
Investigation of Drug-Interaction Potential for Arthritis Dietary Supplements: Chondroitin Sulfate, Glucosamine, and Methylsulfonylmethane

 , , , ,
, , , ,  and
and 
Abstract
:1. Introduction
2. Results
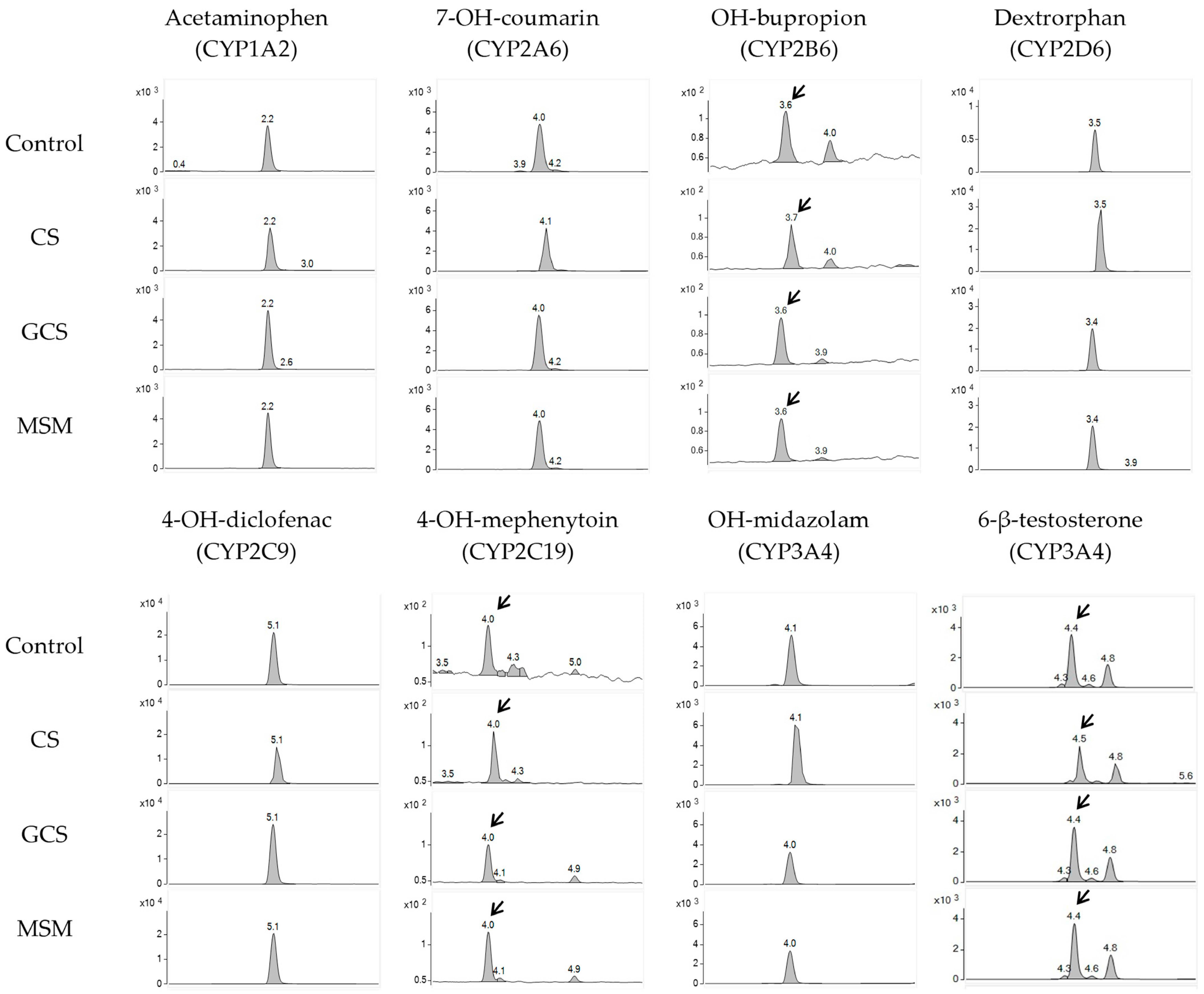
2.1. CYP Inhibition Assay
2.2. Method Validation
2.2.1. Linearity
2.2.2. Selectivity
2.2.3. Accuracy and Precision
3. Discussion
4. Materials and Methods
4.1. Chemicals and Materials
4.2. Microsomal Incubation
4.3. LC-MS/MS Analysis
4.4. Method Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Persiani, S.; Canciani, L.; Larger, P.; Rotini, R.; Trisolino, G.; Antonioli, D.; Rovati, L.C. In vitro study of the inhibition and induction of human cytochromes P450 by crystalline glucosamine sulfate. Drug Metab. Drug Interact. 2009, 24, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Machado, G.C.; Eyles, J.P.; Ravi, V.; Hunter, D.J. Dietary supplements for treating osteoarthritis: A systematic review and meta-analysis. Br. J. Sports Med. 2018, 52, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Neogi, T.; Zhang, Y.Q. Epidemiology of Osteoarthritis. Clin. Geriatr. Med. 2013, 39, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Primorac, D.; Molnar, V.; Matisic, V.; Hudetz, D.; Jelec, Z.; Rod, E.; Cukelj, F.; Vidovic, D.; Vrdoljak, T.; Dobricic, B.; et al. Comprehensive Review of Knee Osteoarthritis Pharmacological Treatment and the Latest Professional Societies’ Guidelines. Pharmaceuticals 2021, 14, 205. [Google Scholar] [CrossRef] [PubMed]
- da Costa, B.R.; Reichenbach, S.; Keller, N.; Nartey, L.; Wandel, S.; Jüni, P.; Trelle, S. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: A network meta-analysis. Lancet 2016, 387, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Machado, G.C.; Maher, C.G.; Ferreira, P.H.; Pinheiro, M.B.; Lin, C.W.C.; Day, R.O.; McLachlan, A.J.; Ferreira, M.L. Efficacy and safety of paracetamol for spinal pain and osteoarthritis: Systematic review and meta-analysis of randomised placebo controlled trials. BMJ-Brit. Med. J. 2015, 350, h1225. [Google Scholar] [CrossRef] [PubMed]
- Hathcock, J.N.; Shao, A. Risk assessment for glucosamine and chondroitin sulfate. Regul. Toxicol. Pharmacol. 2007, 47, 78–83. [Google Scholar] [CrossRef]
- Simental-Mendía, M.; Sánchez-García, A.; Vilchez-Cavazos, F.; Acosta-Olivo, C.A.; Peña-Martínez, V.M.; Simental-Mendía, L.E. Effect of glucosamine and chondroitin sulfate in symptomatic knee osteoarthritis: A systematic review and meta-analysis of randomized placebo-controlled trials. Rheumatol. Int. 2018, 38, 1413–1428. [Google Scholar] [CrossRef]
