.; Porreca, F.; Hruby, V.J. Synthesis and evaluation of new endomorphin analogues modified at the Pro2 residue. Bioorg. Med. Chem. Lett. 2009, 19, 4115–4118. [Google Scholar] [CrossRef]
- Dubois, M.A.J.; Smith, M.A.; White, A.J.P.; Jie, A.L.W.; Mousseau, J.J.; Choi, C.; Bull, J.A. Short synthesis of oxetane and azetidine 3-Aryl-3-carboxylic acid derivatives by selective furan oxidative cleavage. Org. Lett. 2020, 22, 5279–5283. [Google Scholar] [CrossRef]
- He, Z.-T.; Hartwig, J.F. Palladium-catalyzed α-arylation for the addition of small rings to aromatic compounds. Nat. Commun. 2019, 10, 4083. [Google Scholar] [CrossRef]
- Latta, K.S.; Ginsberg, B.; Barkin, R.L. Meperidine: A critical review. Am. J. Ther. 2002, 9, 53–68. [Google Scholar] [CrossRef]
- Lerner, I.; Michaeli, A. Positive Allosteric Modulators of GABAA Receptor. U.S. Patent 2021/035595 A1, 18 November 2021. [Google Scholar]
- Chalyk, B.A.; Kandaurova, I.Y.; Hrebeniuk, K.V.; Manoilenko, O.V.; Kulik, I.B.; Iminov, R.T.; Kubyshkin, V.; Tverdokhlebov, A.V.; Ablialimov, O.K.; Mykhailiuk, P.K. A base promoted multigram synthesis of aminoisoxazoles: Valuable building blocks for drug discovery and peptidomimetics. RSC Adv. 2016, 6, 25713–25723. [Google Scholar] [CrossRef]
- Callery, P.S.; Geelhaar, L.A. 1-Piperideine as an in vivo precursor of the gamma-aminobutyric acid homologue 5-aminopentanoic acid. J. Neurochem. 1985, 45, 946–948. [Google Scholar] [CrossRef]
- Bruzgulienė, J.; Račkauskienė, G.; Bieliauskas, A.; Milišiūnaitė, V.; Dagilienė, M.; Matulevičiūtė, G.; Martynaitis, V.; Krikštolaitytė, S.; Sløk, F.A.; Šačkus, A. Regioselective synthesis of methyl 5-(N-Boc-cycloaminyl)-1,2-oxazole-4-carboxylates as new amino acid-like building blocks. Beilstein J. Org. Chem. 2022, 18, 102–109. [Google Scholar] [CrossRef]
- Iškauskienė, M.; Ragaitė, G.; Sløk, F.A.; Šačkus, A. Facile synthesis of novel amino acid-like building blocks by N-alkylation of heterocyclic carboxylates with N-Boc-3-iodoazetidine. Mol. Divers 2020, 24, 1235–1251. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, J.A.; Wuitschik, G.; Rogers-Evans, M.; Müller, K.; Carreira, E.M. Oxetanes as versatile elements in drug discovery and synthesis. Angew. Chem. Int. Ed. 2010, 49, 9052–9067. [Google Scholar] [CrossRef] [PubMed]
- Kawahata, Y.; Takatsuto, S.; Ikekawa, N.; Murata, M.; Omura, S. Synthesis of a new amino acid-antibiotic, oxetin and its three stereoisomers. Chem. Pharm. Bull. 1986, 34, 3102–3110. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell. 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Seki, J.-I.; Shimada, N.; Takakashi, K.; Takita, T.; Takeuchi, T.; Hoshino, H. Inhibition of infectivity of human immunodeficiency virus by a novel nucleoside, oxetanocin, and related compounds. Antimicrob. Agents Chemother. 1989, 33, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Fauq, A.H. Synthesis of novel four-membered ring amino acids as modulators of the N-Methyl-D-Aspartate(NMDA) receptor complex. Synlett 1991, 783–784. [Google Scholar] [CrossRef]
- Wuitschik, G.; Carreira, E.M.; Wagner, B.; Fisher, H.; Parrilla, I.; Schuler, F.; Rogers-Evans, M.; Muller, K. Oxetanes in drug discovery: Structural and synthetic insights. J. Med. Chem. 2010, 53, 3227–3246. [Google Scholar] [CrossRef]
