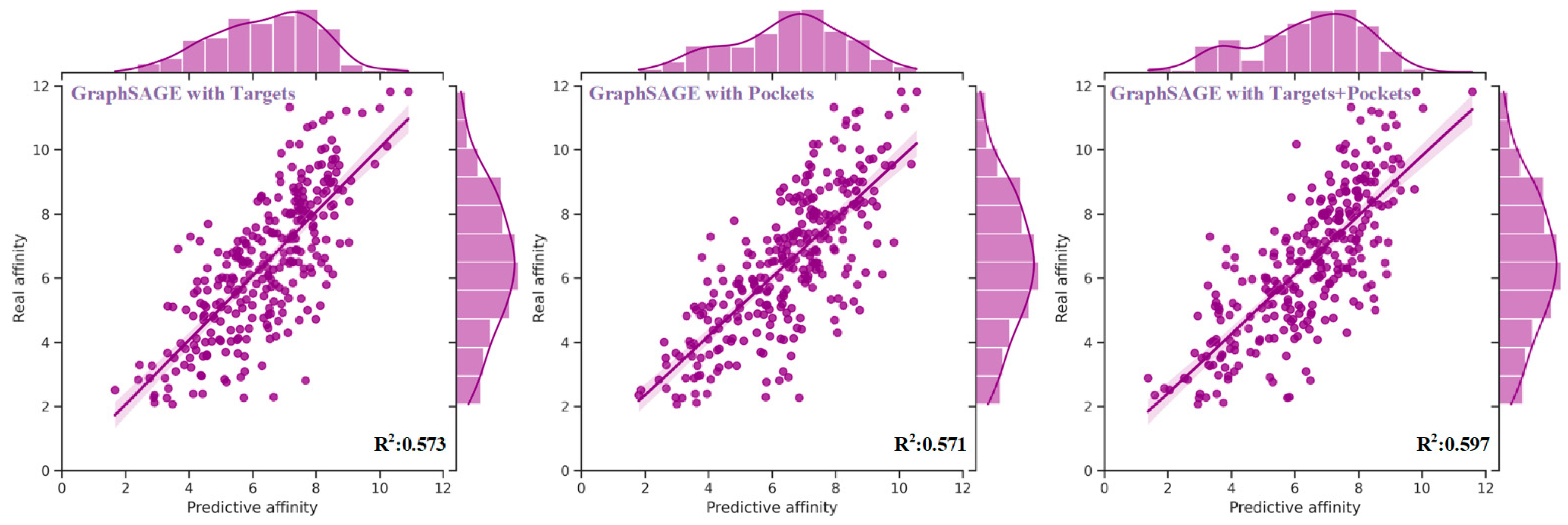
Figure 1.
The distribution of real and predictive affinity of GraphSAGE with targets, pockets, and their combination based on the test set. The performance of GraphSAGE, when harnessed in conjunction with both the sequence and structural knowledge of targets and pockets, surpassed that achieved by solely relying on the sequence and structural features of targets or pockets individually. Specifically, as compared to using only the features of targets, the MAE and RMSE exhibited reductions of 0.047 and 0.04, respectively. Moreover, improvements of 0.018 in PCC, 0.028 in Spearman, 0.013 in CI, and 0.024 in R2 were realized. Similarly, in comparison to using only the features of pockets, the MAE and RMSE experienced decreases of 0.021 and 0.43, while enhancements of 0.017 in PCC, 0.018 in Spearman, 0.009 in CI, and 0.026 in R2 were achieved.
Figure 1.
The distribution of real and predictive affinity of GraphSAGE with targets, pockets, and their combination based on the test set. The performance of GraphSAGE, when harnessed in conjunction with both the sequence and structural knowledge of targets and pockets, surpassed that achieved by solely relying on the sequence and structural features of targets or pockets individually. Specifically, as compared to using only the features of targets, the MAE and RMSE exhibited reductions of 0.047 and 0.04, respectively. Moreover, improvements of 0.018 in PCC, 0.028 in Spearman, 0.013 in CI, and 0.024 in R2 were realized. Similarly, in comparison to using only the features of pockets, the MAE and RMSE experienced decreases of 0.021 and 0.43, while enhancements of 0.017 in PCC, 0.018 in Spearman, 0.009 in CI, and 0.026 in R2 were achieved.
Figure 2.