- Usha, P.R.; Naidu, M.U.R. Randomised, double-blind, parallel, placebo-controlled study of oral glucosamine, methylsulfonylmethane and their combination in osteoarthritis. Clin. Drug Investig. 2004, 24, 353–363. [Google Scholar] [CrossRef]
- Sun, P.; Cao, Y.; Qiu, J.; Kong, J.; Zhang, S.; Cao, X. Inhibitory Mechanisms of Lekethromycin in Dog Liver Cytochrome P450 Enzymes Based on UPLC-MS/MS Cocktail Method. Molecules 2023, 28, 7193. [Google Scholar] [CrossRef]
- Bloomer, R.J.; Butawan, M.; Lin, L.; Ma, D.; Yates, C.R. Blood MSM concentrations following escalating dosages of oral MSM in men and women. J. Nutr. Food Sci. 2019, 9, 1000748. [Google Scholar] [CrossRef]
- Persiani, S.; Roda, E.; Rovati, L.C.; Locatelli, M.; Giacovelli, G.; Roda, A. Glucosamine oral bioavailability and plasma pharmacokinetics after increasing doses of crystalline glucosamine sulfate in man. Osteoarthr. Cartil. 2005, 13, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.G.; Plaas, A.H.; Sandy, J.D.; Hua, C.; Kim-Rolands, S.; Barnhill, J.G.; Harris, C.L.; Clegg, D.O. The human pharmacokinetics of oral ingestion of glucosamine and chondroitin sulfate taken separately or in combination. Osteoarthr. Cartil. 2010, 18, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Ahemad, N.; Pan, Y.; Palanisamy, U.D.; Othman, I.; Ong, C.E. In vitro inhibitory effects of glucosamine, chondroitin and diacerein on human hepatic CYP2D6. Drug Metab. Pers. Ther. 2021, 36, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Ahemad, N.; Pan, Y.; Palanisamy, U.D.; Othman, I.; Yiap, B.C.; Ong, C.E. Cytochrome P450 2C9-natural antiarthritic interactions: Evaluation of inhibition magnitude and prediction from in vitro data. Biopharm. Drug Dispos. 2018, 39, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Kutanzi, K.R.; Ewing, L.E.; Skinner, C.M.; Quick, C.M.; Kennon-McGill, S.; McGill, M.R.; Walker, L.A.; ElSohly, M.A.; Gurley, B.J.; Koturbash, I. Safety and Molecular-Toxicological Implications of Cannabidiol-Rich Cannabis Extract and Methylsulfonylmethane Co-Administration. Int. J. Mol. Sci. 2020, 21, 7808. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Sangkuhl, K.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 2015, 25, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.O.; Attwood, J.; Birkett, D.J. Determinants of Acetaminophen Metabolism—Effect of Inducers and Inhibitors of Drug-Metabolism on Acetaminophens Metabolic Pathways. Clin. Pharmacol. Ther. 1984, 35, 480–486. [Google Scholar] [CrossRef]
