

Recent Progress in CDK4/6 Inhibitors and PROTACs
 and
and
Abstract
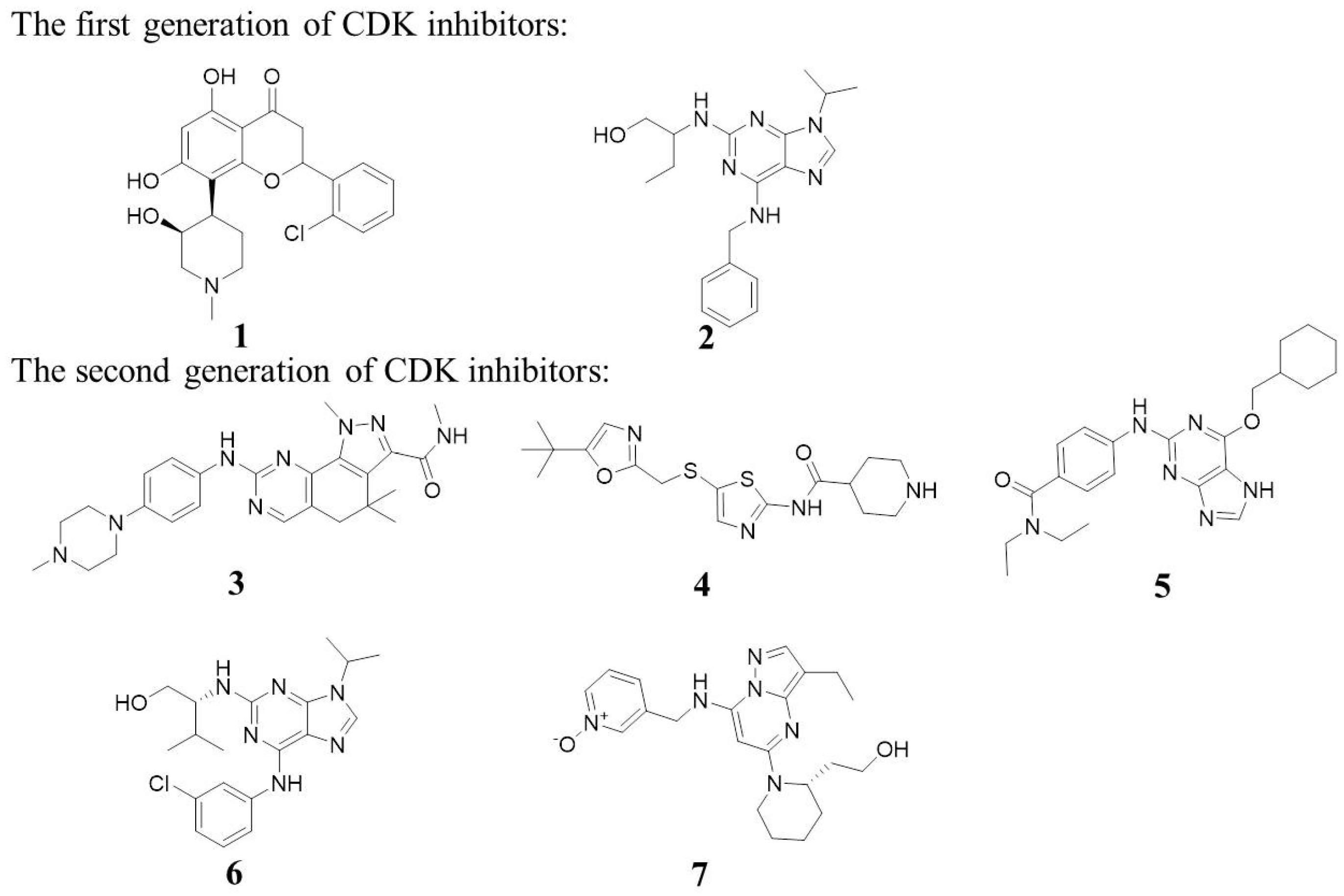
:1. Introduction
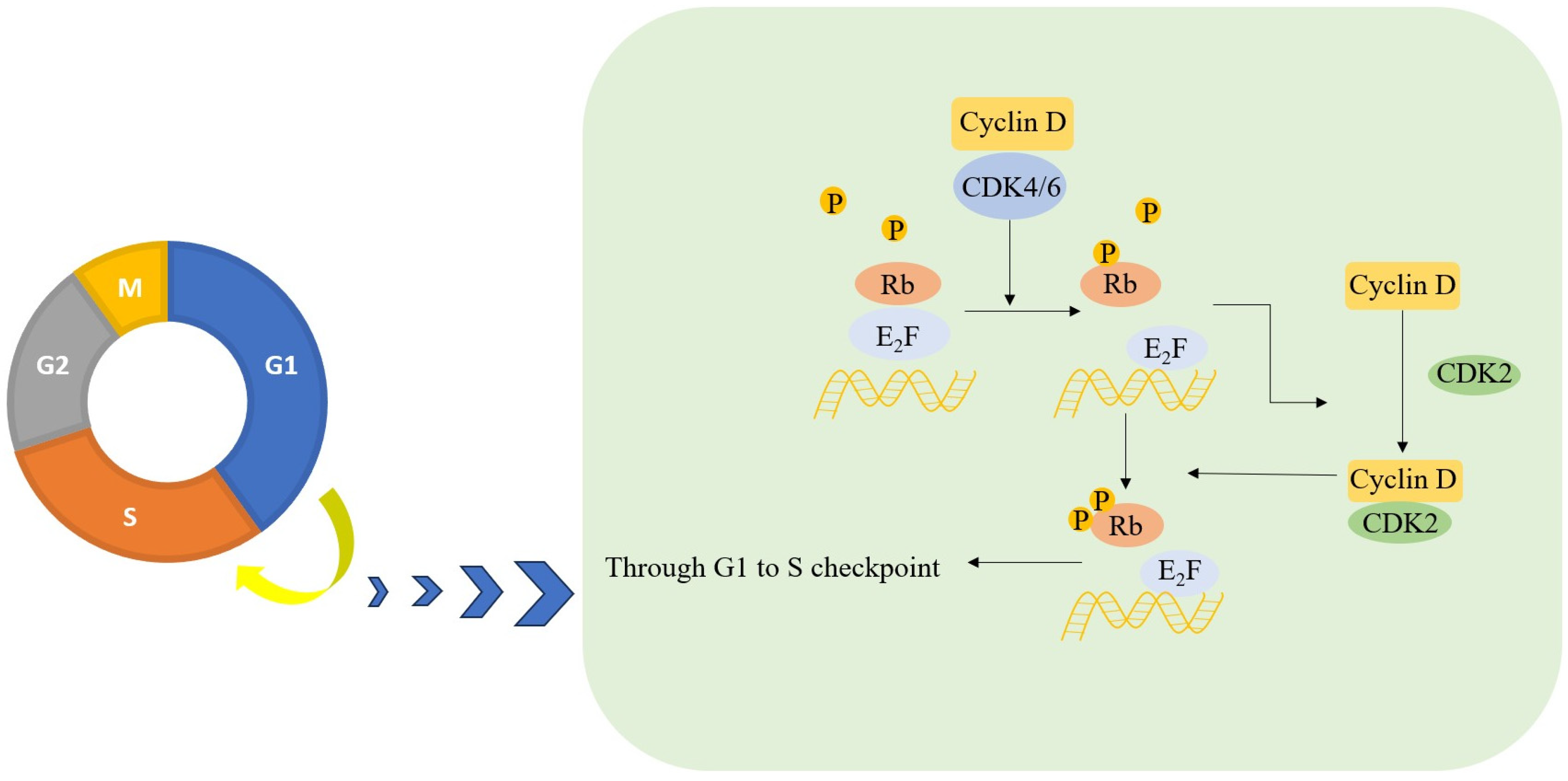
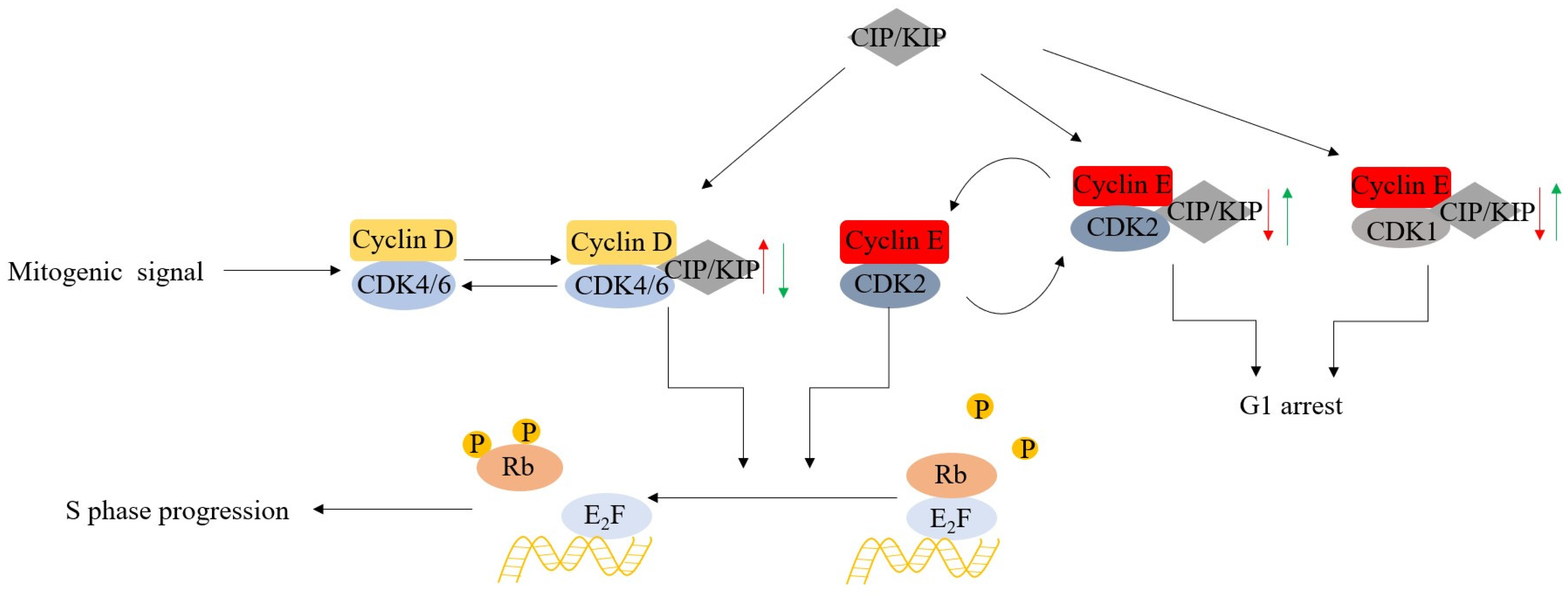
2. The Biological Rationale for Targeting CDK4/6
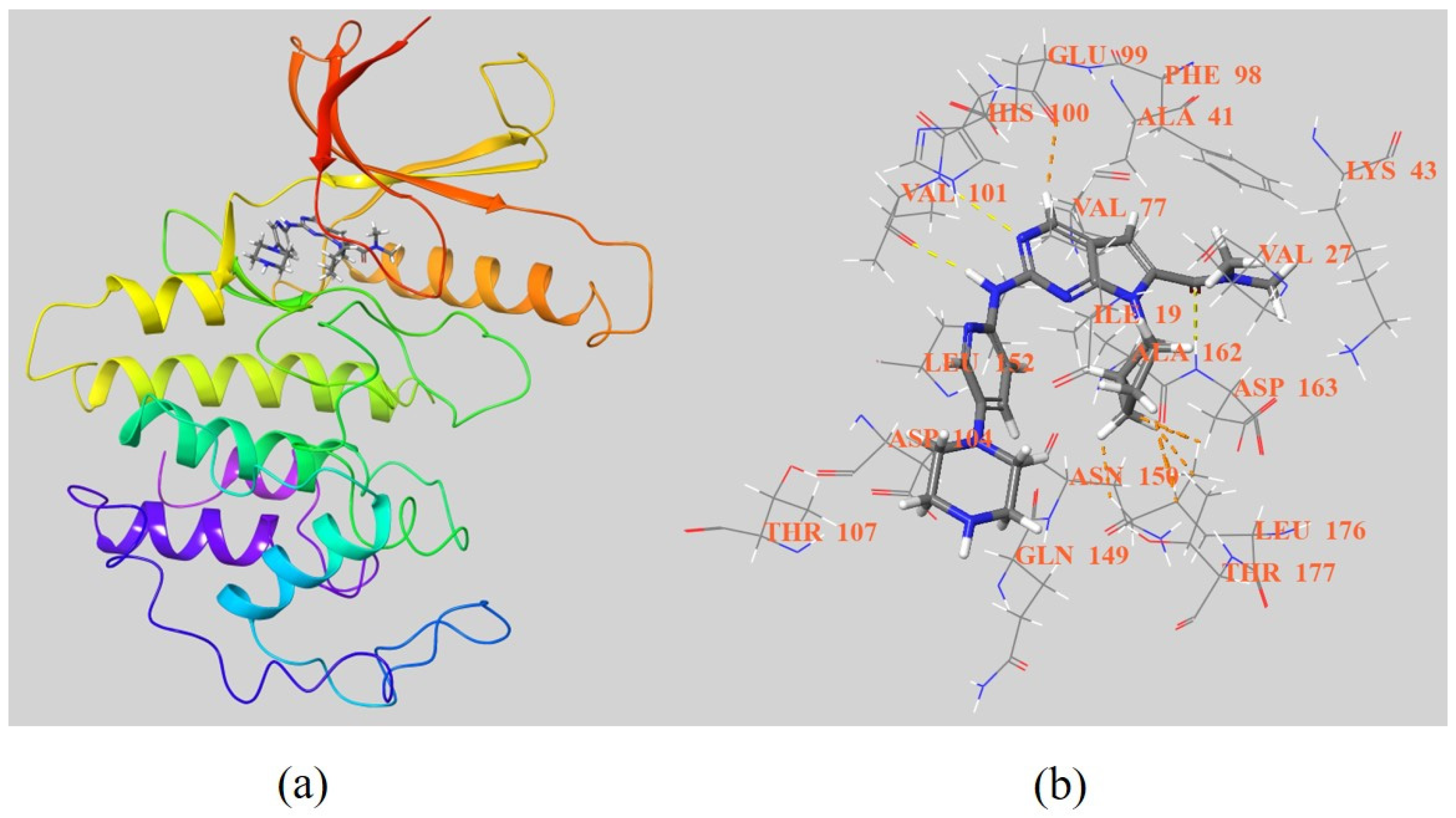
3. The Overview of CDK4/6 Sites
4. Approved CDK4/6 Inhibitors for Marketing
5. Synthesized CDK4/6 Inhibitors
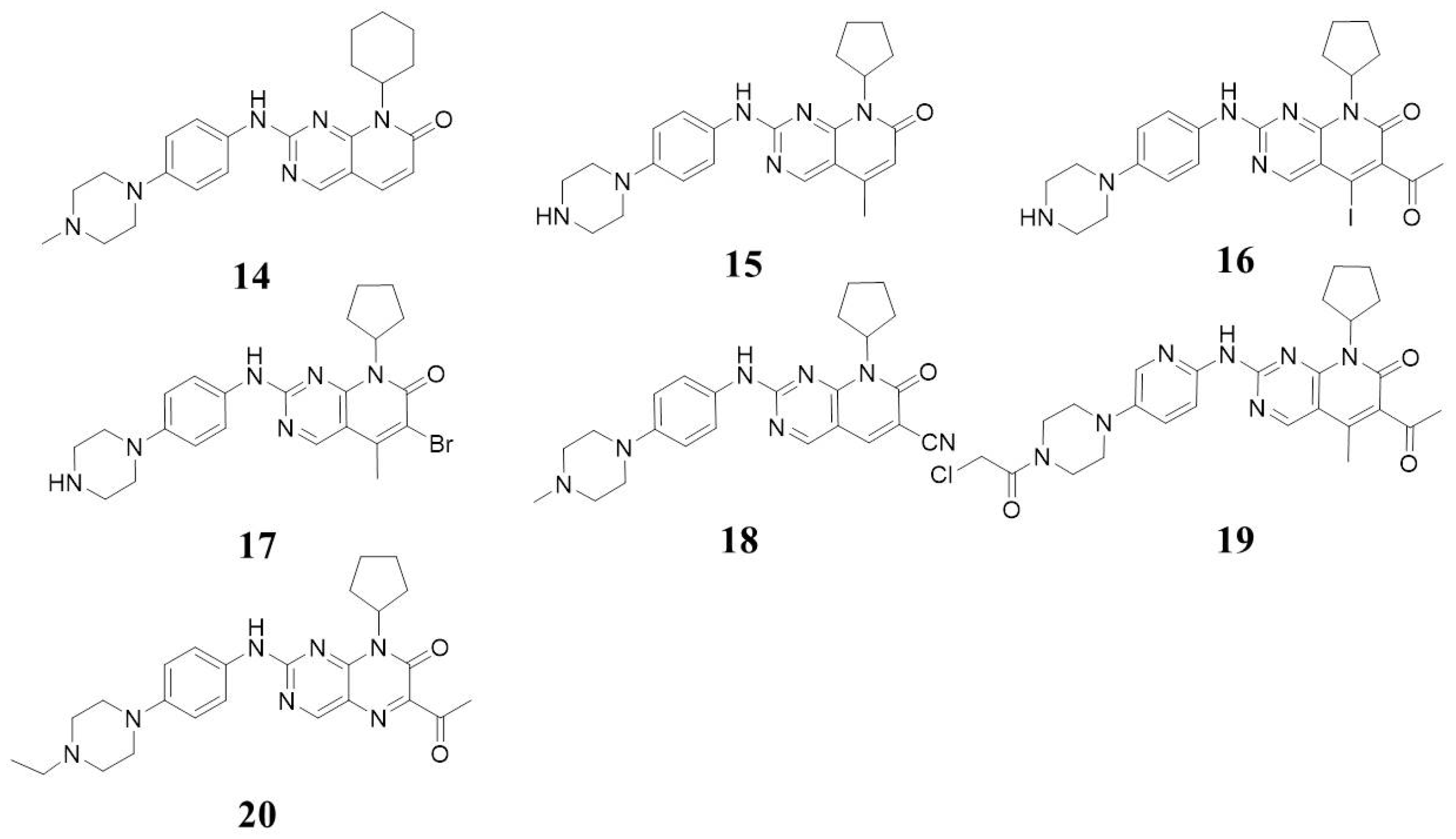
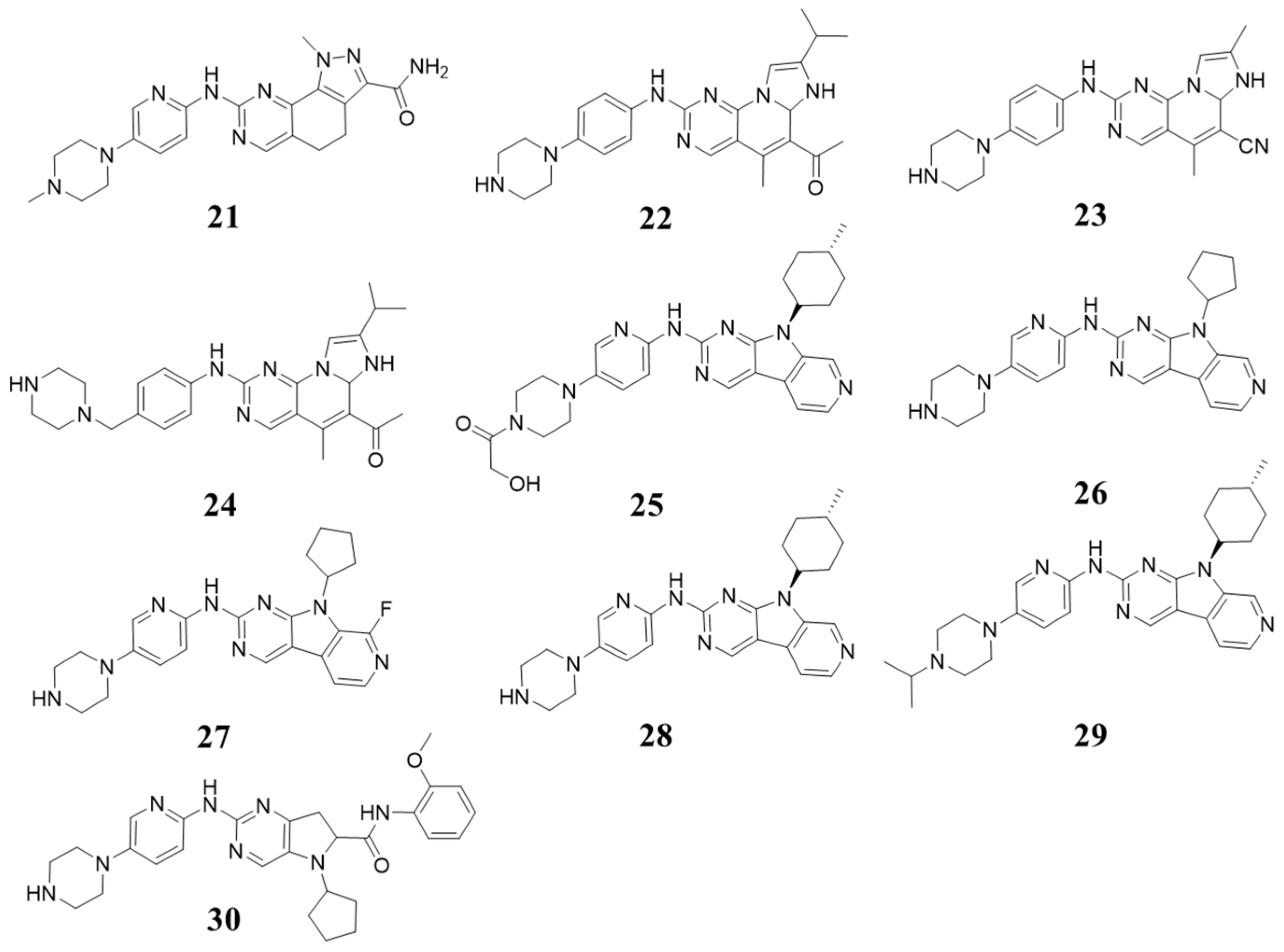
5.1. Structures with 8-Alkyl-2-(arylamino)pyrido [2,3-d]pyrimidin-7(8H)-one
5.2. Structures with a 2-Aminopyrimidin-Containing Tricycle Moiety
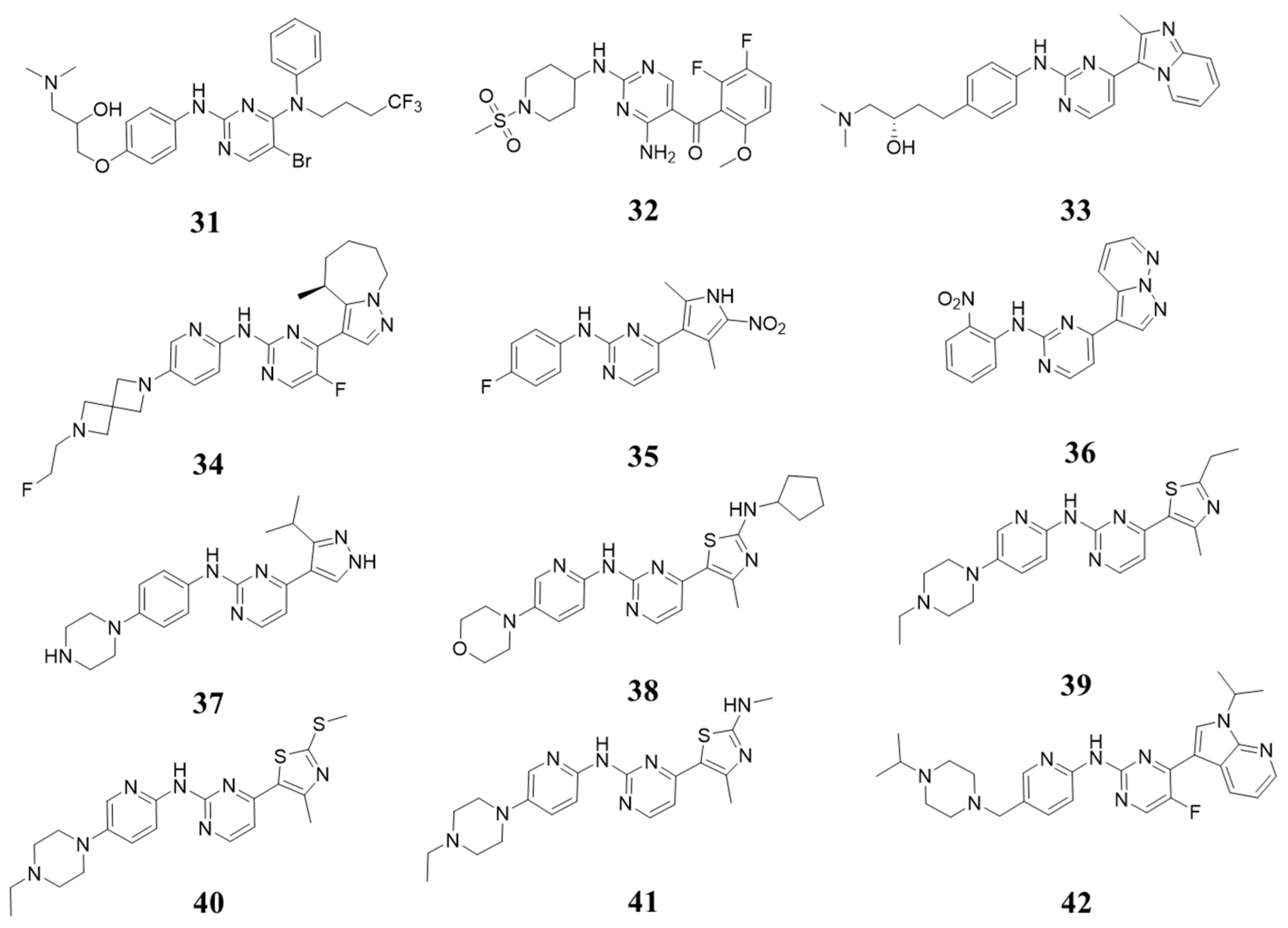
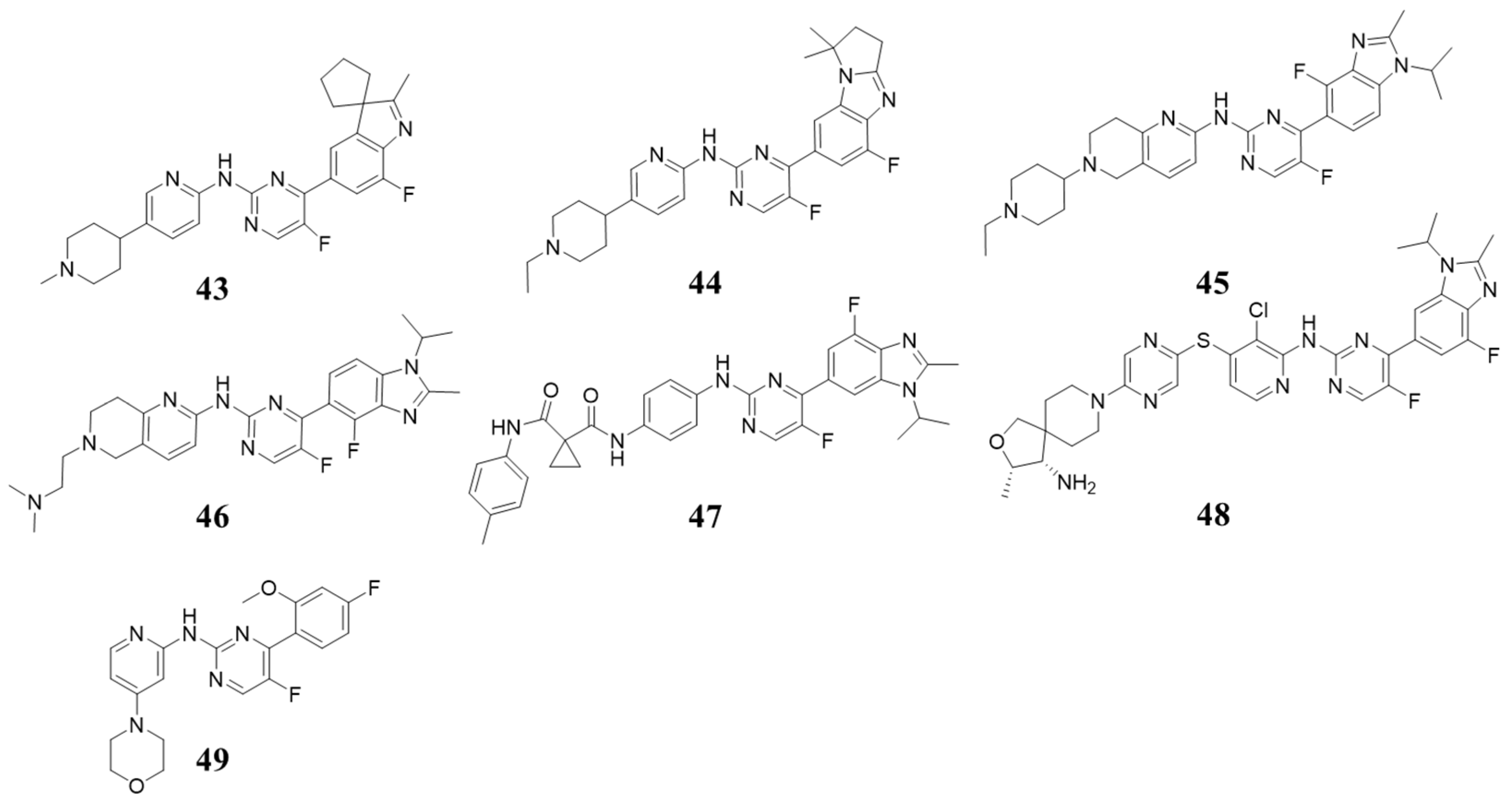
5.3. Structures with 2-(Arylamino)-4-aryl Pyrimidin
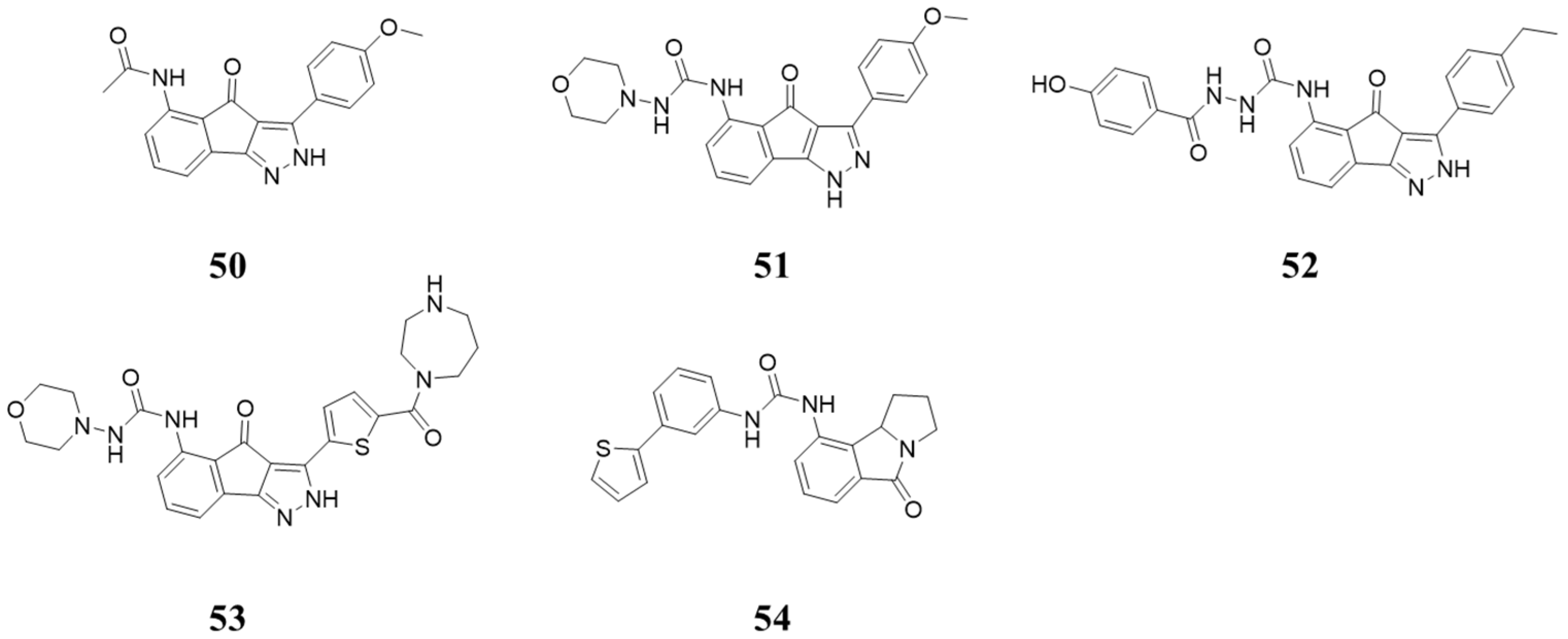
5.4. Structures Containing 5-Amino-3-arylindeno[1,2-c]pyrazol-4(2H)-one
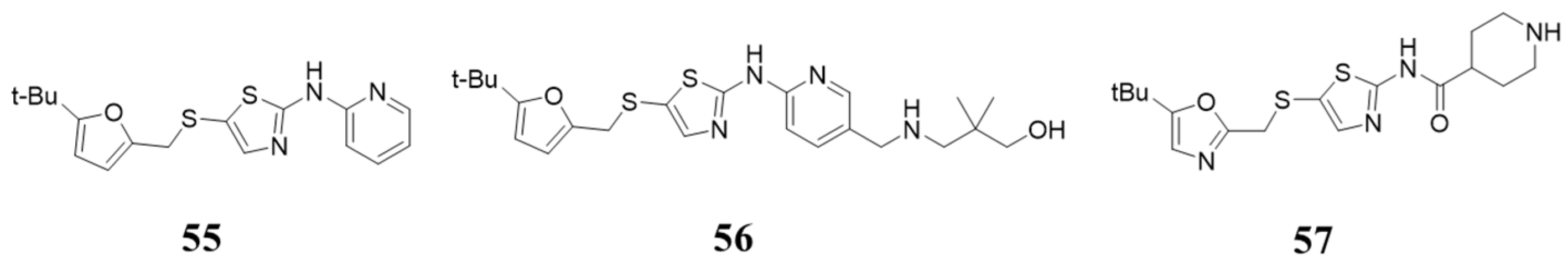
5.5. Structures Containing 2-Aminothiazole
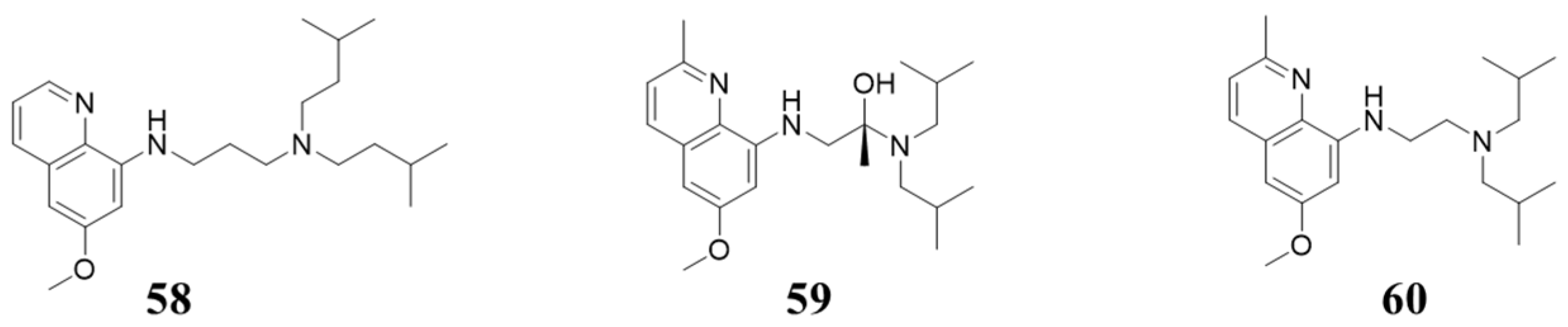
5.6. Structures with 8-Alkylamino-quinolin
5.7. Structures Containing (Z)-4-(Aminomethylene)isoquinoline-1,3(2H,4H)-dione
5.8. Structures Derived from High-Throughput Screening and Natural Products
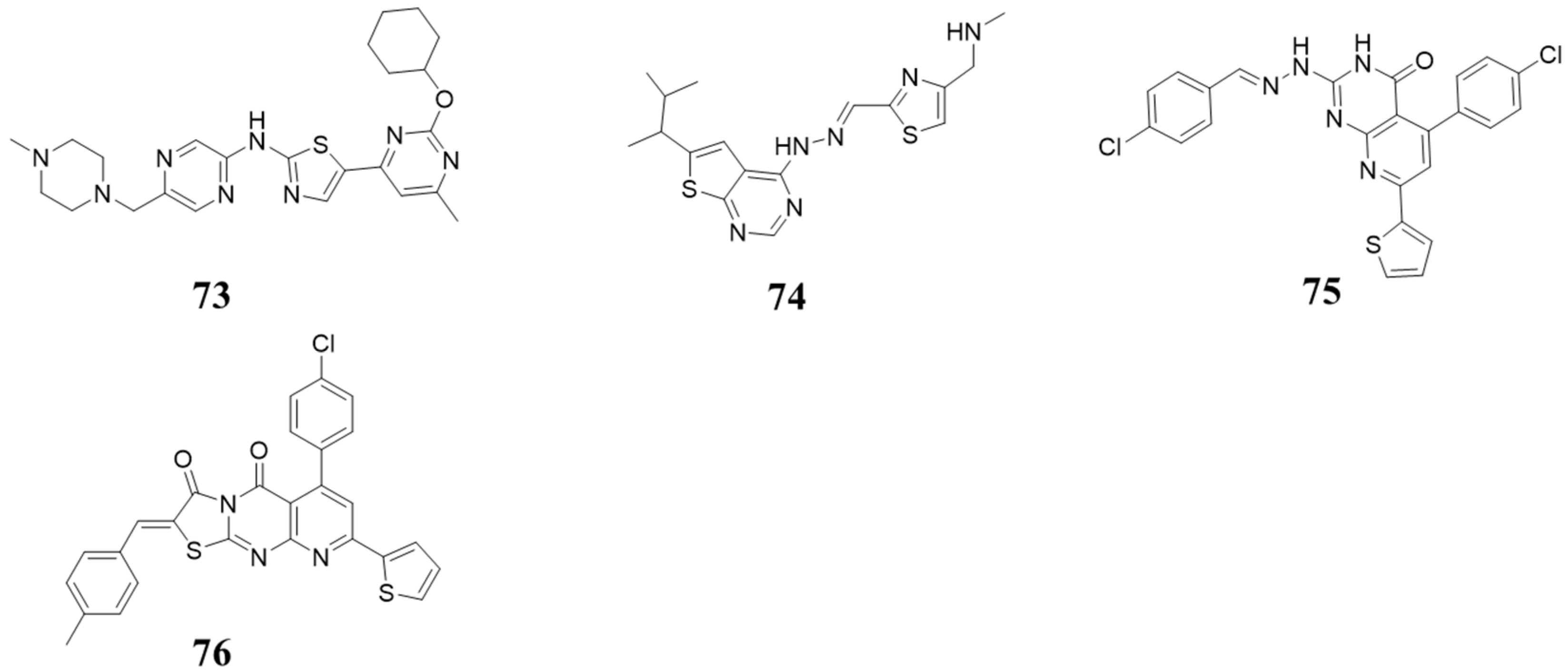
5.9. Other Structures
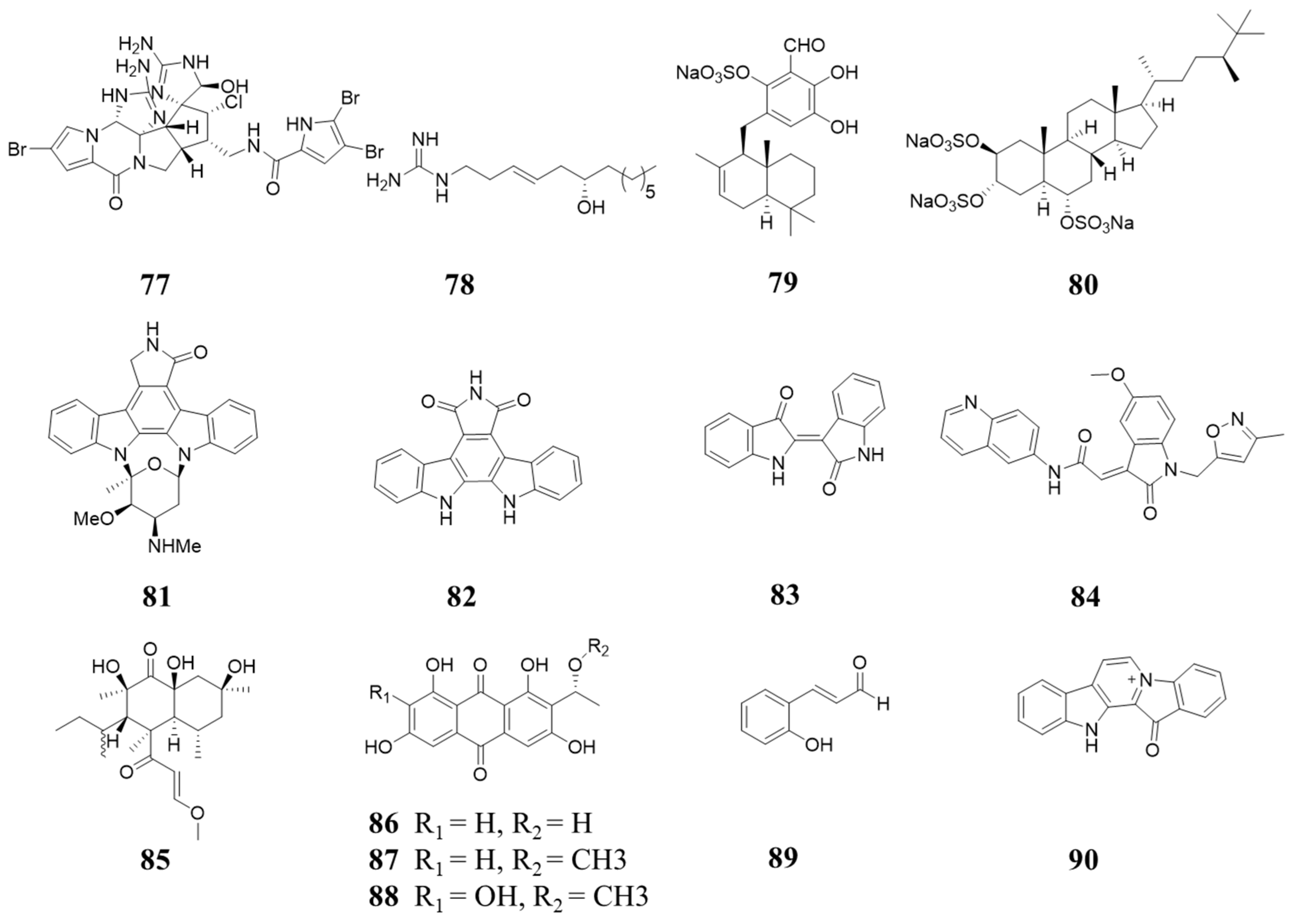
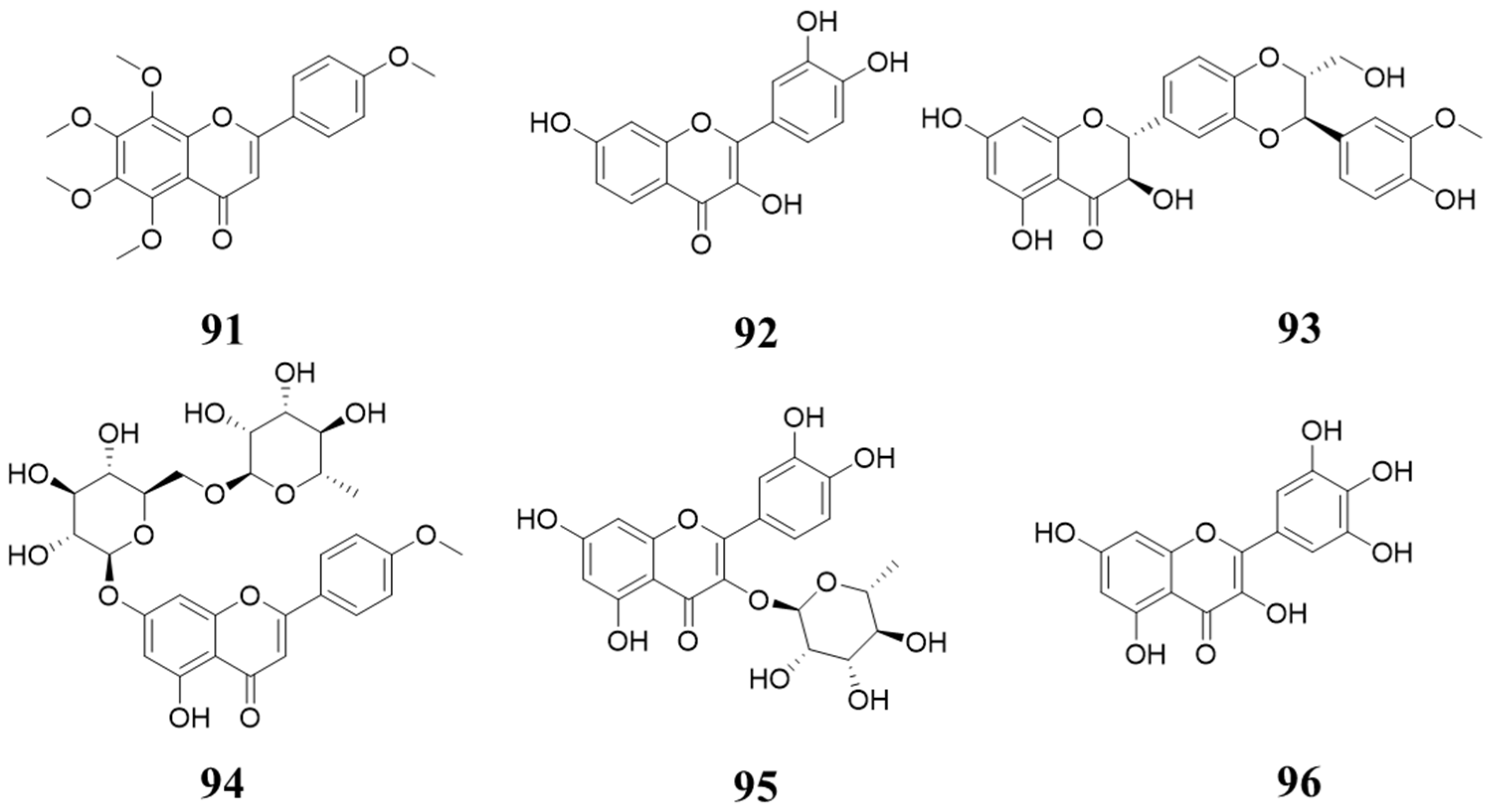
6. Natural Product Inhibiting CDK4/6