- Bull, J.A.; Croft, R.A.; Davis, O.A.; Doran, R.; Morgan, K.F. Oxetanes: Recent advances in synthesis, reactivity, and medicinal chemistry. Chem. Rev. 2016, 116, 12150–12233. [Google Scholar] [CrossRef]
- Powell, N.H.; Clarkson, G.J.; Notman, R.; Raubo, P.; Martin, N.G.; Shipman, M. Synthesis and structure of oxetane containing tripeptide motifs. Chem. Commun. 2014, 50, 8797–8800. [Google Scholar] [CrossRef]
- Lucas, S.D.; Fischer, H.; Alker, A.; Rauter, A.P.; Wessel, H.P. Libraries on oxetane δ-amino acid scaffolds: Syntheses and evaluation of physicochemical and metabolic properties. Int. J. Carbohydr. Chem. 2011, 30, 498–548. [Google Scholar] [CrossRef]
- Madsen, D.; Azevedo, C.; Micco, I.; Petersen, L.K.; Hansen, N.J.V. Chapter four—An overview of DNA-encoded libraries: A versatile tool for drug discovery. Prog. Med. Chem. 2020, 59, 181–249. [Google Scholar] [CrossRef] [PubMed]
- Bisceglia, J.A.; Orelli, L.R. Recent progress in the Horner-Wadsworth-Emmons reaction. Curr. Org. Chem. 2015, 19, 744–775. [Google Scholar] [CrossRef]
- Yang, X.; Kong, W.-Y.; Gao, J.-N.; Cheng, L.; Li, N.-N.; Li, M.; Li, H.-T.; Fan, J.; Gao, J.-M.; Ouyang, Q.; et al. Rhodium catalyzed C-C bond cleavage/coupling of 2-(Azetidin-3-ylidene)acetates and analogs. Chem. Commun. 2019, 55, 12707–12710. [Google Scholar] [CrossRef] [PubMed]
- Harwood, L.M.; Moody, C.J. Experimental Organic Chemistry: Principles and Practice; Blackwell Scientific Publications: Oxford, UK, 1989; p. 778. [Google Scholar]
- Rulev, A.Y.; Tyumentsev, I.A. Pull-pull alkenes in the aza-Michael reaction. Adv. Synth. Catal. 2022, 364, 1622–1642. [Google Scholar] [CrossRef]
- Vicario, J.L.; Badía, D.; Carrillo, L.; Etxebarria, J.; Reyes, E.; Ruiz, N. The asymmetric aza-Michael reaction. A review. Org. Prep. Proced. Int. 2005, 37, 513–538. [Google Scholar] [CrossRef]
- Lin, Y.; Hirschi, W.J.; Kunadia, A.; Paul, A.; Ghiviriga, I.; Abboud, K.A.; Karugu, R.W.; Vetticatt, M.J.; Hirschi, J.S.; Seidel, D. A Selenourea-Thiourea Brønsted acid catalyst facilitates asymmetric conjugate additions of amines to α,β-unsaturated esters. J. Am. Chem. Soc. 2020, 142, 5627–5635. [Google Scholar] [CrossRef]
- Gilfillan, L.; Artschwager, R.; Harkiss, A.H.; Liskamp, R.M.J.; Sutherland, A. Synthesis of pyrazole containing α-amino acids via a highly regioselective condensation/aza-Michael reaction of β-aryl α,β-unsaturated ketones. Org. Biomol. Chem. 2015, 13, 4514–4523. [Google Scholar] [CrossRef]
- Małolepsza, J.; Joachimiak, Ł.; Blazewska, K. Aza-Michael addition of imidazole analogues. Synthesis 2016, 48, 2681–2704. [Google Scholar] [CrossRef]
- Kodolitsch, K.; Gobec, F.; Slugovc, C. Solvent- and Catalyst-Free Aza-Michael Addition of Imidazoles and Related Heterocycles. Solvent- and Catalyst-Free Aza-Michael Addition of Imidazoles and Related Heterocycles. EurJOC 2020, 19, 2973–2978. [Google Scholar] [CrossRef]
- Hou, X.; Hemit, H.; Yong, J.; Nie, L.; Aisa, H.A. Mild and efficient procedure for Michael addition of N-heterocycles to α,β-unsaturated compounds using anhydrous K3PO4 as catalyst. Synth. Commun. 2021, 40, 973–979. [Google Scholar] [CrossRef]
- Yang, J.; Bao, Y.; Zhou, H.; Li, T.; Li, N.; Li, Z. Highly efficient synthesis of N 1-substituted 1H-indazoles by DBU-catalyzed aza-Michael reaction of indazole with enones. Synthesis 2016, 48, 1139–1146. [Google Scholar] [CrossRef]