- Frost, D.A.; Soric, M.M.; Kaiser, R.; Neugebauer, R.E. Efficacy of Tramadol for Pain Management in Patients Receiving Strong Cytochrome P450 2D6 Inhibitors. Pharmacotherapy 2019, 39, 724–729. [Google Scholar] [CrossRef]
- Saarikoski, T.; Saari, T.I.; Hagelberg, N.M.; Backman, J.T.; Neuvonen, P.J.; Scheinin, M.; Olkkola, K.T.; Laine, K. Effects of terbinafine and itraconazole on the pharmacokinetics of orally administered tramadol. Eur. J. Clin. Pharmacol. 2015, 71, 321–327. [Google Scholar] [CrossRef]
- Saarikoski, T.; Saari, T.I.; Hagelberg, N.M.; Neuvonen, M.; Neuvonen, P.J.; Scheinin, M.; Olkkola, K.T.; Laine, K. Rifampicin markedly decreases the exposure to oral and intravenous tramadol. Eur. J. Clin. Pharmacol. 2013, 69, 1293–1301. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Woodall, K.L.; Solbeck, P.; Ross, C.J.; Carleton, B.C.; Hayden, M.R.; Koren, G.; Madadi, P. Codeine-related deaths: The role of pharmacogenetics and drug interactions. Forensic Sci. Int. 2014, 239, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.J.; Svensson, C.K.; Visco, J.P.; Lalka, D. Clinical Pharmacokinetics of Pethidine: 1982. Clin. Pharmacokinet. 1982, 7, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Stamer, U.M.; Tzvetkov, M.V.; Altman, R.B.; Klein, T.E. PharmGKB summary: Tramadol pathway. Pharmacogenet. Genom. 2014, 24, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. Codeine and morphine pathway. Pharmacogenet. Genom. 2009, 19, 556–558. [Google Scholar] [CrossRef] [PubMed]
- Moy, K.V.; Ma, J.D.; Best, B.M.; Atayee, R.S. Factors Impacting Variability of the Urinary Normeperidine-to-Meperidine Metabolic Ratio in Patients with Chronic Pain. J. Anal. Toxicol. 2014, 38, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mazaleuskaya, L.L.; Theken, K.N.; Gong, L.; Thorn, C.F.; FitzGerald, G.A.; Altman, R.B.; Klein, T.E. PharmGKB summary: Ibuprofen pathways. Pharmacogenetics Genom. 2015, 25, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Hynninen, V.V.; Olkkola, K.T.; Leino, K.; Lundgren, S.; Neuvonen, P.J.; Rane, A.; Valtonen, M.; Vyyryläinen, H.; Laine, K. Effects of the antifungals voriconazole and fluconazole on the pharmacokinetics of S-(+)- and R-(−)-ibuprofen. Antimicrob. Agents Chemother. 2006, 50, 1967–1972. [Google Scholar] [CrossRef]