The distribution of real and predictive affinity of heterogeneous models to fuse sequence and structural knowledge from drugs, targets, and pockets based on the test set. Among multiple sequence-processing models, including CNN, LSTM, BiLSTM, and their combinations, the end-to-end heterogeneous model composed of CNN and GCN exhibited the best performance. In comparison to the model without CNN, the MAE and RMSE were reduced by 8.3% and 5.7%, respectively. Moreover, the PCC, Spearman, CI, and R2 exhibited improvements of 3.2%, 3.5%, 1.6% and 7.4%, respectively.
Figure 2.
The distribution of real and predictive affinity of heterogeneous models to fuse sequence and structural knowledge from drugs, targets, and pockets based on the test set. Among multiple sequence-processing models, including CNN, LSTM, BiLSTM, and their combinations, the end-to-end heterogeneous model composed of CNN and GCN exhibited the best performance. In comparison to the model without CNN, the MAE and RMSE were reduced by 8.3% and 5.7%, respectively. Moreover, the PCC, Spearman, CI, and R2 exhibited improvements of 3.2%, 3.5%, 1.6% and 7.4%, respectively.
Figure 3.
The visualization results of drug–target binding affinity by a bidirectional self-attention mechanism for explaining the effectiveness of S2DTA. In the upper row, we present the targets of amino acids with higher drug interaction scores. In the middle row, we show the top 10 amino acids (depicted in purple) identified by four drugs (PubChem ID: 43158872, 444128, 44629533, and 53308636) within the pockets of their respective targets (PDBID: 3AO4, 1A30, 2QNQ, and 3O9I). The statistical findings reveal that the top 5, top 10, and top 20 amino acids within the pockets account for 13.7%, 12.8%, and 12.4%, respectively. The lower row presents the visualization results of the top 5 atoms (represented by the purple logo) with the highest scores in the interactions between the amino acids constituting the targets and the corresponding drugs. Statistical analysis was performed on the top 3, top 5, and top 8 atoms in the attention score matrices. These atoms accounted for 48.6%, 56%, and 64.8% of the total number of atoms in the drugs, respectively.
Figure 3.
The visualization results of drug–target binding affinity by a bidirectional self-attention mechanism for explaining the effectiveness of S2DTA. In the upper row, we present the targets of amino acids with higher drug interaction scores. In the middle row, we show the top 10 amino acids (depicted in purple) identified by four drugs (PubChem ID: 43158872, 444128, 44629533, and 53308636) within the pockets of their respective targets (PDBID: 3AO4, 1A30, 2QNQ, and 3O9I). The statistical findings reveal that the top 5, top 10, and top 20 amino acids within the pockets account for 13.7%, 12.8%, and 12.4%, respectively. The lower row presents the visualization results of the top 5 atoms (represented by the purple logo) with the highest scores in the interactions between the amino acids constituting the targets and the corresponding drugs. Statistical analysis was performed on the top 3, top 5, and top 8 atoms in the attention score matrices. These atoms accounted for 48.6%, 56%, and 64.8% of the total number of atoms in the drugs, respectively.
![Molecules 28 08005 g003]()
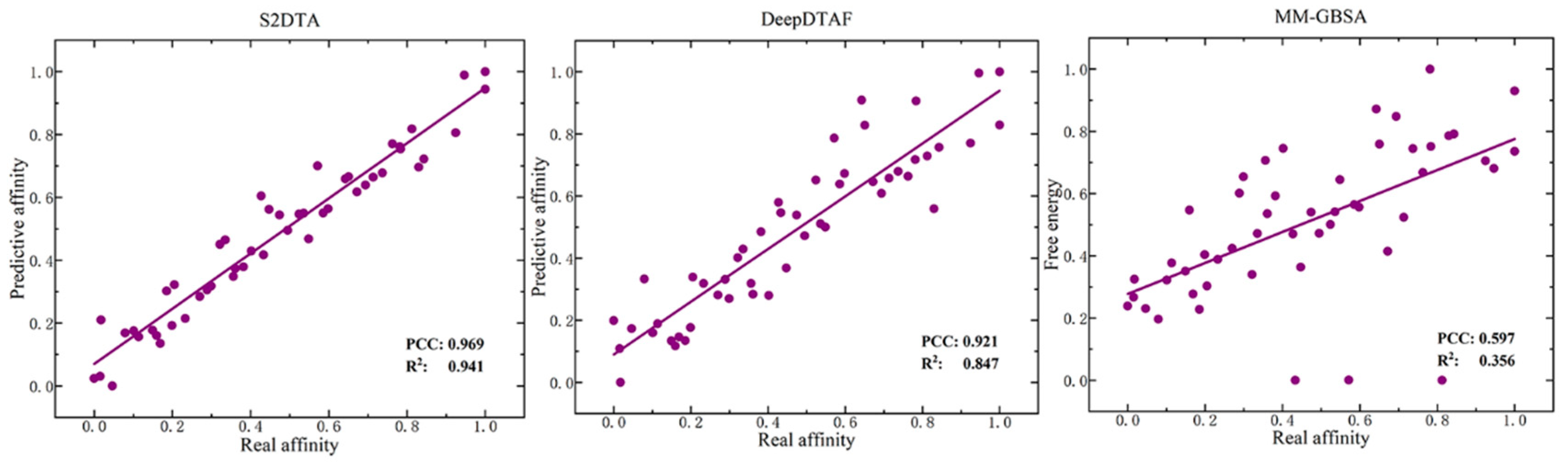
Figure 4.
