
Biological Activity of Hexaazaisowurtzitane Derivatives

 , ,
, ,
Abstract
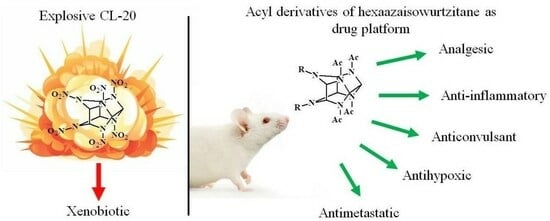
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Analgesic Activity of Hexaazaisowurtzitane Derivatives
3. Anti-Inflammatory Activity of Hexaazaisowurtzitane Derivatives
4. Anticonvulsant Activity of Hexaazaisowurtzitane Derivatives
5. Antihypoxic Activity of Hexaazaisowurtzitane Derivatives
6. Safety in Use of Hexaazaisowurtzitane Derivatives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simpson, R.L.; Urtiew, P.A.; Ornellas, D.L.; Moody, G.L.; Scribner, K.J.; Hoffman, D.M. CL-20 performance exceeds that of HMX and its sensitivity is moderate. Propellants Explos. Pyrotech. 1997, 22, 249–255. [Google Scholar] [CrossRef]
- Nair, U.R.; Sivabalan, R.; Gore, G.M.; Geetha, M.; Asthana, S.N.; Singh, H. Hexanitrohexaazaisowurtzitane (CL-20) and CL-20-based formulations (Review). Combust. Explos. Shock Waves 2005, 41, 121–132. [Google Scholar] [CrossRef]
- Maksimowski, P.; Fabijańska, A.; Adamiak, J. Tetraacetyl-dibenzyl-hexaazaisowurtzitane nitrosation—Studies on scale-up synthesis of HNIW. Propellants Explos. Pyrotech. 2010, 35, 353–358. [Google Scholar] [CrossRef]
- Monteil-Rivera, F.; Paquet, L.; Deschamps, S.; Balakrishnan, V.K.; Beaulieu, C.; Hawari, J. Physico-chemical measurements of CL-20 for environmental applications. J. Chromatogr. A 2004, 1025, 125–132. [Google Scholar] [CrossRef]
- Bhushan, B.; Halasz, A.; Hawari, J. Nitroreductase catalyzed biotransformation of CL-20. Biochem. Biophys. Res. Commun. 2004, 322, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, B.; Halasz, A.; Spain, J.C.; Hawari, J. Initial reaction(s) in biotransformation of CL-20 is catalyzed by salicylate 1-monooxygenase from Pseudomonas Sp. Strain ATCC 29352. Appl. Environ. Microbiol. 2004, 70, 4040–4047. [Google Scholar] [CrossRef] [PubMed]
- Haley, M.V.; Anthony, J.S.; Davis, E.A.; Kurnas, C.W.; Kuperman, R.G.; Checkai, R.T. Toxicity of the Cyclic Nitramine Energetic Material CL-20 to Aquatic Receptors; Defense Technical Information Center: Washington, DC, USA, 2007; 22p. Available online: https://apps.dtic.mil/sti/citations/ADA473749 (accessed on 30 September 2023).