- Tornio, A.; Niemi, M.; Neuvonen, P.J.; Backman, J.T. Stereoselective interaction between the CYP2C8 inhibitor gemfibrozil and racemic ibuprofen. Eur. J. Clin. Pharmacol. 2007, 63, 463–469. [Google Scholar] [CrossRef]
- Moore, N.; Pollack, C.; Butkerait, P. Adverse drug reactions and drug-drug interactions with over-the-counter NSAIDs. Ther. Clin. Risk Manag. 2015, 11, 1061–1075. [Google Scholar]
- Boerma, J.S.; Vermeulen, N.P.E.; Commandeur, J.N.M. One-electron oxidation of diclofenac by human cytochrome P450s as a potential bioactivation mechanism for formation of 2′-(glutathion-S-yl)-deschloro-diclofenac. Chem.-Biol. Interact. 2014, 207, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chesné, C.; Guyomard, C.; Guillouzo, A.; Schmid, J.; Ludwig, E.; Sauter, T. Metabolism of Meloxicam in human liver involves cytochromes P4502C9 and 3A4. Xenobiotica 1998, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, Y.H.; Kim, S.H.; Byeon, J.Y.; Lee, Y.; Lee, Y.J. Drug-Drug Interaction between Meloxicam and Amiodarone. Clin. Ther. 2015, 37, E60–E61. [Google Scholar] [CrossRef]
- Hynninen, V.V.; Olkkola, K.T.; Bertilsson, L.; Kurkinen, K.J.; Korhonen, T.; Neuvonen, P.J.; Laine, K. Voriconazole Increases while Itraconazole Decreases Plasma Meloxicam Concentrations. Antimicrob. Agents Chemother. 2009, 53, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Thorn, C.F.; Bertagnolli, M.M.; Grosser, T.; Altman, R.B.; Klein, T.E. Celecoxib pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genom. 2012, 22, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Garnett, W.R. Clinical implications of drug interactions with coxibs. Pharmacotherapy 2001, 21, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Vo, N.X.; Le, N.N.H.; Chu, T.D.P.; Pham, H.L.; Dinh, K.X.A.; Che, U.T.T.; Ngo, T.T.T.; Bui, T.T. Effectiveness and Safety of Glucosamine in Osteoarthritis: A Systematic Review. Pharmacy 2023, 11, 117. [Google Scholar] [CrossRef] [PubMed]
- Crawford, P.; Crawford, A.; Nielson, F.; Lystrup, R. Methylsulfonylmethane for treatment of low back pain: A safety analysis of a randomized, controlled trial. Complement. Ther. Med. 2019, 45, 85–88. [Google Scholar] [CrossRef]
- Knudsen, J.E.; Sokol, G.H. Potential glucosamine-warfarin interaction resulting in increased international normalized ratio: Case report and review of the literature and MedWatch database. Pharmacotherapy 2008, 28, 540–548. [Google Scholar] [CrossRef]
- Rozenfeld, V.; Crain, J.L.; Callahan, A.K. Possible augmentation of warfarin effect by glucosamine-chondroitin. Am. J. Health-Syst. Pharm. 2004, 61, 306–307. [Google Scholar] [CrossRef]