The correlation analysis between the real affinity values and predictive values of S2DTA, DeepDTAF, and MM-GBSA/PBSA. The results show that the real affinity values of the selected samples exhibited a strong linear relationship with the predictive affinity values of both S2DTA (PCC = 0.969, R2 = 0.941) and DeepDTAF (PCC = 0.921, R2 = 0.847). Notably, S2DTA showed slightly superior performance compared to DeepDTAF. In contrast, we observed that the real affinity values of the selected samples did not align well with the free energy predicted by MM-GBSA/PBSA (PCC = 0.597, R2 = 0.356) in terms of a linear relationship, and its predictive values exhibited a weak correlation with the real affinity values.
Figure 4.
The correlation analysis between the real affinity values and predictive values of S2DTA, DeepDTAF, and MM-GBSA/PBSA. The results show that the real affinity values of the selected samples exhibited a strong linear relationship with the predictive affinity values of both S2DTA (PCC = 0.969, R2 = 0.941) and DeepDTAF (PCC = 0.921, R2 = 0.847). Notably, S2DTA showed slightly superior performance compared to DeepDTAF. In contrast, we observed that the real affinity values of the selected samples did not align well with the free energy predicted by MM-GBSA/PBSA (PCC = 0.597, R2 = 0.356) in terms of a linear relationship, and its predictive values exhibited a weak correlation with the real affinity values.
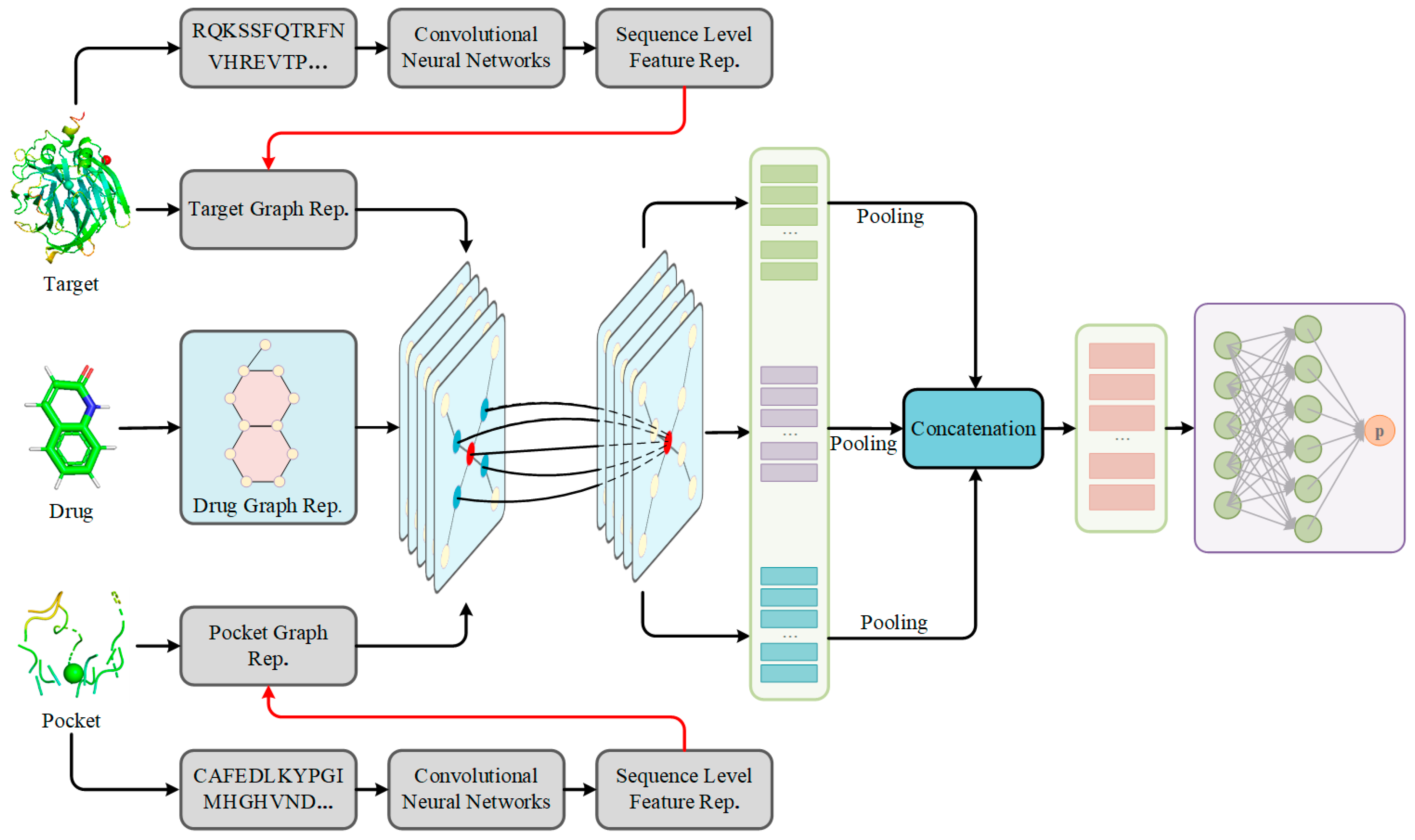
Figure 5.
The architecture of S2DTA. S2DTA is an advanced model designed to predict drug–target affinity (DTA) by leveraging sequence features of drug SMILES, targets, and pockets, and their corresponding structural features. This model comprises four essential modules: (1) The data input module was responsible for representing the sequence and structural data of drugs, targets, and pockets. (2) Within the sequence learning module, a 1D-CNN layer was employed to extract semantic features from both targets and pockets, based on their sequence data. (3) In the structure learning module, three independent Graph Convolutional Networks (GCNs) were employed to extract high-level features from the vertices present in the graph-based representation of drugs, targets, and pockets. (4) The feature fusion module encompasses a two-layer fully connected (FC) network, which played a pivotal role in predicting DTA by integrating the extracted features.