- Robidoux, P.Y.; Sunahara, G.I.; Savard, K.; Berthelot, Y.; Dodard, S.; Martel, M.; Gong, P.; Hawari, J. Acute and chronic toxicity of the new explosive CL-20 to the earthworm (Eisenia andrei) exposed to amended natural soils. Environ. Toxicol. Chem. 2004, 23, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Inouye, L.S.; Perkins, E.J. Comparative neurotoxicity of two energetic compounds, hexanitrohexaazaisowurtzitane and hexahydro-1,3,5-trinitro-1,3,5-triazine, in the earthworm Eisenia fetida. Environ. Toxicol. Chem. 2007, 26, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Basu, N.; Scheuhammer, A.M.; Perkins, E.J. Neurochemical and electrophysiological diagnosis of reversible neurotoxicity in earthworms exposed to sublethal concentrations of CL-20. Environ. Sci. Pollut. Res. 2010, 17, 181–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gong, P.; Guan, X.; Pirooznia, M.; Liang, C.; Perkins, E.J. Gene expression analysis of CL-20-induced reversible neurotoxicity reveals GABAA receptors as potential targets in the earthworm Eisenia fetida. Environ. Sci. Technol. 2012, 46, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Gong, P.; Escalon, B.L.; Hayes, C.A.; Perkins, E.J. Uptake of Hexanitrohexaazaisowurtzitane (CL-20) by the earthworm Eisenia Fetida through dermal contact. Sci. Total Environ. 2008, 390, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.; Sunahara, G.I.; Spear, P.A.; Martel, M.; Gong, P.; Hawari, J. Effects of dietary administration of CL-20 on Japanese Quail Coturnix Coturnix japonica. Arch. Environ. Contam. Toxicol. 2005, 49, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Bardai, G.K.; Halasz, A.; Sunahara, G.I.; Dodard, S.; Spear, P.A.; Grosse, S.; Hoang, J.; Hawari, J. In vitro degradation of hexanitrohexaazaisowurtzitane (CL-20) by cytosolic enzymes of Japanese Quail and the rabbit. Environ. Toxicol. Chem. 2006, 25, 3221–3229. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dang, K.; Li, C.; Gao, J.; Wang, H.; Gao, Y.; Zhao, B.; Fan, P.; Qian, A. Isolation and identification of a novel bacterium, Pseudomonas sp. ZyL-01, involved in the biodegradation of CL-20. AMB Expr. 2020, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, H.; Liu, Z.; Lv, X.; Gao, W.; Gao, Y.; Gao, J.; Hu, L. Salidroside protect Chinese hamster V79 cells from genotoxicity and oxidative stress induced by CL-20. Toxicol. Res. 2023, 12, 133–142. [Google Scholar] [CrossRef]
- Bhushan, B.; Halasz, A.; Hawari, J. Biotransformation of CL-20 by a dehydrogenase enzyme from Clostridium sp. EDB2. Appl. Microbiol. Biotechnol. 2005, 69, 448–455. [Google Scholar] [CrossRef]
- Karakaya, P.; Christodoulatos, C.; Koutsospyros, A.; Balas, W.; Nicolich, S.; Sidhoum, M. Biodegradation of the High Explosive Hexanitrohexaazaisowurtzitane (CL-20). Int. J. Environ. Res. Public Health 2009, 6, 1371–1392. [Google Scholar] [CrossRef]
- Nielsen, A.T.; Nissan, R.A.; Vanderah, D.J.; Coon, C.L.; Gilardi, R.D.; George, C.F.; Flippen-Anderson, J. Poliazapolycyclics by Condensation of Aldehides with Amines. 2. Formation of 2,4,6,8,10,12-Hexabenzil-2,4,6,8,10,12-hexaazatertacyclo[5.5.0.05.9.03,11]dodecanes from Glyoxal and Benzilamines. J. Org. Chem. 1990, 55, 1459–1466. [Google Scholar] [CrossRef]
- Lin, G.; Tsai, H.-J.; Tsai, Y.-H. Cage amines as the stopper inhibitors of cholinesterases. Bioorg. Med. Chem. Lett. 2003, 13, 2887–2890. [Google Scholar] [CrossRef]
- Tolstikova, T.G.; Morozova, E.A.; Sysolyatin, S.V.; Kalashnikov, A.I.; Zhukova, Y.I.; Surmachev, V.N. Synthesis and biological activity of 2,4,6,8,10,12-hexaazatetracyclo[5.5.0.03,11.05,9]dodecane derivatives. Chem. Sustain. Dev. 2010, 18, 431–436. Available online: https://www.sibran.ru/upload/iblock/169/synthesis_and_biological_activity_of_the_derivatives_of_2_4_6_8_10_12_hexaazatetracyclo_5_5_0_0_sup_.pdf (accessed on 12 October 2023).