- Qinna, N.A.; Shubbar, M.H.; Matalka, K.Z.; Al-Jbour, N.; Ghattas, M.A.; Badwan, A.A. Glucosamine Enhances Paracetamol Bioavailability by Reducing Its Metabolism. J. Pharm. Sci. 2015, 104, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.F.; Liu, J.P.; Chowbay, B. Polymorphism of human cytochrome P450 enzymes and its clinical impact. Drug Metab. Rev. 2009, 41, 89–295. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Worldwide Distribution of Cytochrome P450 Alleles: A Meta-analysis of Population-scale Sequencing Projects. Clin. Pharmacol. Ther. 2017, 102, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Choi, M.S.; Park, J.S.; Rehman, S.U.; Nakamura, K.; Yoo, H.H. Inhibitory Effects of Garcinia cambogia Extract on CYP2B6 Enzyme Activity. Planta Med. 2017, 83, 895–900. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Ji, Y.S.; Jo, S.Y.; Piao, X.-L.; Yoo, H.H. Evaluation of the inhibitory effect of Gynostemma pentaphyllum extracts on CYP450 enzyme activities using LC-MS/MS. Mass Spectrom. Lett. 2023, 14, 116–119. [Google Scholar]
- Bu, H.Z.; Knuth, K.; Magis, L.; Teitelbaum, P. High-throughput cytochrome P450 inhibition screening via cassette probe-dosing strategy. IV. Validation of a direct injection on-line guard cartridge extraction/tandem mass spectrometry method for simultaneous CYP3A4, 2D6 and 2E1 inhibition assessment. Rapid Commun. Mass Spectrom. 2000, 14, 1943–1948. [Google Scholar] [CrossRef]

{kind=link}
| CYP Isozyme | Remaining Activities (% of Control, n = 3) | |||||||
|---|---|---|---|---|---|---|---|---|
| CS (μM) | ||||||||
| 0.1 | 0.3 | 1 | 3 | 10 | 30 | 100 | 200 | |
| CYP1A2 | 99.5 ± 4.0 | 96.9 ± 10.5 | 95.1 ± 13.3 | 106.8 ± 4.1 | 109.6 ± 3.7 | 108.6 ± 2.3 | 113.4 ± 16.5 | 100.3 ± 4.9 |
| CYP2A6 | 101.2 ± 0.9 | 99.8 ± 7.1 | 99.5 ± 10.7 | 109.6 ± 1.8 | 111.1 ± 0.7 | 116.6 ± 4.6 | 124.9 ± 9.4 | 116.4 ± 1.4 |
| CYP2B6 | 94.9 ± 3.9 | 99.0 ± 3.3 | 89.7 ± 14.4 | 99.1 ± 1.9 | 99.0 ± 6.2 | 100.6 ± 8.6 | 100.1 ± 8.0 | 106.4 ± 9.3 |
| CYP2C9 | 105.3 ± 2.7 | 105.7 ± 2.5 | 102.4 ± 11.2 | 111.1 ± 1.0 | 115.6 ± 4.7 | 117.3 ± 4.2 | 121.3 ± 9.2 | 117.3 ± 2.4 |
| CYP2C19 | 88.4 ± 6.2 | 90.6 ± 7.0 | 86.7 ± 9.6 | 94.7 ± 8.3 | 98.2 ± 10.2 | 106.0 ± 5.9 | 112.7 ± 12.6 | 119.9 ± 17.0 |
| CYP2D6 | 100.5 ± 1.4 | 102.2 ± 5.8 | 99.0 ± 9.6 | 100.8 ± 1.1 | 105.7 ± 4.4 | 103.0 ± 3.7 | 104.4 ± 8.6 | 103.5 ± 6.4 |
| CYP3A4(M) * | 97.6 ± 3.8 | 99.1 ± 3.1 | 91.2 ± 10.6 | 87.9 ± 27.9 | 101.4 ± 4.9 | 106.6 ± 5 | 113.6 ± 8.4 | 114.6 ± 2.8 |