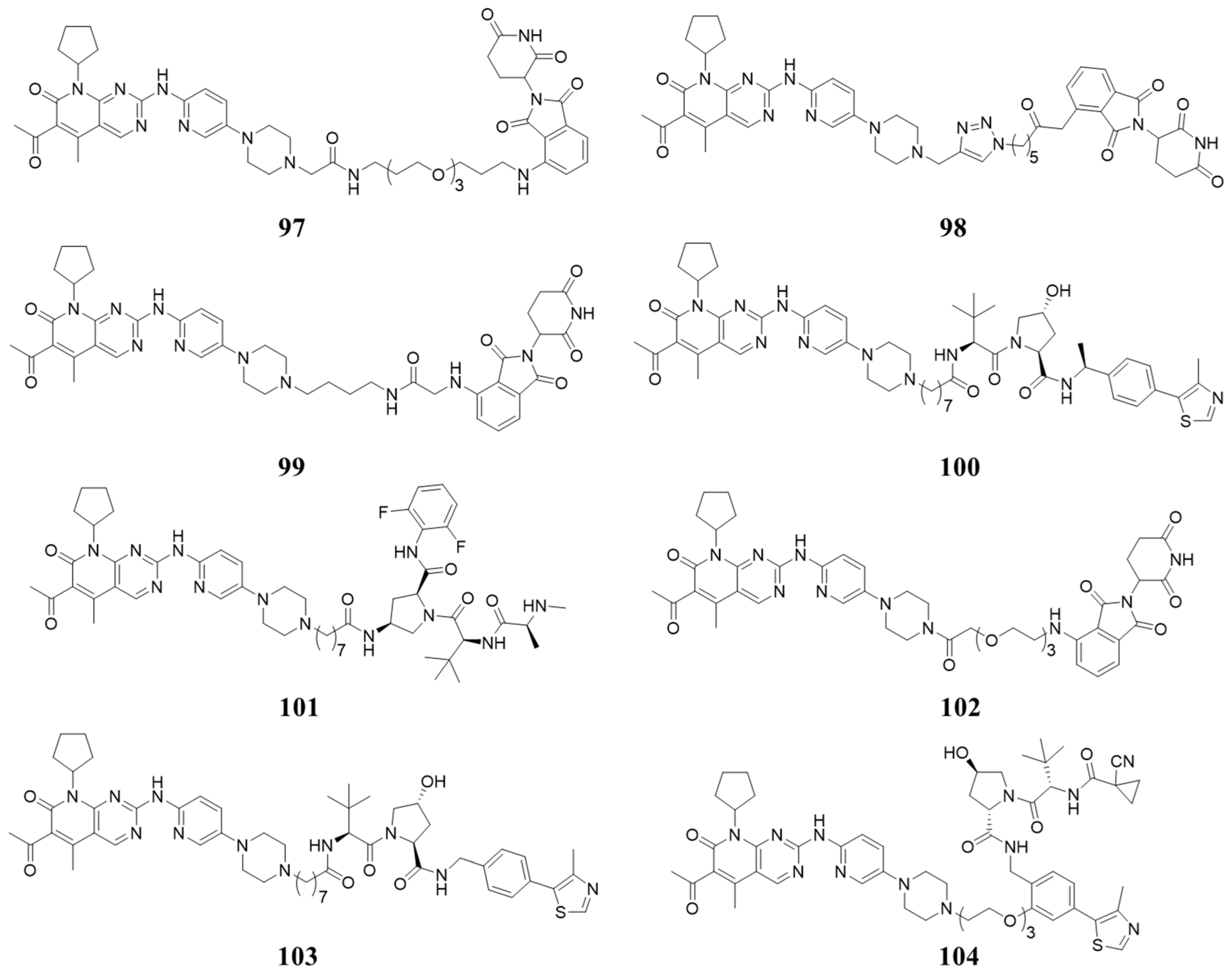
7. PROTAC Targeting CDK4/6 and Cyclins
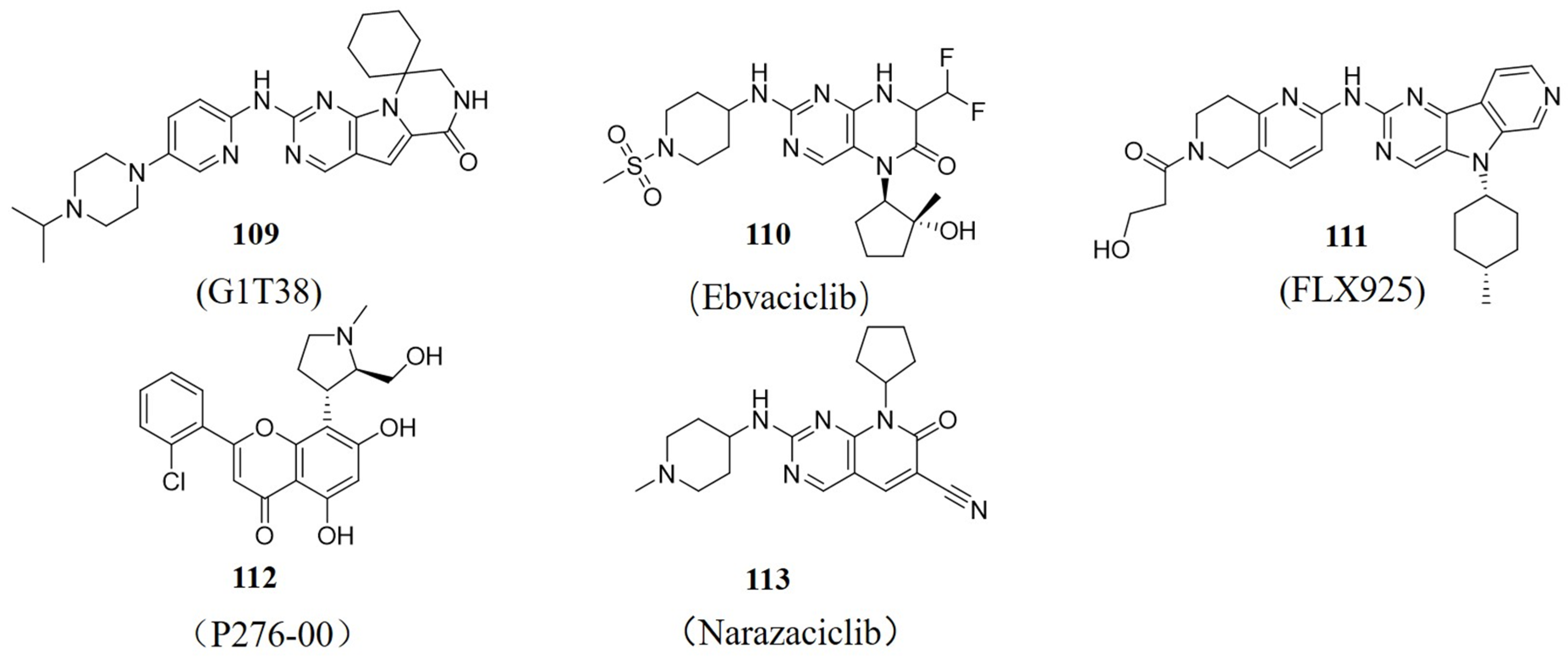
8. CDK4/6 Inhibitors in Clinical Research
9. Summary and Prospect
Author Contributions
Funding
Conflicts of Interest
References
- Fisher, R.P. A cell cycle regulator branches out: A key G1 kinase targets transcription to drive cell cycle commitment. Science 2021, 374, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Michowski, W.; Chick, J.M.; Chu, C.; Kolodziejczyk, A.; Wang, Y.; Suski, J.M.; Abraham, B.; Anders, L.; Day, D.; Dunkl, L.M.; et al. Cdk1 Controls Global Epigenetic Landscape in Embryonic Stem Cells. Mol. Cell 2020, 78, 459–476.e13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, X. DEAD-box RNA helicases in cell cycle control and clinical therapy. Cells 2021, 10, 1540. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, A.; You, Z.; Xu, J.; Zhu, S. Epigenetic silencing of CDKN1A and CDKN2B by SNHG1 promotes the cell cycle, migration and epithelial-mesenchymal transition progression of hepatocellular carcinoma. Cell Death Dis. 2020, 11, 823. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, S.; Wang, G.; Zhao, W.; Zhang, D.; Wang, F.; Li, W.; Sun, L. Study of the mechanism by which dinaciclib induces apoptosis and cell cycle arrest of lymphoma Raji cells through a CDK1-involved pathway. Cancer Med. 2019, 8, 4348–4358. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Pfizer Inc. IBRANCE® (Palbociclib): US Prescribing Information. 2015. Available online: http://www.accessdata.fda.gov. (accessed on 12 February 2015).