- Qiu, W.; Chen, S. Oxidation of N-benzyl groups. Chin. J. Chem. 2000, 18, 756–758. [Google Scholar] [CrossRef]
- Chung, K.; Kil, H.; Choi, I.; Chu, C.; Lee, I. New Precursors for hexanitrohexaazaisowurtzitane (HNIW, CL-20). J. Heterocycl. Chem. 2000, 37, 1647–1649. [Google Scholar] [CrossRef]
- Kubasova, V.A.; Surmachev, V.N. Acylation of 4,10-dibenzyl-2,6,8,12-tetraacetyl2,4,6,8,10,12-hexaazaisowurtzitane. Polzunovsky Vestn. 2016, 4, 89–91. Available online: https://elib.altstu.ru/journals/Files/archive/pv/2016/PV_4_1_2016.pdf (accessed on 15 August 2023). (In Russian).
- Golding, P.; Maccuish, A.J.; Bellamy, A.J. Protection of Amine and Hydroxyl Groups Using Fluoroacylation. Patent WO2004076384A2, 10 September 2003. [Google Scholar]
- Bellamy, A.J.; MacCuish, A.; Golding, P.; Mahon, M.F. The Use of trifluoroacetyl as an N- and O-protecting group during the synthesis of energetic compounds containing nitramine and/or nitrate ester groups. Propellants Explos. Pyrotech. 2007, 32, 20–31. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Lu, H.; Song, H. N-acylation of tetraacetylhexaazaisowurtzitane. J. Henan Norm. Univ. 2010, 38, 104–106. [Google Scholar]
- Dong, K.; Wang, Y.; Gong, X.-B.; Zhang, J.; Sun, C.-H.; Pang, S.-P. Formyl azido substituted nitro hexaazaisowurtzitane—Synthesis, characterization and energetic properties. New J. Chem. 2013, 37, 3685. [Google Scholar] [CrossRef]
- Kulagina, D.A.; Sysolyatin, S.V.; Malykhin, V.V.; Kalashnikov, A.I. Acylation of 2,6,8,12-tetraacetyl-2,4,6,8,10,12- hexaazatetracyclo[5.5.0.03,11.05,9]Dodecane. Mendeleev Commun. 2016, 26, 139–140. [Google Scholar] [CrossRef]
- Druzhilovskiy, D.S.; Rudik, A.V.; Filimonov, D.A.; Gloriozova, T.A.; Lagunin, A.A.; Dmitriev, A.V.; Pogodin, P.V.; Dubovskaya, V.I.; Ivanov, S.M.; Tarasova, O.A.; et al. Computational platform Way2Drug: From the prediction of biological activity to drug repurposing. Russ. Chem. Bull. 2017, 66, 1832–1841. [Google Scholar] [CrossRef]
- Filz, O.A.; Lagunin, A.A.; Filimonov, D.A.; Poroikov, V.V. In silico fragment-based drug design using a PASS approach. SAR QSAR Environ. Res. 2012, 23, 279–296. [Google Scholar] [CrossRef]
- David-Pereira, A.; Dickenson, A.H. Issues in the future development of new analgesic drugs. Curr. Opin. Support. Palliat. Care 2019, 13, 107–110. [Google Scholar] [CrossRef]
- Kissin, I. Progress in Analgesic Development: How to Assess its Real Merits? Curr. Rev. Clin. Exp. Pharmacol. 2022, 17, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Sandri, A. Diclofenac: Novità su tollerabilità ed azione antinfiammatoria spinale. Minerva Medica 2014, 105, 313–318. [Google Scholar] [PubMed]
- Available online: https://www.drugs.com/diclofenac.html (accessed on 30 November 2023).