- Xie, S.-S.; Hui, Y.-H.; Long, X.-J.; Wang, C.-C.; Xie, Z.-F. Aza-Michael addition reactions between nitroolefins and benzotriazole catalyzed by MCM-41 immobilized heteropoly acids in water. Chin. Chem. Lett. 2013, 24, 28–30. [Google Scholar] [CrossRef]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangs, K.K.; Sreekantha, B.; Jonnalagadda, S.B. A review on recent advances in nitrogen-containing molecules and their biological applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Kachaeva, M.V.; Obernikhina, N.V.; Veligina, E.S.; Zhuravlova, M.Y.; Prostota, Y.O.; Kachkovsky, O.D.; Brovarets, V.S. Estimation of biological affinity of nitrogen-containing conjugated heterocyclic pharmacophores. Chem. Heterocycl. Compd. 2019, 55, 448–454. [Google Scholar] [CrossRef]
- Chen, S.-W.; Zhang, G.-C.; Lou, Q.-X.; Cui, W.; Zhang, S.-S.; Hu, W.-H.; Zhao, J.-L. Organocatalytic enantioselective aza-Michael reaction of benzotriazole to β,β-disubstituted nitroalkenes. ChemCatChem 2015, 7, 1935–1938. [Google Scholar] [CrossRef]
- Li, Z.; Li, T.; Fu, R.; Yang, J. Regioselective 1,4-conjugate aza-Michael addition of dienones with benzotriazole. Heterocycl. Commun. 2017, 23, 287–291. [Google Scholar] [CrossRef]
- Azizi, N.; Mohammad, R.; Saidi, M.R. LiClO4 Accelerated Michael addition of amines to α,β-unsaturated olefins under solvent-free condition. Tetrahedron 2004, 60, 383–387. [Google Scholar] [CrossRef]
- Dutt, S.; Goel, V.; Garg, N.; Choudhury, D.; Mallick, D.; Tyagi, V. Biocatalytic aza-Michael addition of aromatic amines to enone using α-amylase in water. Adv. Synth. Catal. 2020, 362, 858–866. [Google Scholar] [CrossRef]
- Ying, A.-G.; Wang, L.-M.; Deng, H.-X.; Chen, J.-H.; Chen, X.-Z.; Yeb, W.-D. Green and efficient aza-Michael additions of aromatic amines to α,β-unsaturated ketones catalyzed by DBU based task-specific ionic liquids without solvent. Arkivoc 2009, 11, 288–298. [Google Scholar] [CrossRef]
- Nyoni, D.; Lobb, K.A.; Kaye, P.T.; Cairab, M.R. DBU-Mediated cleavage of aryl- and heteroaryl disulfides. Arkivoc 2012, 6, 245–252. [Google Scholar] [CrossRef]
- Ying, A.G.; Liu, L.; Wu, G.F.; Chen, G.; Chen, X.Z.; Ye, W.D. Aza-Michael addition of aliphatic or aromatic amines to α,β-unsaturated compounds catalyzed by a DBU-derived ionic liquid under solvent-free conditions. Tetrahedron Lett. 2009, 50, 1653–1657. [Google Scholar] [CrossRef]
- Yeom, C.-E.; Kim, M.J.; Kim, B.M. 1,8-Diazabicyclo [5.4.0]undec-7-ene (DBU)-promoted efficient and versatile aza-Michael addition. Tetrahedron 2007, 63, 904–909. [Google Scholar] [CrossRef]
- Xu, J.; Cai, J.; Chen, J.; Zong, X.; Wu, X.; Ji, M.; Wang, P. An efficient synthesis of baricitinib. J. Chem. Res. 2016, 40, 205–208. [Google Scholar] [CrossRef]
- Colella, M.; Musci, P.; Cannillo, D.; Spennacchio, M.; Aramini, A.; Degennaro, L.; Luisi, R. Flow synthesis of 2-substituted azetines and 3-substituted azetidines by using a common synthetic precursor. J. Org. Chem. 2021, 86, 13943–13954. [Google Scholar] [CrossRef] [PubMed]
- Musci, P.; Colella, M.; Altomare, A.; Romanazzi, G.; Sheikh, N.S.; Degennaro, L.; Luisi, R. Dynamic phenomena and complexation effects in the α-lithiation and asymmetric functionalization of azetidines. Molecules 2022, 27, 2847. [Google Scholar] [CrossRef]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Rainer, M.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and tuning physicochemical properties in lead optimization: Amine basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef]