- Xiang, R.; Fan, Y.; Li, Y.; Guo, Q.; Huang, Z. Heterocyclic Derivative with CDK4/6 and HDAC Inhibitory Activity. CN106831780, 13 June 2017. [Google Scholar]
- Eli Lilly and Company. VERZENIOTM (Abemaciclib): US Prescribing Information. 2017. Available online: https://www.fda.gov. (accessed on 17 October 2017).
- Dhillon, S. Trilaciclib: First Approval. Drugs 2021, 81, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Mariaule, G.; Belmont, P. Cyclin-dependent kinase inhibitors as marketed anticancer drugs: Where are we now? A short survey. Molecules 2014, 19, 14366. [Google Scholar] [CrossRef]
- Cheng, W.; Yang, Z.; Wang, S.; Li, Y.; Wei, H.; Tian, X.; Kan, Q. Recent development of CDK inhibitors: An overview of CDK/inhibitor co-crystal structures. Eur. J. Med. Chem. 2019, 164, 615–639. [Google Scholar] [CrossRef]
- Ammazzalorso, A.; Agamennone, M.; De Filippis, B.; Fantacuzzi, M. Development of CDK4/6 inhibitors: A five years update. Molecules 2021, 26, 1488. [Google Scholar] [CrossRef]
- Susanti, N.M.P.; Tjahjono, D.H. Cyclin-Dependent Kinase 4 and 6 Inhibitors in Cell Cycle Dysregulation for Breast Cancer Treatment. Molecules 2021, 26, 4462. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Wang, X.; Dong, H.; Min, W.; Hao, H.; Yang, P. Selective inhibition of CDK4/6: A safe and effective strategy for developing anticancer drugs. Acta Pharm. Sin. B 2021, 11, 30–54. [Google Scholar] [CrossRef] [PubMed]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef]
- Cheng, M.; Olivier, P.; Diehl, J.A.; Fero, M.; Roussel, M.F.; Roberts, J.M.; Sherr, C.J. The p21Cip1 and p27Kip1 CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999, 18, 1571–1583. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. Systematic Screen for CDK4/6 Substrates Links FOXM1 Phosphorylation to Senescence Suppression in Cancer Cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Matsushime, H.; Ewen, M.E.; Strom, D.K.; Kato, J.Y.; Hanks, S.K.; Roussel, M.F.; Sherr, C.J. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell 1992, 71, 323. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef]
- Uras, I.Z.; Walter, G.J.; Scheicher, R.; Bellutti, F.; Prchal-Murphy, M.; Tigan, A.S.; Valent, P.; Heidel, F.H.; Kubicek, S.; Scholl, C.; et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood 2016, 127, 2890–2902. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Kalra, S.; Joshi, G.; Munshi, A.; Kumar, R. Structural insights of cyclin dependent kinases: Implications in design of selective inhibitors. Eur. J. Med. Chem. 2017, 142, 424–458. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.V.; Yuan, J.; Eisele, K. Abstract LB-136: Mechanistic exploration of combined CDK4/6 and ER inhibition in ER-positive breast cancer. Cancer Res. 2014, 74 (Suppl. 19), LB-136. [Google Scholar] [CrossRef]