- Mallinson, T.E. A review of ketorolac as a prehospital analgesic. J. Paramed. Pract. 2017, 9, 522–526. [Google Scholar] [CrossRef]
- Available online: https://www.medizzine.com/en/patients/drugs/K/ketorolac.php#d (accessed on 30 November 2023).
- Available online: https://www.drugs.com/cdi/tramadol-tablets.html (accessed on 30 November 2023).
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef] [PubMed]
- Gates, B.J.; Nguyen, T.T.; Setter, S.M.; Davies, N.M. Meloxicam: A reappraisal of pharmacokinetics, efficacy and safety. Expert. Opin. Pharmacother. 2005, 6, 2117–2140. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.drugs.com/cdi/meloxicam-tablets.html (accessed on 30 November 2023).
- Krylova, S.G.; Amosova, E.N.; Zueva, E.P.; Razina, T.G.; Rybalkina, O.Y.; Lopatina, K.A.; Sysolyatin, S.V.; Kalashnikov, A.I.; Malykhin, V.V.; Dygai, A.M.; et al. 4-(3,4-Dibromothiophenecarbonyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,3,11,5,9]dodecane as Analgesic Agent and Method for Production Thereof. RU Patent 2565766C1, 20 October 2015. Available online: https://patents.google.com/patent/RU2565766C1/en?oq=RU2565766 (accessed on 12 October 2023).
- Aguero, S.; Megy, S.; Eremina, V.V.; Kalashnikov, A.I.; Krylova, S.G.; Kulagina, D.A.; Lopatina, K.A.; Fournier, M.; Povetyeva, T.N.; Vorozhtsov, A.B.; et al. Discovery of a novel non-narcotic analgesic derived from the CL-20 explosive: Synthesis, pharmacology, and target identification of Thiowurtzine, a potent inhibitor of the opioid receptors and the ion channels. ACS Omega 2021, 6, 15400–15411. [Google Scholar] [CrossRef]
- Bryushinina, O.S.; Yanovskaya, E.A.; Abdrashitova, N.Y.; Zyuz’kova, Y.G.; Frelikh, G.A.; Lopatina, K.A.; Zueva, E.P.; Krylova, S.G.; Kulagina, D.A.; Sysolyatin, S.V.; et al. Pharmacokinetics of a new analgesic on the basis of hexaazaisowurtzitane derivative. Bull. Exp. Biol. Med. 2021, 170, 448–452. [Google Scholar] [CrossRef]
- Krylova, S.G.; Amosova, E.N.; Zueva, E.P.; Razina, T.G.; Rybalkina, O.Y.; Lopatina, K.A.; Sysolyatin, S.V.; Kalashnikov, A.I.; Malykhin, V.V.; Dygai, A.M.; et al. 4,10-Bis((±)-5-Benzoyl-2,3-Dihydro-1h-Pyrrolo[1,2-a]pirrol-1-Carbonyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,3,11,5,9]dodecane as an Analgesic and Method for Its Obtaining. RU Patent 2558148C1, 27 July 2015. Available online: https://patents.google.com/patent/RU2558148C1/en?oq=RU2558148C1 (accessed on 18 August 2023).
- Kulagina, D.A.; Sysolyatin, S.V.; Kalashnikov, A.I.; Malykhin, V.V.; Sonina, E.G.; Shevchenko, A.V.; Krylova, S.G.; Lopatina, K.A.; Zueva, E.P.; Razina, T.G.; et al. Synthesis and analgesic activity of 4,10-bis((±)-5-benzoyl-2,3-dihydro-1hpyrrolo[1,2-a]pyrrole-1-carbonyl)-2,6,8,12-tetraacetyl-2,4,6,8,10,12-hexaazatetracyclo[5,5,3,11,5,9]dodecane. Pharm. Chem. J. 2021, 54, 1140–1144. [Google Scholar] [CrossRef]
- Krylova, S.G.; Povetyeva, E.N.; Suslov, N.I.; Zueva, E.P.; Nesterova, Y.V.; Afanasyeva, O.G.; Kulpin, P.V.; Amosova, E.N.; Razina, T.G.; Rybalkina, O.Y.; et al. 4,10-Di(ethoxyacetyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,3,11,5,9] Dodecane as an Analgesic Agent and a Method for Production Thereof. RU Patent 2736936C1, 23 November 2020. Available online: https://patents.google.com/patent/RU2736936C1/en?oq=RU2736936C1 (accessed on 18 September 2023).