- Li, G.; Kakarla, R.; Gerritz, S.W. A fast and efficient bromination of isoxazoles and pyrazoles by microwave irradiation. Tetrahedron Lett. 2007, 48, 4595–4599. [Google Scholar] [CrossRef]
- Ríos, M.-C.; Portilla, J. Recent advances in synthesis and properties of pyrazoles. Chemistry 2022, 4, 940–968. [Google Scholar] [CrossRef]
- Mykhailiuk, P.K. Fluorinated pyrazoles: From synthesis to applications. Chem. Rev. 2021, 121, 1670–1715. [Google Scholar] [CrossRef]
- Lipunova, G.N.; Nosova, E.V.; Charushin, V.N.; Chupakhin, O.N. Fluorine-containing pyrazoles and their condensed derivatives: Synthesis and biological activity. J. Fluor. Chem. 2015, 175, 84–109. [Google Scholar] [CrossRef]
- Lee, L.F.; Schleppnik, F.M.; Schneider, R.W.; Campbell, D.H. Synthesis and 13C NMR of (trifluoromethyl)hydroxypyrazoles. J. Het. Chem. 1990, 27, 243–245. [Google Scholar] [CrossRef]
- Jiang, Z.-Y.; Huang, Z.-Y.; Yang, H.; Zhou, L.; Li, Q.-H.; Zhao, Z.-G. Cs2CO3 catalyzed direct aza-Michael addition of azoles to α,β-unsaturated malonates. RSC Adv. 2022, 12, 19265–19269. [Google Scholar] [CrossRef] [PubMed]
- Bulger, P.G.; Cottrell, I.F.; Cowden, C.J.; Davies, A.J.; Dolling, U.-H. An investigation into the alkylation of 1,2,4-triazole. Tetrahedron Lett. 2000, 41, 1297–1301. [Google Scholar] [CrossRef]
- Ma, B.; Wang, G.; Zhou, H.; Yang, J. Alkali salt-catalyzed aza-Michael addition of 1,2,4-triazole to α,β-unsaturated ketones and imides. Chin. J. Org. Chem. 2020, 40, 115–124. [Google Scholar] [CrossRef]
- Gerencsér, J.; Balázs, Á.; Dormán, G. Transition metal-catalyzed coupling reactions in library synthesis. In Synthesis and Modification of Heterocycles by Metal-Catalyzed Cross-coupling Reactions; Patonay, T., Kónya, K., Eds.; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Zhang, Y.; Han, J.; Liu, Z.-J. Palladium-catalyzed double-Suzuki–Miyaura reactions using cyclic dibenziodoniums: Synthesis of o-tetraaryls. J. Org. Chem. 2016, 81, 1317–1323. [Google Scholar] [CrossRef]
- Žukauskaitė, Ž.; Buinauskaitė, V.; Solovjova, J.; Malinauskaitė, L.; Kveselytė, A.; Bieliauskas, A.; Ragaitė, G.; Šačkus, A. Microwave-assisted synthesis of new fluorescent indoline-based building blocks by ligand free Suzuki-Miyaura cross-coupling reaction in aqueous media. Tetrahedron 2016, 72, 2955–2963. [Google Scholar] [CrossRef]
- Voiciuk, V.; Redeckas, K.; Martynaitis, V.; Steponavičiūtė, R.; Šačkus, A.; Vengris, A. Improving the photochromic properties of indolo [2,1-b][1,3]benzoxazines with phenylic substituents. J. Photochem. Photobiol. A Chem. 2014, 278, 60–68. [Google Scholar] [CrossRef]
- Malik, A.; Rasool, N.; Kanwal, I.; Hashmi, M.A.; Zahoor, A.F.; Ahmad, G.; Altaf, A.A.; Shah, S.A.A.; Sultan, S.; Zakaria, Z.A. Suzuki–Miyaura reactions of (4-bromophenyl)-4,6-dichloropyrimidine through commercially available palladium catalyst: Synthesis, optimization and their structural aspects identification through computational studies. Processes 2020, 8, 1342. [Google Scholar] [CrossRef]
- Talele, T.T. The “cyclopropyl fragment” is a versatile player that frequently appears in preclinical/clinical drug. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef]
- Wallace, D.J.; Chen, C.-y. Cyclopropylboronic acid: Synthesis and Suzuki cross-coupling reactions. Tetrahedron Lett. 2002, 39, 6987–6990. [Google Scholar] [CrossRef]