- O’Brien, N.T.; Emmanuelle, E.D.; Ayala, R. In vivo efficacy of combined targeting of CDK4/6, ER and PI3K signaling in ER+ breast cancer. Cancer Res. 2014, 74 (Suppl. 19), 4756. [Google Scholar] [CrossRef]
- Eli Lilly and Company. FDA Grants Priority Review for Potential New Indication for Lilly’s VerzenioTM (Abemaciclib) As Initial Treatment of Advanced Breast Cancer [Media Release]. Available online: https://www.lilly.com (accessed on 12 October 2017).
- US FDA. FDA Approves New Treatment for Certain Advanced or Metastatic Breast Cancers [Media Release]. 2017. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatment-certain-advanced-or-metastatic-breast-cancers (accessed on 28 September 2017).
- Finn, R.S.; Aleshin, A.; Slamon, D.J. Targeting the cyclin-dependent kinases (CDK) 4/6 in estrogen receptor-positive breast cancers. Breast Cancer Res. 2016, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- US FDA. FDA Approves Drug to Reduce Bone Marrow Suppression Caused by Chemotherapy; Media Release; U.S. FDA: Washington, DC, USA, 2021.
- G1 Therapeutics. COSELATM (Trilaciclib): US Prescribing Information. 2021. Available online: https://www.g1therapeutics.com/consela/pi/ (accessed on 25 February 2021).