- Krylova, S.G.; Kiseleva, E.A.; Povet’eva, T.N.; Nesterova, Y.V.; Rybalkina, O.Y.; Kul’pin, P.V.; Afanas’eva, O.G.; Kulagina, D.A.; Eremina, V.V.; Baibakova, O.V.; et al. Antinociceptive effect of Ethowurtzine, a new compound from the class of hexaazaizowurzitane. Bull. Exp. Biol. Med. 2022, 174, 230–235. [Google Scholar] [CrossRef]
- Krylova, S.G.; Povetyeva, E.N.; Suslov, N.I.; Zueva, E.P.; Nesterova, Y.V.; Zhdanov, V.V. 4-(3,4-Dibromothiophenylcarbonyl)-2,6,8,10,12-Pentaacetyl-2,4,6,8,10,12-Hexaazaisowurtzitane as an Analgesic Agent and Method for Its Preparation. RU Patent 2769523C1, 1 April 2022. Available online: https://patents.google.com/patent/RU2769523C1/en?oq=RU2769523C1 (accessed on 18 September 2023).
- Eremina, V.V.; Kulagina, D.A.; Krylova, S.G.; Kiseleva, E.A.; Povetyeva, T.N.; Zueva, E.P.; Suslov, N.I.; Amosova, E.N.; Razina, T.G.; Rybalkina, O.Y.; et al. Synthesis and biological activity of 4-(3,4-dibromothiophenecarbonyl)-2,6,8,10,12-pentaacetyl-2,4,6,8,10,12-hexaazaisowurtzitane: A new non-narcotic analgesic of the hexaazaisowurtzitane class. Pharm. Chem. J. 2023, 57, 40–45. [Google Scholar] [CrossRef]
- Krylova, S.G.; Povetyeva, E.N.; Suslov, N.I.; Zueva, E.P.; Amosova, E.N.; Zhdanov, V.V.; Baibakova, O.V.; Eremina, V.V.; Kulagina, D.A.; Sysolyatin, S.V. 4,10-Di(Ethoxyacetyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo [5,5,0,03.11,05.9]dodecane as an Anti-Inflammatory Agent. RU Patent 2798994C1, 30 June 2023. Available online: https://patents.google.com/patent/RU2798994C1/en?oq=RU2798994C1 (accessed on 18 September 2023).
- Available online: https://www.drugs.com/monograph/carbamazepine.html (accessed on 1 December 2023).
- Powell, G.; Saunders, M.; Rigby, A.; Marson, A.G. Immediate-release versus controlled-release carbamazepine in the treatment of epilepsy. Cochrane Database Syst. Rev. 2016, 12, CD007124. [Google Scholar] [CrossRef] [PubMed]
- Krylova, S.G.; Povetyeva, E.N.; Sysolyatin, S.V.; Suslov, N.I.; Zueva, E.P.; Nesterova, Y.V.; Afanasyeva, O.G.; Lopatina, K.A.; Kulpin, P.V.; Zhdanov, V.V.; et al. 4-(3,4-Dibromothiophenecarbonyl)-2,6,8,12-Tetraacetyl-2,4,6,8,10,12-Hexaazatetracyclo[5,5,0,3,11,5,9]dodecane Used as an Anticonvulsant. RU Patent 2684107C1, 4 April 2019. Available online: https://patents.google.com/patent/RU2684107C1/en?oq=RU2684107C1 (accessed on 18 September 2023).