Figure 5.
The architecture of S2DTA. S2DTA is an advanced model designed to predict drug–target affinity (DTA) by leveraging sequence features of drug SMILES, targets, and pockets, and their corresponding structural features. This model comprises four essential modules: (1) The data input module was responsible for representing the sequence and structural data of drugs, targets, and pockets. (2) Within the sequence learning module, a 1D-CNN layer was employed to extract semantic features from both targets and pockets, based on their sequence data. (3) In the structure learning module, three independent Graph Convolutional Networks (GCNs) were employed to extract high-level features from the vertices present in the graph-based representation of drugs, targets, and pockets. (4) The feature fusion module encompasses a two-layer fully connected (FC) network, which played a pivotal role in predicting DTA by integrating the extracted features.
Table 1.
Performance of machine learning and deep learning models using only sequence knowledge compared to graph-based models fusing structure knowledge.
Table 1.
Performance of machine learning and deep learning models using only sequence knowledge compared to graph-based models fusing structure knowledge.
| Category | Model | Feature Representation | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| Machine learning models | RR | label encoding | 1.471 | 1.864 | 0.515 | 0.521 | 0.685 | 0.261 |
| KNN | label encoding | 1.377 | 1.732 | 0.604 | 0.565 | 0.703 | 0.362 |
| DT | label encoding | 1.409 | 1.882 | 0.590 | 0.577 | 0.707 | 0.247 |
| SVR | label encoding | 1.749 | 2.150 | 0.156 | 0.231 | 0.527 | 0.017 |
| RF | label encoding | 1.221 | 1.527 | 0.726 | 0.713 | 0.760 | 0.504 |
| Sequence-based deep learning models | 1D-CNN | label encoding | 1.569 | 1.938 | 0.535 | 0.531 | 0.683 | 0.201 |
| one-hot encoding | 1.159 | 1.473 | 0.752 | 0.741 | 0.776 | 0.539 |
| RNN | label encoding | 1.815 | 2.227 | 0.175 | 0.329 | 0.611 | −0.054 |
| one-hot encoding | 1.669 | 2.045 | 0.459 | 0.460 | 0.657 | 0.111 |
| Structure-based deep learning models | GraphSAGE | label encoding | 1.288 | 1.599 | 0.675 | 0.664 | 0.739 | 0.449 |
| one-hot encoding | 1.127 | 1.417 | 0.758 | 0.745 | 0.778 | 0.573 |
Table 2.