- Zhang, H.; Yan, S.; Zhan, Y.; Ma, S.; Bian, Y.; Li, S.; Tian, J.; Li, G.; Zhong, D.; Diao, X.; et al. A mass balance study of [14C] SHR6390 (dalpiciclib), a selective and potent CDK4/6 inhibitor in humans. Front. Pharmacol. 2023, 14, 1116073. [Google Scholar] [CrossRef] [PubMed]
- Kan, Q.; Tian, X.; Zhang, X.; Yang, Z.; Du, Y.; Cheng, W.; Yuan, Y.; Wang, S. Pyrimidine Derivative Useful in Treatment of Cancer and Its Preparation. CN108299312, 20 June 2018. [Google Scholar]
- Barvian, M.; Boschelli, D.; Cossrow, J.; Dobrusin, E.; Fattaey, A.; Fritsch, A.; Fry, D.; Harvey, P.; Keller, P.; Garrett, M.; et al. Pyrido[2,3-d]pyrimidin-7-one Inhibitors of Cyclin-Dependent Kinases. J. Med. Chem. 2000, 43, 4606–4616. [Google Scholar] [CrossRef] [PubMed]
- Bathini, Y.; Singh, I.; Harvey, P.J.; Keller, P.R.; Singh, R.; Micetich, R.G.; Fry, D.W.; Dobrusin, E.M.; Toogood, P.L. 2-Aminoquinazoline inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2005, 15, 3881–3885. [Google Scholar] [CrossRef] [PubMed]
- VanderWel, S.N.; Harvey, P.J.; McNamara, D.J.; Repine, J.T.; Keller, P.R.; Quin, J., III; Booth, R.J.; Elliott, W.L.; Dobrusin, E.M.; Fry, D.W.; et al. Pyrido[2,3-d]pyrimidin-7-ones as Specific Inhibitors of Cyclin-Dependent Kinase 4. J. Med. Chem. 2005, 48, 2371–2387. [Google Scholar] [CrossRef]
- Sanchez-Martinez, C.; Gelbert, L.M.; Lallena, M.J.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs. Bioorg. Med. Chem. Lett. 2015, 25, 3420–3435. [Google Scholar] [CrossRef]
- Shan, H.; Ma, X.; Yan, G.; Luo, M.; Zhong, X.; Lan, S.; Yang, J.; Liu, Y.; Pu, C.; Tong, Y.; et al. Discovery of a novel covalent CDK4/6 inhibitor based on palbociclib scaffold. Eur. J. Med. Chem. 2021, 219, 113432. [Google Scholar] [CrossRef]
- He, H.; Liu, Q.; Chen, L.; Wang, J.; Yuan, Y.; Li, H.; Qian, X.; Zhao, Z.; Chen, Z. Design, synthesis and biological evaluation of pteridine-7(8H)-one derivatives as potent and selective CDK4/6 inhibitors. Bioorg. Med. Chem. Lett. 2022, 76, 128991. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, X.; Cao, K.; Zhang, Y.; Zhao, K.; Tang, C.; Feng, B. Synthesis and SAR of 4,5-dihydro-1H-pyrazolo[4,3-h]quinazoline derivatives as potent and selective CDK4/6 inhibitors. Eur. J. Med. Chem. 2018, 157, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Wang, Q.; Liao, X.; Ge, H.; Huo, G.; Zhang, L.; Chen, N.; Zhai, X.; Hong, Y.; Wang, L.; et al. Discovery of a novel series of imidazo[1′,2′:1,6]pyrido[2,3-d]pyrimidin derivatives as potent cyclin-dependent kinase 4/6 inhibitors. Eur. J. Med. Chem. 2020, 193, 112239. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Eksterowicz, J.; Gribble, M.W.; Alba, G.Q.; Ayres, M.; Carlson, T.J.; Chen, A.; Chen, X.; Cho, R.; et al. Discovery of AMG 925, a FLT3 and CDK4 Dual Kinase Inhibitor with Preferential Affinity for the Activated State of FLT3. J. Med. Chem. 2014, 57, 3430–3449. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, C.; Lallena, M.J.; Sanfeliciano, S.G.; de Dios, A. Cyclin dependent kinase (CDK) inhibitors as anticancer drugs: Recent advances (2015–2019). Bioorg. Med. Chem. Lett. 2019, 29, 126637. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Quan, Y.; Wang, Y.; Wang, Y.; Li, Y. Design, synthesis, and biological evaluation of 2,6,7-substituted pyrrolo[2,3-d]pyrimidines as cyclin dependent kinase inhibitor in pancreatic cancer cells. Bioorg. Med. Chem. Lett. 2021, 33, 127725. [Google Scholar] [CrossRef] [PubMed]
- Beattie, J.F.; Breault, G.A.; Ellston, R.P.A.; Green, S.; Jewsbury, P.J.; Midgley, C.J.; Naven, R.T.; Minshull, C.A.; Pauptit, R.A.; Tucker, J.A.; et al. Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 1: Identification and optimization of substituted 4,6-Bis anilino pyrimidines. Bioorg. Med. Chem. Lett. 2003, 13, 2955–2960. [Google Scholar] [CrossRef]
- Breault, G.A.; Ellston, R.P.A.; Green, S.; James, S.R.; Jewsbury, P.J.; Midgley, C.J.; Pauptit, R.A.; Minshull, C.A.; Tucker, J.A.; Pease, J.E. Cyclin-dependent kinase 4 inhibitors as a treatment for cancer. Part 2: Identification and optimization of substituted 2,4-bis anilino pyrimidines. Bioorg. Med. Chem. Lett. 2003, 13, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.-J.; DePinto, W.; Bartkovitz, D.; So, S.-S.; Vu, B.T.; Packman, K.; Lukacs, C.; Ding, Q.; Jiang, N.; Wang, K.; et al. Discovery of [4-Amino-2-(1-methanesulfonylpiperidin-4-ylamino)pyrimidin-5-yl](2,3-difluoro-6- methoxyphenyl)methanone (R547), A Potent and Selective Cyclin-Dependent Kinase Inhibitor with Significant in Vivo Antitumor Activity. J. Med. Chem. 2006, 49, 6549–6560. [Google Scholar] [CrossRef]
- Anderson, M.; Beattie, J.F.; Breault, G.A.; Breed, J.; Byth, K.F.; Culshaw, J.D.; Ellston, R.P.A.; Green, S.; Minshull, C.A.; Norman, R.A.; et al. Imidazo[1,2-a]pyridines: A potent and selective class of cyclin-Dependent kinase inhibitors identified through structure-Based hybridization. Bioorg. Med. Chem. Lett. 2003, 13, 3021–3026. [Google Scholar] [CrossRef]
- Bronner, S.M.; Merrick, K.A.; Murray, J.; Salphati, L.; Moffat, J.G.; Pang, J.; Sneeringer, C.J.; Dompe, N.; Cyr, P.; Purkey, H.; et al. Design of a brain-penetrant CDK4/6 inhibitor for glioblastoma. Bioorg. Med. Chem. Lett. 2019, 29, 2294–2301. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wood, G.; Meades, C.; Griffiths, G.; Midgley, C.; McNae, I.; McInnes, C.; Anderson, S.; Jackson, W.; Mezna, M.; et al. Synthesis and biological activity of 2-anilino-4-(1H-pyrrol-3-yl)pyrimidine CDK inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 4237–4240. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.L.; Reno, M.J.; Alberti, J.B.; Price, D.J.; Kane-Carson, L.S.; Knick, V.B.; Shewchuk, L.M.; Hassell, A.M.; Veal, J.M.; Davis, S.T.; et al. Synthesis and evaluation of pyrazolo[1,5-b]pyridazines as selective cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5758–5762. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.-S.; Borland, M.; Brain, C.; Chen, C.H.T.; Cheng, H.; Chopra, R.; Chung, K.; Groarke, J.; He, G.; Hou, Y.; et al. 4-(Pyrazol-4-yl)-pyrimidines as Selective Inhibitors of Cyclin-Dependent Kinase 4/6. J. Med. Chem. 2010, 53, 7938–7957. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Yu, M.; Mekonnen, L.B.; Lam, F.; Islam, S.; Tomusange, K.; Rahaman, M.H.; Noll, B.; Basnet, S.K.C.; Teo, T.; et al. Highly Potent, Selective, and Orally Bioavailable 4-Thiazol-N-(pyridin-2-yl)pyrimidin-2-amine Cyclin-Dependent Kinases 4 and 6 Inhibitors as Anticancer Drug Candidates: Design, Synthesis, and Evaluation. J. Med. Chem. 2017, 60, 1892–1915. [Google Scholar] [CrossRef]
- Tadesse, S.; Zhu, G.; Mekonnen, L.B.; Lenjisa, J.L.; Yu, M.; Brown, M.P.; Wang, S. A novel series of N-(pyridin-2-yl)-4-(thiazol-5-yl)pyrimidin-2-amines as highly potent CDK4/6 inhibitors. Future Med. Chem. 2017, 9, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Bantie, L.; Tomusange, K.; Yu, M.; Islam, S.; Bykovska, N.; Noll, B.; Zhu, G.; Li, P.; Lam, F.; et al. Discovery and pharmacological characterization of a novel series of highly selective inhibitors of cyclin-dependent kinases 4 and 6 as anticancer agents. Br. J. Pharmacol. 2018, 175, 2399–2413. [Google Scholar] [CrossRef]
- Yuan, K.; Kuang, W.; Chen, W.; Ji, M.; Min, W.; Zhu, Y.; Hou, Y.; Wang, X.; Li, J.; Wang, L.; et al. Discovery of novel and orally bioavailable CDK 4/6 inhibitors with high kinome selectivity, low toxicity and long-acting stability for the treatment of multiple myeloma. Eur. J. Med. Chem. 2022, 228, 114024. [Google Scholar] [CrossRef]
- Yin, L.; Li, H.; Liu, W.; Yao, Z.; Cheng, Z.; Zhang, H.; Zou, H. A highly potent CDK4/6 inhibitor was rationally designed to overcome blood brain barrier in gliobastoma therapy. Eur. J. Med. Chem. 2018, 144, 1–28. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, W.-J.; Yin, L.; Li, H.; Chen, Z.-H.; Zhu, D.-X.; Song, X.-Q.; Cheng, Z.-Z.; Song, P.; Wang, Z.; et al. Design and synthesis of 4-(2,3-dihydro-1H-benzo[d]pyrrolo[1,2-a]imidazol-7-yl)-N-(5-(piperazin-1-ylmethyl)pyridine-2-yl)pyrimidin-2-amine as a highly potent and selective cyclin-dependent kinases 4 and 6 inhibitors and the discovery of structure-activity relationships. Bioorg. Med. Chem. Lett. 2018, 28, 974–978. [Google Scholar]
- Zha, C.; Deng, W.; Fu, Y.; Tang, S.; Lan, X.; Ye, Y.; Su, Y.; Jiang, L.; Chen, Y.; Huang, Y.; et al. Design, synthesis and biological evaluation of tetrahydronaphthyridine derivatives as bioavailable CDK4/6 inhibitors for cancer therapy. Eur. J. Med. Chem. 2018, 148, 140–153. [Google Scholar] [CrossRef]
- Shi, C.; Wang, Q.; Liao, X.; Ge, H.; Huo, G.; Zhang, L.; Chen, N.; Zhai, X.; Hong, Y.; Wang, L.; et al. Discovery of 6-(2-(dimethylamino)ethyl)-N-(5-fluoro-4-(4-fluoro-1-isopropyl-2-methyl-1H-benzo[d]imidazole-6-yl)pyrimidin-2-yl)-5,6,7,8-tetrahydro-1,6-naphthyridin-2-amine as a highly potent cyclin-dependent kinase 4/6 inhibitor for treatment of cancer. Eur. J. Med. Chem. 2019, 178, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhao, B.; Qin, Z.; Li, Y.; Wang, T.; Zhou, W.; Zheng, J.; Yang, S.; Shi, Y.; Fan, Y.; et al. Novel dual inhibitors targeting CDK4 and VEGFR2 synergistically suppressed cancer progression and angiogenesis. Eur. J. Med. Chem. 2019, 181, 111541. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shu, C.; Li, W.; Hou, Q.; Luo, G.; Yang, K.; Wu, X. Discovery of a Novel Src Homology-2 Domain Containing Protein Tyrosine Phosphatase-2 (SHP2) and Cyclin-Dependent Kinase 4 (CDK4) Dual Inhibitor for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2022, 65, 6729–6747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; Liu, D.; Zheng, D.; Li, X.; Li, C.; Huang, C.; Wang, Y.; Wang, X.; Li, Q.; et al. A Potential Anti-Glioblastoma Compound LH20 Induces Apoptosis and Arrest of Human Glioblastoma Cells via CDK4/6 Inhibition. Molecules 2023, 28, 5047. [Google Scholar] [CrossRef] [PubMed]
- Nugiel, D.A.; Etzkorn, A.-M.; Vidwans, A.; Benfield, P.A.; Boisclair, M.; Burton, C.R.; Cox, S.; Czerniak, P.M.; Doleniak, D.; Seitz, S.P. Indenopyrazoles as Novel Cyclin Dependent Kinase (CDK) Inhibitors. J. Med. Chem. 2001, 44, 1334–1336. [Google Scholar] [CrossRef] [PubMed]