- Edwards, A.-M.; Ahmed, M.; Pulz, R.A.; Rooney, L.A.; Smith, N.; Troxler, T.J. Carboxamide Derivates. Patent US 9.403,833 B2, 28 February 2017. [Google Scholar]
- Gładkowski, W.; Siepka, M.; Janeczko, T.; Kostrzewa-Susłow, E.; Popłoński, J.; Mazur, M.; Żarowska, B.; Łaba, W.; Maciejewska, G.; Wawrzeńczyk, C. Synthesis and antimicrobial activity of methoxy-substituted γ-oxa-ε-lactones derived from flavanones. Molecules 2019, 24, 4151. [Google Scholar] [CrossRef] [PubMed]
- Eicher-Lorka, O.; Kuodis, Z.; Matijoška, A.; Rutavičius, A. Synthesis of 4-cyclo(propyl- and butyl)-1-ethylpyridinium bromides and calculation of their proton and carbon chemical shifts. Arkivoc 2010, 11, 114–132. [Google Scholar] [CrossRef]
- Gobbi, L.; Jaeschke, G.; Rodriguez, S.R.M.; Steward, L. D3 and 5-HT2A Receptor Modulators. U.S. Patent 2010/75983 A1, 25 March 2010. [Google Scholar]
- Arrebola-Liébanas, F.J.; Romero-González, R.; Garrido-Frenich, A. HRMS: Fundamentals and basic concepts. In Applications in High Resolution Mass Spectrometry: Food Safety and Pesticide Residue Analysis, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017; Chapter 1; pp. 1–14. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Base or Salt | Solvent | Temp. °C | t (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | DBU | MeCN | 65 | 16 | 46 |
| 2 | - | MeCN | 65 | 16 | - |
| 3 | LiF | MeCN | 65 | 16 | - |
| 4 | LiCl | MeCN | 65 | 16 | - |
| 5 | Cs2CO3 | MeCN | 65 | 16 | 56 |
| 6 | KOAc | MeCN | 65 | 16 | 58 |
| 7 | K3PO4 | MeCN | 65 | 16 | 61 |
| 8 | K2CO3 | MeCN | 65 | 16 | 65 |
| 9 | DBU | EtOH | Reflux | 24 | 39 |
| 10 | K2CO3 | EtOH | Reflux | 24 | 44 |
| 11 | K2CO3 | Dioxane | 65 | 16 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gudelis, E.; Krikštolaitytė, S.; Stančiauskaitė, M.; Šachlevičiūtė, U.; Bieliauskas, A.; Milišiūnaitė, V.; Jankauskas, R.; Kleizienė, N.; Sløk, F.A.; Šačkus, A. Synthesis of New Azetidine and Oxetane Amino Acid Derivatives through Aza-Michael Addition of NH-Heterocycles with Methyl 2-(Azetidin- or Oxetan-3-Ylidene)Acetates. Molecules 2023, 28, 1091. https://doi.org/10.3390/molecules28031091
Gudelis E, Krikštolaitytė S, Stančiauskaitė M, Šachlevičiūtė U, Bieliauskas A, Milišiūnaitė V, Jankauskas R, Kleizienė N, Sløk FA, Šačkus A. Synthesis of New Azetidine and Oxetane Amino Acid Derivatives through Aza-Michael Addition of NH-Heterocycles with Methyl 2-(Azetidin- or Oxetan-3-Ylidene)Acetates. Molecules. 2023; 28(3):1091. https://doi.org/10.3390/molecules28031091
Chicago/Turabian StyleGudelis, Emilis, Sonata Krikštolaitytė, Monika Stančiauskaitė, Urtė Šachlevičiūtė, Aurimas Bieliauskas, Vaida Milišiūnaitė, Rokas Jankauskas, Neringa Kleizienė, Frank A. Sløk, and Algirdas Šačkus. 2023. "Synthesis of New Azetidine and Oxetane Amino Acid Derivatives through Aza-Michael Addition of NH-Heterocycles with Methyl 2-(Azetidin- or Oxetan-3-Ylidene)Acetates" Molecules 28, no. 3: 1091. https://doi.org/10.3390/molecules28031091
APA StyleGudelis, E., Krikštolaitytė, S., Stančiauskaitė, M., Šachlevičiūtė, U., Bieliauskas, A., Milišiūnaitė, V., Jankauskas, R., Kleizienė, N., Sløk, F. A., & Šačkus, A. (2023). Synthesis of New Azetidine and Oxetane Amino Acid Derivatives through Aza-Michael Addition of NH-Heterocycles with Methyl 2-(Azetidin- or Oxetan-3-Ylidene)Acetates. Molecules, 28(3), 1091. https://doi.org/10.3390/molecules28031091