
Synthesis and Antiproliferative Potential of Thiazole and 4-Thiazolidinone Containing Motifs as Dual Inhibitors of EGFR and BRAFV600E


 , , , , and
, , , , and
Abstract
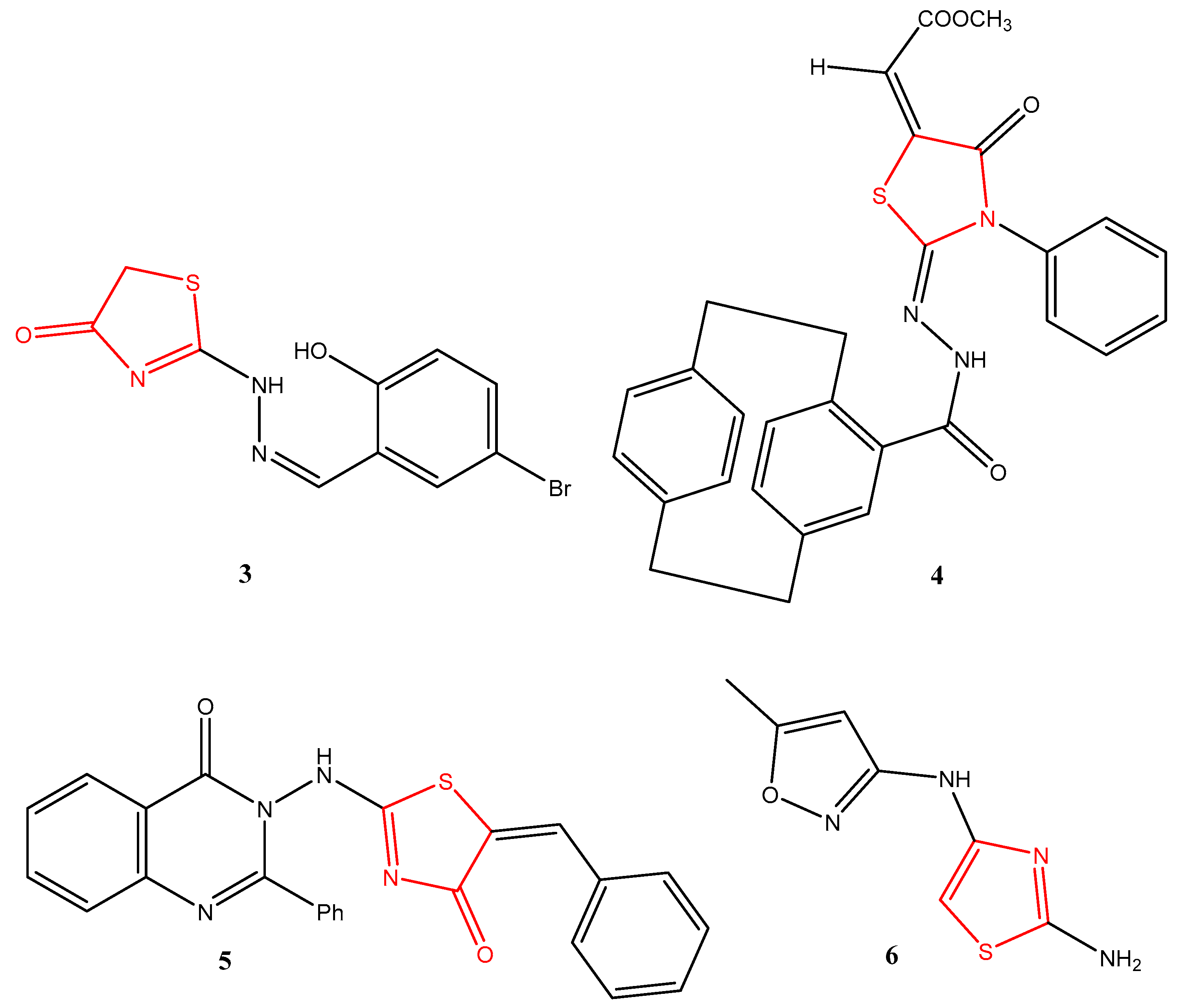
:1. Introduction
2. Results and Discussion
2.1. Biology
2.1.1. Cell Viability Assay
2.1.2. Antiproliferative Assay
2.1.3. EGFR Inhibitory Assay
2.1.4. BRAFV600E Inhibitory Assay
2.1.5. Docking Study
3. Materials and Methods
4. Biology Section
4.1. Assay for the Effect of 9a–f and 12a–f on Cell Viability
4.2. Assay for Antiproliferative Activity
4.3. Assay for EGFR Inhibitory Effect
4.4. Assay for BRAFV600E Inhibitory Effect
5. Protocol of Docking Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Who Report on Cancer: Setting Priorities, Investing Wisely and Providing Care for All; Global Report; World Health Organization: Geneva, Switzerland, 2020; pp. 1–160. [Google Scholar]
- Youssif, B.G.; Abdelrahman, M.H.; Abdelazeem, A.H.; Ibrahim, H.M.; Salem, O.I.; Mohamed, M.F.; Treambleau, L.; Bukhari, S.N.A. Design, synthesis, mechanistic and histopathological studies of small-molecules of novel indole-2-carboxamides and pyrazino [1, 2-a] indol-1 (2h)-ones as potential anticancer agents effecting the reactive oxygen species production. Eur. J. Med. Chem. 2018, 146, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.M.; Olson, K.; Catton, P.; Catton, C.N.; Fleshner, N.E.; Krzyzanowska, M.K.; McCready, D.R.; Wong, R.K.; Jiang, H.; Howell, D. Cancer-related fatigue and associated disability in post-treatment cancer survivors. J. Cancer Surviv. 2016, 10, 51–61. [Google Scholar] [CrossRef]
- Riechelmann, R.P.; Krzyzanowska, M.K. Drug interactions and oncological outcomes: A hidden adversary. Ecancermedicalscience 2019, 13, ed88. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.; Youssif, B.G. Design and synthesis of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as antiproliferative egfr and brafv600e dual inhibitors. Bioorganic Chem. 2020, 104, 104260. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef] [PubMed]