- Nugiel, D.A.; Vidwans, A.; Etzkorn, A.-M.; Rossi, K.A.; Benfield, P.A.; Burton, C.R.; Cox, S.; Doleniak, D.; Seitz, S.P. Synthesis and Evaluation of Indenopyrazoles as Cyclin-Dependent Kinase Inhibitors. 2. Probing the Indeno Ring Substituent Pattern. J. Med. Chem. 2002, 45, 5224–5232. [Google Scholar] [CrossRef] [PubMed]
- Nugiel, D.A.; Vidwans, A.; Dzierba, C.D. Parallel synthesis of acylsemicarbazide libraries: Preparation of potent cyclin dependent kinase (cdk) inhibitors. Bioorg. Med. Chem. Lett. 2004, 14, 5489–5491. [Google Scholar] [CrossRef]
- Singh, S.K.; Dessalew, N.; Bharatam, P.V. 3D-QSAR CoMFA study on indenopyrazole derivatives as cyclin dependent kinase 4 (CDK4) and cyclin dependent kinase 2 (CDK2) inhibitors. Eur. J. Med. Chem. 2006, 41, 1310–1319. [Google Scholar] [CrossRef]
- Honma, T.; Hayashi, K.; Aoyama, T.; Hashimoto, N.; Machida, T.; Fukasawa, K.; Iwama, T.; Ikeura, C.; Ikuta, M.; Suzuki-Takahashi, I.; et al. Structure-Based Generation of a New Class of Potent Cdk4 Inhibitors: New de Novo Design Strategy and Library Design. J. Med. Chem. 2001, 44, 4615–4627. [Google Scholar] [CrossRef]
- Misra, R.N.; Xiao, H.-y.; Williams, D.K.; Kim, K.S.; Lu, S.; Keller, K.A.; Mulheron, J.G.; Batorsky, R.; Tokarski, J.S.; Sack, J.S.; et al. Synthesis and biological activity of N-aryl-2-aminothiazoles: Potent pan inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2004, 14, 2973–2977. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.N.; Xiao, H.-y.; Kim, K.S.; Lu, S.; Han, W.-C.; Barbosa, S.A.; Hunt, J.T.; Rawlins, D.B.; Shan, W.; Ahmed, S.Z.; et al. N-(cycloalkylamino)acyl-2-aminothiazole inhibitors of cyclin-dependent kinase 2. N-[5-[[[5-(1,1-dimethylethyl)-2-oxazolyl]methyl]thio]-2-thiazolyl]-4- piperidinecarboxamide (BMS-387032), a highly efficacious and selective antitumor agent. J. Med. Chem. 2004, 47, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Klein, M.A.; Mayo, K.H.; Kratzke, R.A. p16INK4a Peptide mimetics identified via virtual screening. Bioorg. Med. Chem. Lett. 2010, 20, 403–405. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.-R.; Otteng, M.; Tran, T.; Floyd, M.B., Jr.; Reich, M.; Birnberg, G.; Kutterer, K.; Ayral-Kaloustian, S.; Ravi, M.; Nilakantan, R.; et al. 4-(Phenylaminomethylene)isoquinoline-1,3(2H,4H)-diones as Potent and Selective Inhibitors of the Cyclin-Dependent Kinase 4 (CDK4). J. Med. Chem. 2008, 51, 3507–3525. [Google Scholar] [CrossRef] [PubMed]
- Tsou, H.-R.; Liu, X.; Birnberg, G.; Kaplan, J.; Otteng, M.; Tran, T.; Kutterer, K.; Tang, Z.; Suayan, R.; Zask, A.; et al. Discovery of 4-(Benzylaminomethylene)isoquinoline-1,3-(2H,4H)-diones and 4-[(Pyridylmethyl)aminomethylene]isoquinoline-1,3-(2H,4H)-diones as Potent and Selective Inhibitors of the Cyclin-Dependent Kinase 4. J. Med. Chem. 2009, 52, 2289–2310. [Google Scholar] [CrossRef]
- Ryu, C.-K.; Kang, H.-Y.; Lee, S.K.; Nam, K.A.; Hong, C.Y.; Ko, W.-G.; Lee, B.-H. 5-arylamino-2-methyl-4,7-dioxobenzothiazoles as inhibitors of cyclin-dependent kinase 4 and cytotoxic agents. Bioorg. Med. Chem. Lett. 2000, 10, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Carini, D.J.; Kaltenbach, R.F.; Liu, J.; Benfield, P.A.; Boylan, J.; Boisclair, M.; Brizuela, L.; Burton, C.R.; Cox, S.; Grafstrom, R.; et al. Identification of selective inhibitors of cyclin dependent kinase 4. Bioorg. Med. Chem. Lett. 2001, 11, 2209–2211. [Google Scholar] [CrossRef]
- Markwalder, J.A.; Arnone, M.R.; Benfield, P.A.; Boisclair, M.; Burton, C.R.; Chang, C.-H.; Cox, S.S.; Czerniak, P.M.; Dean, C.L.; Doleniak, D.; et al. Synthesis and Biological Evaluation of 1-Aryl-4,5-dihydro-1H-pyrazolo[3,4-d]pyrimidin-4-one Inhibitors of Cyclin-Dependent Kinases. J. Med. Chem. 2004, 47, 5894–5911. [Google Scholar] [CrossRef]
- Dessalew, N.; Bharatam, P.V. 3D-QSAR and molecular docking study on bisarylmaleimide series as glycogen synthase kinase 3, cyclin dependent kinase 2 and cyclin dependent kinase 4 inhibitors: An insight into the criteria for selectivity. Eur. J. Med. Chem. 2007, 42, 1014–1027. [Google Scholar] [CrossRef]
- Horiuchi, T.; Chiba, J.; Uoto, K.; Soga, T. Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based inhibitors of Cyclin D1-CDK4: Synthesis, biological evaluation, and structure-activity relationships. Bioorg. Med. Chem. Lett. 2009, 19, 305–308. [Google Scholar] [CrossRef]
- Mahale, S.; Bharate, S.B.; Manda, S.; Joshi, P.; Bharate, S.S.; Jenkins, P.R.; Vishwakarma, R.A.; Chaudhuri, B. Biphenyl-4-carboxylic Acid [2-(1H-Indol-3-yl)-ethyl]-methylamide (CA224), a Nonplanar Analogue of Fascaplysin, Inhibits Cdk4 and Tubulin Polymerization: Evaluation of in Vitro and in Vivo Anticancer Activity. J. Med. Chem. 2014, 57, 9658–9672. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Mahesh, R.; Nayak, V.L.; Babu, K.S.; Kumar, G.B.; Shaik, A.B.; Kapure, J.S.; Alarifi, A. Discovery of pyrrolospirooxindole derivatives as novel cyclin dependent kinase 4 (CDK4) inhibitors by catalyst-free, green approach. Eur. J. Med. Chem. 2016, 108, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Al-Warhi, T.; El Kerdawy, A.M.; Aljaeed, N.; Ismael, O.E.; Ayyad, R.R.; Eldehna, W.M.; Abdel-Aziz, H.A.; Al-Ansary, G.H. Synthesis, biological evaluation and in silico studies of certain oxindole-indole conjugates as anticancer CDK inhibitors. Molecules 2020, 25, 2031. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.M.; El-Moselhy, T.; El-Bastawissy, E.; Ibrahim, M.A.A.; Sayed, S.M.; Hegazy, M.-E.F.; Efferth, T.; Jaragh, L.A.; Sidhom, P.A. The mystery of titan hunter: Rationalized striking of the MAPK pathway via Newly synthesized 6-Indolylpyridone-3-Carbonitrile derivatives. Eur. J. Med. Chem. 2023, 259, 115675. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Shibata, J.; Kurihara, H.; Mita, T.; Otsuki, S.; Sagara, T.; Hirai, H.; Iwasawa, Y. Identification of potent 5-pyrimidinyl-2-aminothiazole CDK4, 6 inhibitors with significant selectivity over CDK1, 2, 5, 7, and 9. Bioorg. Med. Chem. Lett. 2006, 16, 3751–3754. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, T.; Takeda, Y.; Haginoya, N.; Miyazaki, M.; Nagata, M.; Kitagawa, M.; Akahane, K.; Uoto, K. Discovery of novel thieno[2,3-d]pyrimidin-4-yl hydrazone-based cyclin-dependent kinase 4 inhibitors: Synthesis, biological evaluation and structure-activity relationships. Chem. Pharm. Bull. 2011, 59, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.E.S.; George, R.F.; Samir, E.M.; Aref, M.M.A.; Abdel-Aziz, H.A. Synthesis and anticancer activity of some pyrido[2,3-d]pyrimidine derivatives as apoptosis inducers and cyclin-dependent kinase inhibitors. Future Med. Chem. 2019, 11, 2395–2414. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, J.i.; Suzuki, M.; Tsuda, M. Konbu’acidin A, a new bromopyrrole alkaloid with cdk4 inhibitory activity from Hymeniacidon sponge. Tetrahedron 1997, 53, 15681–15684. [Google Scholar] [CrossRef]
- Moussa, A.Y.; Mostafa, N.M.; Singab, A.N.B. Pulchranin A: First report of isolation from an endophytic fungus and its inhibitory activity on cyclin dependent kinases. Nat. Prod. Res. 2020, 34, 2715–2722. [Google Scholar] [CrossRef]
- Furusaki, A.; Hashiba, N.; Matsumoto, T.; Hirano, A.; Iwai, Y.; Omura, S. X-ray structure of staurosporine: A new alkaloid from a Streptomyces strain. J. Chem. Soc. Chem. Commun. 1978, 18, 800. [Google Scholar] [CrossRef]
- Sanchez-Martinez, C.; Shih, C.; Faul, M.M.; Zhu, G.; Paal, M.; Somoza, C.; Li, T.; Kumrich, C.A.; Winneroski, L.L.; Xun, Z.; et al. Aryl[a]pyrrolo[3,4-c]carbazoles as selective cyclin D1-CDK4 inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 3835–3839. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.A.; Longley, R.E.; McConnell, O.J.; Ballas, L.M. Staurosporine aglycon (K252-c) and arcyriaflavin A from the marine ascidian, Eudistoma sp. Experientia 1994, 50, 843. [Google Scholar] [CrossRef] [PubMed]
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Chiou, C.-T.; Lee, W.-C.; Liao, J.-H.; Cheng, J.-J.; Lin, L.-C.; Chen, C.-Y.; Song, J.-S.; Wu, M.-H.; Shia, K.-S.; Li, W.-T. Synthesis and evaluation of 3-ylideneoxindole acetamides as potent anticancer agents. Eur. J. Med. Chem. 2015, 98, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brauers, G.; Edrada, R.A.; Ebel, R.; Proksch, P.; Wray, V.; Berg, A.; Graefe, U.; Schaechtele, C.; Totzke, F.; Finkenzeller, G.; et al. Anthraquinones and betaenone derivatives from the sponge-associated fungus Microsphaeropsis species: Novel inhibitors of protein kinases. J. Nat. Prod. 2000, 63, 739–745. [Google Scholar] [CrossRef]
- Jeong, H.W.; Kim, M.R.; Son, K.H.; Young Han, M.; Ha, J.H.; Garnier, M.; Meijer, L.; Kwon, B.M. Cinnamaldehydes inhibit cyclin dependent kinase 4/cyclin D1. Bioorg. Med. Chem. Lett. 2000, 10, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Muller, L.; Furet, P.; Schoepfer, J.; Stephan, C.; Zumstein-Mecker, S.; Fretz, H.; Chaudhuri, B. Inhibition of Cyclin-Dependent Kinase 4 (Cdk4) by Fascaplysin, a Marine Natural Product. Biochem. Biophys. Res. Commun. 2000, 275, 877–884. [Google Scholar] [CrossRef]
- Zupan, A.; Mikulic-Petkovsek, M.; Slatnar, A.; Stampar, F.; Veberic, R. Individual phenolic response and peroxidase activity in peel of differently sun-exposed apples in the period favorable for sunburn occurrence. J. Plant Physiol. 2014, 171, 1706–1712. [Google Scholar] [CrossRef]
- Pan, M.-H.; Chen, W.-J.; Lin-Shiau, S.-Y.; Ho, C.-T.; Lin, J.-K. Tangeretin induces cell-cycle G1 arrest through inhibiting cyclin-dependent kinases 2 and 4 activities as well as elevating Cdk inhibitors p21 and p27 in human colorectal carcinoma cells. Carcinogenesis 2002, 23, 1677–1684. [Google Scholar] [CrossRef]
- Lu, X.; Jung, J.I.; Cho, H.J.; Lim, D.Y.; Lee, H.S.; Chun, H.S.; Kwon, D.Y.; Park, J.H.Y. Fisetin inhibits the activities of cyclin-dependent kinases leading to cell cycle arrest in HT-29 human colon cancer cells. J. Nutr. 2005, 135, 2884–2890. [Google Scholar] [CrossRef]