Performance comparison of different graph-based models.
Table 2.
Performance comparison of different graph-based models.
| Model | Feature Representation | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| GCN | one-hot encoding | 1.393 | 1.743 | 0.601 | 0.589 | 0.707 | 0.354 |
| GIN | one-hot encoding | 1.524 | 1.842 | 0.532 | 0.549 | 0.692 | 0.279 |
| GAT | one-hot encoding | 1.708 | 2.091 | 0.296 | 0.313 | 0.606 | 0.070 |
| GraphSAGE | one-hot encoding | 1.127 | 1.417 | 0.758 | 0.745 | 0.778 | 0.573 |
Table 3.
Performance comparison of GraphSAGE with different feature representations.
Table 3.
Performance comparison of GraphSAGE with different feature representations.
| Feature Representation | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| one-hot encoding | 1.127 | 1.417 | 0.758 | 0.745 | 0.778 | 0.573 |
| physical–chemical properties | 1.444 | 1.770 | 0.583 | 0.585 | 0.706 | 0.334 |
| PSSM | 1.236 | 1.526 | 0.722 | 0.713 | 0.761 | 0.505 |
| HMM | 1.342 | 1.664 | 0.644 | 0.639 | 0.729 | 0.411 |
| semantic feature (Protbert) | 1.139 | 1.485 | 0.734 | 0.720 | 0.768 | 0.531 |
| semantic feature (CNN) | 1.158 | 1.477 | 0.733 | 0.723 | 0.769 | 0.536 |
| semantic feature (Word2Vec) | 1.136 | 1.411 | 0.760 | 0.746 | 0.777 | 0.577 |
Table 4.
Performance comparison of GraphSAGE with targets or combining the knowledge of pockets.
Table 4.
Performance comparison of GraphSAGE with targets or combining the knowledge of pockets.
| Name | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| Targets | 1.127 | 1.417 | 0.758 | 0.745 | 0.778 | 0.573 |
| Pockets | 1.101 | 1.420 | 0.759 | 0.755 | 0.782 | 0.571 |
| Targets + Pockets | 1.080 | 1.377 | 0.776 | 0.773 | 0.791 | 0.597 |
Table 5.
Performance comparison of heterogeneous models with fused sequence and structural knowledge.
Table 5.
Performance comparison of heterogeneous models with fused sequence and structural knowledge.
| Sequence Processing Model | Structural Model | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| LSTM | GraphSAGE | 1.084 | 1.397 | 0.766 | 0.749 | 0.781 | 0.585 |
| BiLSTM | 1.100 | 1.399 | 0.764 | 0.765 | 0.785 | 0.584 |
| CNN + LSTM | 1.075 | 1.370 | 0.776 | 0.771 | 0.789 | 0.601 |
| CNN | 0.990 | 1.299 | 0.801 | 0.800 | 0.804 | 0.641 |
Table 6.
Performance comparison of S2DTA with three state-of-the-art methods.
Table 6.
Performance comparison of S2DTA with three state-of-the-art methods.
| Model | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| DeepDTA | 1.323 | 1.641 | 0.667 | 0.681 | 0.744 | 0.428 |
| GraphDTA | 1.367 | 1.715 | 0.618 | 0.612 | 0.717 | 0.374 |
| DeepDTAF | 1.327 | 1.625 | 0.670 | 0.654 | 0.734 | 0.429 |
| S2DTA | 0.990 | 1.299 | 0.801 | 0.800 | 0.804 | 0.641 |
Table 7.
Performance comparison of S2DTA based on the real and predictive structures of targets.
Table 7.
Performance comparison of S2DTA based on the real and predictive structures of targets.
| Model | Structures of Targets | MAE | RMSE | PCC | Spearman | CI | R2 |
|---|
| S2DTA | Real structures | 1.046 | 1.316 | 0.792 | 0.787 | 0.799 | 0.627 |
| Predictive structures | 1.133 | 1.432 | 0.748 | 0.749 | 0.780 | 0.558 |
Table 8.
Data statistics for interpretability analysis of S2DTA.
Table 8.
Data statistics for interpretability analysis of S2DTA.
| Atoms of Drugs | Amino Acids of Targets |
|---|
| Top 5 | Top 10 | Top 20 | Top 3 | Top 5 | Top 8 |
|---|
| 13.7% | 12.8% | 12.4% | 48.6% | 56% | 64.8% |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}