| CYP3A4(T) * | 101.7 ± 1.4 | 96.9 ± 7.0 | 97.8 ± 6.1 | 100.9 ± 0.6 | 107.5 ± 3.2 | 102.9 ± 0.7 | 109.3 ± 7.7 | 98.8 ± 2.6 |
| CYP Isozyme | Remaining Activities (% of Control, n = 3) | |||||||
|---|---|---|---|---|---|---|---|---|
| MSM (μM) | ||||||||
| 0.3 | 1 | 3 | 10 | 30 | 100 | 300 | 1000 | |
| CYP1A2 | 95.5 ± 12.9 | 103.7 ± 3.5 | 98.7 ± 5.1 | 103.0 ± 5.8 | 99.8 ± 6.6 | 101.8 ± 9.0 | 104.2 ± 7.5 | 101.2 ± 7.3 |
| CYP2A6 | 98.7 ± 7.7 | 103.1 ± 3.3 | 101.1 ± 4.3 | 103.5 ± 5.1 | 102.1 ± 3.3 | 102.5 ± 5.0 | 103.5 ± 4.1 | 100.8 ± 3.6 |
| CYP2B6 | 98.9 ± 8.0 | 104.9 ± 4.2 | 103.3 ± 6.0 | 104.3 ± 4.3 | 106.0 ± 4.1 | 105.00 ± 1.3 | 108.5 ± 7.5 | 106.9 ± 6.0 |
| CYP2C9 | 106.7 ± 10.0 | 111.4 ± 3.4 | 108.1 ± 3.5 | 111.6 ± 6.8 | 110.5 ± 4.3 | 109.8 ± 4.3 | 110.8 ± 4.3 | 109.3 ± 6.1 |
| CYP2C19 | 101.1 ± 11.0 | 108.2 ± 0.9 | 101.5 ± 9.2 | 105.7 ± 6.4 | 102.1 ± 7.1 | 102.7 ± 8.1 | 106.3 ± 9.5 | 102.6 ± 11.6 |
| CYP2D6 | 96.8 ± 6.3 | 100.9 ± 1.6 | 98.0 ± 3.4 | 100.4 ± 2.5 | 98.5 ± 3.6 | 98.9 ± 3.9 | 100.8 ± 3.9 | 101.1 ± 3.7 |
| CYP3A4(M) * | 93.2 ± 13.2 | 97.8 ± 4.3 | 96.2 ± 4.3 | 96.8 ± 4.2 | 96.9 ± 7.2 | 96.7 ± 6.2 | 97.9 ± 4.4 | 99.7 ± 4.1 |
| CYP3A4(T) * | 95.3 ± 2.9 | 98.5 ± 4.2 | 95.1 ± 3.5 | 98.0 ± 2.4 | 95.3 ± 1.7 | 90.8 ± 3.4 | 95.9 ± 4.2 | 95.2 ± 0.5 |
| CYP Isozyme | Remaining Activities (% of Control, n = 3) | |||||||
|---|---|---|---|---|---|---|---|---|
| GCS (μM) | ||||||||
| 0.3 | 1 | 3 | 10 | 30 | 100 | 300 | 1000 | |
| CYP1A2 | 110.2 ± 20.5 | 103.1 ± 1.5 | 101.1 ± 7.5 | 94.2 ± 1.8 | 94.1 ± 3.0 | 97.2 ± 3.7 | 86.7 ± 2.8 | 82.5 ± 12.0 |
| CYP2A6 | 107.6 ± 5.2 | 105.3 ± 0.4 | 106.2 ± 2.3 | 101.5 ± 1.4 | 102.1 ± 2.5 | 103.4 ± 2.7 | 97.9 ± 0.4 | 94.1 ± 7.3 |
| CYP2B6 | 121.0 ± 11.6 | 116.2 ± 5.8 | 114.3 ± 3.5 | 108.2 ± 3.1 | 107.8 ± 1.3 | 111.2 ± 2.3 | 105.6 ± 1.8 | 103.5 ± 9.2 |
| CYP2C9 | 115.5 ± 15.2 | 108.7 ± 1.7 | 109.2 ± 2.4 | 104.0 ± 2.3 | 105.8 ± 2.7 | 101.2 ± 4.8 | 97.2 ± 2.8 | 91.1 ± 10.1 |
| CYP2C19 | 128.4 ± 27.9 | 102.5 ± 3.0 | 102.6 ± 8.8 | 101.9 ± 4.3 | 90.5 ± 8.7 | 100.3 ± 3.7 | 95.5 ± 5.6 | 88.7 ± 7.3 |
| CYP2D6 | 107.6 ± 7.4 | 104.9 ± 1.0 | 106.2 ± 3.7 | 101.6 ± 0.5 | 100.4 ± 1.4 | 102.6 ± 1.7 | 100.6 ± 1.3 | 99.5 ± 4.8 |
| CYP3A4(M) * | 104.4 ± 13.5 | 103.5 ± 3.2 | 98.3 ± 1.9 | 97.2 ± 1.5 | 96.9 ± 6.1 | 95.2 ± 8.2 | 91.0 ± 3.3 | 88.7 ± 12.1 |
| CYP3A4(T) * | 92.8 ± 0.6 | 92.4 ± 0.9 | 91.1 ± 3.1 | 91.7 ± 4.4 | 91.8 ± 3.0 | 84.8 ± 7.8 | 82.1 ± 3.4 | 80.0 ± 6.6 |