- Varghese, L.; Agarwal, C.; Tyagi, A.; Singh, R.P.; Agarwal, R. Silibinin Efficacy against Human Hepatocellular Carcinoma. Clin. Cancer Res. 2005, 11, 8441–8448. [Google Scholar] [CrossRef] [PubMed]
- Sivashanmugam, M.; Raghunath, C.; Vetrivel, U. Virtual screening studies reveal linarin as a potential natural inhibitor targeting CDK4 in retinoblastoma. J. Pharmacol. Pharmacother. 2013, 4, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, R.M.P.; Hoyo-Vadillo, C. Anti-inflammatory Potential of Petiveria alliacea on Activated RAW264.7 Murine Macrophages. Pharmacogn. Mag. 2017, 13 (Suppl. 2), S174–S178. [Google Scholar] [CrossRef] [PubMed]
- Bodiba, D.C.; Prasad, P.; Srivastava, A.; Crampton, B.; Lall, N.S. Antibacterial activity of Azadirachta indica, Pongamia pinnata, Psidium guajava, and Mangifera indica and their mechanism of action against Streptococcus mutans. Pharmacogn. Mag. 2018, 14, 76–80. [Google Scholar] [CrossRef]
- Rana, S.; Bendjennat, M.; Kour, S.; King, H.M.; Kizhake, S.; Zahid, M.; Natarajan, A. Selective degradation of CDK6 by a palbociclib based PROTAC. Bioorg. Med. Chem. Lett. 2019, 29, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Burgess, K. PROTACs suppression of CDK4/6, crucial kinases for cell cycle regulation in cancer. Chem. Commun. 2019, 55, 2704–2707. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.A.; Cryan, J.; Ahmed, A.; Dai, H.; McGonagle, G.A.; Rozier, C.; Benowitz, A.B. Selective CDK6 degradation mediated by cereblon, VHL, and novel IAP-recruiting PROTACs. Bioorg. Med. Chem. Lett. 2020, 30, 127106. [Google Scholar] [CrossRef] [PubMed]
- Steinebach, C.; Ng, Y.L.D.; Sosic, I.; Lee, C.-S.; Chen, S.; Lindner, S.; Vu, L.P.; Bricelj, A.; Haschemi, R.; Monschke, M.; et al. Systematic exploration of different E3 ubiquitin ligases: An approach towards potent and selective CDK6 degraders. Chem. Sci. 2020, 11, 3474–3486. [Google Scholar] [CrossRef]
- Verano, A.L.; You, I.; Donovan, K.A.; Mageed, N.; Yue, H.; Nowak, R.P.; Fischer, E.S.; Wang, E.S.; Gray, N.S. Redirecting the Neo-Substrate Specificity of Cereblon-Targeting PROTACs to Helios. ACS Chem. Biol. 2022, 17, 2404–2410. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhong, Y.; Yim, H.; Yang, X.; Park, K.-S.; Xie, L.; Poulikakos, P.I.; Han, X.; Xiong, Y.; Chen, X.; et al. Bridged Proteolysis Targeting Chimera (PROTAC) Enables Degradation of Undruggable Targets. J. Am. Chem. Soc. 2022, 144, 22622–22632. [Google Scholar] [CrossRef]
- Li, Z.; Ma, S.; Zhang, L.; Zhang, S.; Ma, Z.; Du, L.; Li, M. Targeted Protein Degradation Induced by HEMTACs Based on HSP90. J. Med. Chem. 2023, 66, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Liu, Y.; Deng, R.; Xu, Q.; Wang, S.; Zhang, H.; Luo, D.; Ma, X.; Tong, Y.; Li, R. Development of PROTAC degrader probe of CDK4/6 based on DCAF16. Bioorg. Chem. 2023, 138, 106637. [Google Scholar] [CrossRef]
- Bisi, J.E.; Sorrentino, J.A.; Jordan, J.L.; Darr, D.D.; Roberts, P.J.; Tavares, F.X.; Strum, J.C. Preclinical development of G1T38: A novel, potent and selective inhibitor of cyclin dependent kinases 4/6 for use as an oral antineoplastic in patients with CDK4/6 sensitive tumors. Oncotarget 2017, 8, 42343–42358. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Hoffman, R.L.; Behenna, D.C.; Boras, B.; Carelli, J.; Diehl, W.; Ferre, R.A.; He, Y.-A.; Hui, A.; Huang, B.; et al. Discovery of PF-06873600, a CDK2/4/6 Inhibitor for the Treatment of Cancer. J. Med. Chem. 2021, 64, 9056–9077. [Google Scholar] [CrossRef] [PubMed]
- Bozhenko, V.K.; Kulinich, T.M.; Kudinova, E.A.; Boldyrev, A.; Solodkij, V.A. New targeted anti CDK4/6 peptide MM-D37K. J. Clin. Oncol. 2013, 31, e13545. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, J.; Ding, L. Preparation of (Heterocyclyl)azinylaminopyrimidinyl Tetrahydrobenzimidazopyridines and Dihydrobenzimidazooxazines as Inhibitors of CDK4 and CDK6 for Use as Anticancer Agents. WO2018113771, 28 June 2018. [Google Scholar]
- Available online: https://www.medicilon.com.cn//index.php?keywords=RGT-419B&c=list&pagelen=20&tempid=59&catid=48 (accessed on 5 December 2023).
- O’Rourke, K.A.T.E. Highlights from the 2022 Annual ASCO Meeting. Cancer 2022, 128, 3010–3011. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.tykmedicines.com/ty-302/ (accessed on 5 December 2023).
- Available online: https://db.yaozh.com/linchuangshiyan/bJaRbmNnZ2hqlWRilJaWlg==.html (accessed on 5 December 2023).
- Available online: http://www.bebettermed.cn/goods-25-view.html (accessed on 5 December 2023).
- Carlino, L.; Christodoulou, M.S.; Restelli, V.; Caporuscio, F.; Foschi, F.; Semrau, M.S.; Costanzi, E.; Tinivella, A.; Pinzi, L.; Lo Presti, L.; et al. Structure-activity relationships of hexahydrocyclopenta[c]quinoline derivatives as allosteric inhibitors of CDK2 and EGFR. ChemMedChem 2018, 13, 2627–2634. [Google Scholar] [CrossRef]
- Jain, S.K.; Bharate, S.B.; Vishwakarma, R.A. Cyclin-dependent kinase inhibition by flavoalkaloids. Mini-Rev. Med. Chem. 2012, 12, 632–649. [Google Scholar] [CrossRef]
- Available online: https://www.sphchina.com/main_business/pharmaceutical_R&D.html (accessed on 5 December 2023).
- Liu, C.; Liu, B.; Xu, C.; Zhang, P.; Li, B.; Ji, B.; Zhang, B.; Liu, Q.; Zhang, J.; Yu, C. ETH-155008, a Novel Selective Dual Inhibitor of FLT3 and CDK4/6 in Preclinical Treatment of Acute Myeloid Leukemia. Blood 2019, 134, 5114. [Google Scholar] [CrossRef]
- Profitós-Pelejà, N.; Ribeiro, M.L.; Parra, R.J.; Fernández-Serrano, M.; Marín-Escudero, P.; Makovski-Silverstein, A.; Cosenza, S.; Esteller, M.; Roué, G. Prolonged cell cycle arrest by the CDK4/6 antagonist narazaciclib restores ibrutinib response in preclinical models of BTKi-resistant mantle cell lymphoma. Hematol. Oncol. 2023, 553–554. [Google Scholar] [CrossRef]
- Available online: http://www.sipac.gov.cn/szgyyq/jsdt/202212/fb4e3e65573e454d8487929ee03b74be.shtml (accessed on 5 December 2023).
- Bessudo, A.; Cohen, E.E.W.; Gutierrez, R.; Johnson, D.H.; Rosenberg, A.; Sukari, A.; Stemmer, S.M.; Weinberg, B.A.; Reuveni, H.; Schickler, M.; et al. A phase 1/2 study with open-label, dose escalation phase followed by single-arm expansion at the maximum tolerated dose to assess the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of NT219 injection alone and in combination with cetuximab in adults with advanced solid tumors and head and neck cancer. J. Clin. Oncol. 2021, 39, TPS3156. [Google Scholar]
- Available online: http://www.jrfbio.com/list-12-1.html (accessed on 5 December 2023).
- Available online: http://www.changbaicao.cn/trialsdetail-id-1493.html (accessed on 5 December 2023).
- Available online: https://www.medicilon.com.cn/tindex.html/QHRD110 (accessed on 5 December 2023).
- Available online: https://investingnews.com/nuvation-bio-announces-discontinuation-of-nuv-422-clinical-development-program/ (accessed on 5 December 2023).
- Kong, T.; Xue, Y.; Cencic, R.; Zhu, X.; Monast, A.; Fu, Z.; Pilon, V.; Sangwan, V.; Guiot, M.-C.; Foulkes, W.D.; et al. eIF4A inhibitors suppress cell-cycle feedback response and acquired resistance to CDK4/6 inhibition in cancer. Mol. Cancer Ther. 2019, 18, 2158–2170. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-Q.; Pan, X.-H.; Wang, T.-T.; Wang, J.; Yang, B.; He, Q.-J.; Ding, L. Intrinsic and acquired resistance to CDK4/6 inhibitors and potential overcoming strategies. Acta Pharmacol. Sin. 2021, 42, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and letrozole in advanced breast cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | R&D Company | Stage of Development | Reference |
|---|---|---|---|
| Lerociciclib (G1T38) | G1 Therapeutics (North Carolina, NC, USA) | Phase II | [111] |
| Ebvaciclib (PF-06873600) | PFIZER (New York, NY, USA) | Phase I/II | [112] |
| MM-D37K | MetaMax Ltd. (Moscow, Russia) | Phase I/II | [113] |
| BPI-16350 | Beida Pharmaceutical Company (Hangzhou, China) | Phase III | [114] |
| RGT-419B | Shanghai Qilu Ruige Pharmaceutical R&D Co., Ltd. (Shanghai, Chian) | Phase I | [115] |
| FCN-437c | Fuchuang Pharmaceutical Company (Chongqing, China) | Phase I | [116] |
| TY-302 | Zhengzhou Taiji Hongnuo Pharmaceutical Co., Ltd. (Zhengzhou, China) | Phase I | [117] |
| TQB3616 | Zhengda Tianqing Pharmaceutical Co., Ltd. (Lianyungang, China) | Phase III | [118] |
| BEBT-209 | Guangzhou Beibeite Pharmaceutical Co., Ltd. (Guangzhou, China) | Phase II | [119] |
| AMG925(FLX925) | Amgen (FLX BIO) (San Fernando, CA, USA) | Phase I | [120] |
| P-276-00 | Piramal (Mumbai, India) | Phase II | [121] |
| SPH4366 | Shanghai Pharmaceutical Group (Shanghai, China) | Phase II/III | [122] |
| ETH-155008 | Shengke Pharmaceutical (Suzhou, China) | Phase I | [123] |
| Narazaciclib (ON123300) | Onconova Therapeutics Inc. (Newtown, PA, USA) | Phase I/II | [124] |
| CGT-1967 | Suzhou Shengshi Taike Company (Suzhou, China) | Phase I | [125] |
| Auceliciclib | Aucentra Therapeutics (Adelaide, Australia) | Phase I/II | [126] |
| XH-30002 | Shanghai Xunhe Pharmaceutical Technology Co., Ltd. (Shanghai, China) | Phase I | [127] |
| HS-10342 | Hansen Company (Shanghai, China) | Phase I | [128] |
| QHRD110 | Changzhou Qianhong Biochemical Pharmaceutical Co., Ltd. (Changzhou, China) | Phase I | [129] |
| NUV-422 | Nuvation Bio. (New York, NY, USA) | Phase II | [130] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Ba, J.; Kang, Y.; Gong, Z.; Liang, T.; Zhang, Y.; Qi, J.; Wang, J. Recent Progress in CDK4/6 Inhibitors and PROTACs. Molecules 2023, 28, 8060. https://doi.org/10.3390/molecules28248060
Wang H, Ba J, Kang Y, Gong Z, Liang T, Zhang Y, Qi J, Wang J. Recent Progress in CDK4/6 Inhibitors and PROTACs. Molecules. 2023; 28(24):8060. https://doi.org/10.3390/molecules28248060
Chicago/Turabian StyleWang, Hao, Jianfei Ba, Yue Kang, Zeqiao Gong, Tingting Liang, Yahong Zhang, Jianguo Qi, and Jianhong Wang. 2023. "Recent Progress in CDK4/6 Inhibitors and PROTACs" Molecules 28, no. 24: 8060. https://doi.org/10.3390/molecules28248060
APA StyleWang, H., Ba, J., Kang, Y., Gong, Z., Liang, T., Zhang, Y., Qi, J., & Wang, J. (2023). Recent Progress in CDK4/6 Inhibitors and PROTACs. Molecules, 28(24), 8060. https://doi.org/10.3390/molecules28248060