- Winblad, B. Piracetam: A review of pharmacological properties and clinical uses. CNS Drug Rev. 2005, 11, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.drugs.com/uk/nootropil-800mg-tablets-leaflet.html (accessed on 1 December 2023).
- Kulagina, D.A. Development of Methods for Synthesis of Acyl Derivatives of Hexaazaisowurtzitane. Ph.D. Thesis, Tomsk Polytechnic University, Tomsk, Russia, 2018. Available online: https://www.dissercat.com/content/razrabotka-metodov-sinteza-atsilnykh-proizvodnykh-geksaazaizovyurtsitana (accessed on 18 September 2023). (In Russian).
- Caraceni, A.; Shkodra, M. Cancer pain assessment and classification. Cancers 2019, 11, 510. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, S. The need of a comprehensive approach in a condition of poorly opioid-responsive cancer pain. J. Pain Symptom Manag. 2023, 66, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Glavas Tahtler, J.; Djapic, D.; Neferanovic, M.; Miletic, J.; Milosevic, M.; Kralik, K.; Neskovic, N.; Tomas, I.; Mesaric, D.; Marjanovic, K.; et al. Long-term outcomes of breast cancer patients receiving levobupivacaine wound infiltration or diclofenac for postoperative pain relief. Pharmaceutics 2023, 15, 2183. [Google Scholar] [CrossRef]
- Burnod, A.; Maindet, C.; George, B.; Minello, C.; Allano, G.; Lemaire, A. A clinical approach to the management of cancer-related pain in emergency situations. Support. Care Cancer 2019, 27, 3147–3157. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.; Brodsky, R. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Available online: https://www.drugs.com/international/cyclophosphamide.html (accessed on 1 December 2023).
- Lopatina, K.A.; Krylova, S.G.; Safonova, E.A.; Zueva, E.P.; Kulagina, D.A.; Churin, A.A.; Fomina, T.I.; Sysolyatin, S.V. A New analgesic agent based on hexaazaisowurzitan: Feasibility of using in managing patients with cancer. Sib. J. Oncol. 2020, 19, 76–81. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulagina, D.A.; Sysolyatin, S.V.; Krylova, S.G.; Kiseleva, E.A.; Povetyeva, T.N.; Zueva, E.P.; Eremina, V.V.; Alekseeva, N.A.; Strokova, S.V.; Suslov, N.I.; et al. Biological Activity of Hexaazaisowurtzitane Derivatives. Molecules 2023, 28, 8084. https://doi.org/10.3390/molecules28248084
Kulagina DA, Sysolyatin SV, Krylova SG, Kiseleva EA, Povetyeva TN, Zueva EP, Eremina VV, Alekseeva NA, Strokova SV, Suslov NI, et al. Biological Activity of Hexaazaisowurtzitane Derivatives. Molecules. 2023; 28(24):8084. https://doi.org/10.3390/molecules28248084
Chicago/Turabian StyleKulagina, Daria A., Sergey V. Sysolyatin, Svetlana G. Krylova, Elena A. Kiseleva, Tatiana N. Povetyeva, Elena P. Zueva, Valeria V. Eremina, Natalia A. Alekseeva, Svetlana V. Strokova, Nikolai I. Suslov, and et al. 2023. "Biological Activity of Hexaazaisowurtzitane Derivatives" Molecules 28, no. 24: 8084. https://doi.org/10.3390/molecules28248084
APA StyleKulagina, D. A., Sysolyatin, S. V., Krylova, S. G., Kiseleva, E. A., Povetyeva, T. N., Zueva, E. P., Eremina, V. V., Alekseeva, N. A., Strokova, S. V., Suslov, N. I., & Zhdanov, V. V. (2023). Biological Activity of Hexaazaisowurtzitane Derivatives. Molecules, 28(24), 8084. https://doi.org/10.3390/molecules28248084