| Drug Type | Drug | Metabolism-Associated CYPs | CYP-Mediated Drug Interactions | Refs |
|---|---|---|---|---|
| Analgesics | Acetaminophen | CYP3A4 (major), CYP2El, CYP1A2, CYP2D6 | Isoniazid (CYP2E1 inducer) Carbamazepine (CYP3A4 inducer) Rifampicin (CYPs inducer) | [17,18] |
| Tramadol | CYP2D6 (major), CYP3A4, CYP2B6 | Terbinafine (CYP2D6 inhibitor) Escitalopram (CYP2D6 inhibitor) Rifampicin (CYPs inducer) | [19,20,21,24] | |
| Codeine | CYP3A4, CYP2D6 (major), CYP2C8 | Bupropion (CYP2D6 inhibitor) Paroxetine (CYP2D6 inhibitor) Fluoxetine (CYP2D6 inhibitor) | [22,25] | |
| Pethidine | CYP2B6 (major), CYP3A4 (major), CYP2C19 | Phenobarbitone (CYP inducer) Chlorpromazine (CYP inhibitor) | [23,26] | |
| NSAIDs | Ibuprofen | CYP2C8 (major) CYP2C9 (major), CYP2C19, CYP3A4 | Voriconazole (CYP2C9 inhibitor) Fluconazole (CYP2C9 inhibitor) Gemfibrozil (CYP2C8 inhibitor) | [27,28,29] |
| Diclofenac | CYP2C8, CYP2C9 (major), CYP2C18, CYP2C19, CYP2B6, CYP3A4 | Voriconazole (CYP2C9 inhibitor) | [30,31] | |
| Meloxicam | CYP2C9 (major), CYP3A4 | Amiodarone (CYP2C9 inhibitor) Voriconazole (CYP2C9 inhibitor) Itraconazole (CYP3A4 inhibitor) | [32,33,34] | |
| Celecoxib | CYP2C9 (major), CYP3A4 | Fluconazole (CYP2C9 inhibitor) Fluvastatin (CYP2C9 inhibitor) Rifampicin (CYP2C9 inducer) | [35,36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.M.; Jo, S.Y.; Park, H.-Y.; Lee, Y.R.; Yu, J.S.; Yoo, H.H. Investigation of Drug-Interaction Potential for Arthritis Dietary Supplements: Chondroitin Sulfate, Glucosamine, and Methylsulfonylmethane. Molecules 2023, 28, 8068. https://doi.org/10.3390/molecules28248068
Kim SM, Jo SY, Park H-Y, Lee YR, Yu JS, Yoo HH. Investigation of Drug-Interaction Potential for Arthritis Dietary Supplements: Chondroitin Sulfate, Glucosamine, and Methylsulfonylmethane. Molecules. 2023; 28(24):8068. https://doi.org/10.3390/molecules28248068
Chicago/Turabian StyleKim, Su Min, So Young Jo, Ho-Young Park, Yu Ra Lee, Jun Sang Yu, and Hye Hyun Yoo. 2023. "Investigation of Drug-Interaction Potential for Arthritis Dietary Supplements: Chondroitin Sulfate, Glucosamine, and Methylsulfonylmethane" Molecules 28, no. 24: 8068. https://doi.org/10.3390/molecules28248068
APA StyleKim, S. M., Jo, S. Y., Park, H.-Y., Lee, Y. R., Yu, J. S., & Yoo, H. H. (2023). Investigation of Drug-Interaction Potential for Arthritis Dietary Supplements: Chondroitin Sulfate, Glucosamine, and Methylsulfonylmethane. Molecules, 28(24), 8068. https://doi.org/10.3390/molecules28248068