- Al-Wahaibi, L.H.; Mahmoud, M.A.; Mostafa, Y.A.; Raslan, A.E.; Youssif, B.G. Novel piperine-carboximidamide hybrids: Design, synthesis, and antiproliferative activity via a multi-targeted inhibitory pathway. J. Enzym. Inhib. Med. Chem. 2023, 38, 376–386. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Cheng, W.-L.; Feng, P.-H.; Lee, K.-Y.; Chen, K.-Y.; Sun, W.-L.; Van Hiep, N.; Luo, C.-S.; Wu, S.-M. The role of ereg/egfr pathway in tumor progression. Int. J. Mol. Sci. 2021, 22, 12828. [Google Scholar] [CrossRef]
- Gomaa, H.A.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.; Mohamed, F.A.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L. Optimization and sar investigation of novel 2, 3-dihydropyrazino [1, 2-a] indole-1, 4-dione derivatives as egfr and brafv600e dual inhibitors with potent antiproliferative and antioxidant activities. Bioorganic Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Yao, Y.-m.M.; Donoho, G.P.; Iversen, P.W.; Zhang, Y.; Van Horn, R.D.; Forest, A.; Novosiadly, R.D.; Webster, Y.W.; Ebert, P.; Bray, S. Mouse pdx trial suggests synergy of concurrent inhibition of raf and egfr in colorectal cancer with braf or kras mutations. Clin. Cancer Res. 2017, 23, 5547–5560. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, J.; Wang, Y.; Wang, X.; Wang, Y.; Zhang, Z.; Shuai, W.; Wang, G.; Chen, J.; Wang, C. Development of dual inhibitors targeting epidermal growth factor receptor in cancer therapy. J. Med. Chem. 2022, 65, 5149–5183. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Mohammed, A.F.; Abdelrahman, M.H.; Trembleau, L.; Youssif, B.G. Design, synthesis, and antiproliferative activity of new 5-chloro-indole-2-carboxylate and pyrrolo [3, 4-b] indol-3-one derivatives as potent inhibitors of egfrt790m/brafv600e pathways. Molecules 2023, 28, 1269. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, D.S.; Chawla, P.A.; Gurnule, W.B.; Shejul, S.K.; Bumbrah, G.S. An insight into synthesis and anticancer potential of thiazole and 4-thiazolidinone containing motifs. Curr. Org. Chem. 2021, 25, 819–841. [Google Scholar] [CrossRef]
- Sharma, P.C.; Bansal, K.K.; Sharma, A.; Sharma, D.; Deep, A. Thiazole-containing compounds as therapeutic targets for cancer therapy. Eur. J. Med. Chem. 2020, 188, 112016. [Google Scholar] [CrossRef] [PubMed]
- Kundenapally, R. Design Synthesis and Bioactivity of Benzimidazole Oxadiazole Thiazolidinone and Thiazole Containing 1 and 8 Naphthyridines. 2023. Available online: http://hdl.handle.net/20.500.14146/12567 (accessed on 1 November 2023).
- T Chhabria, M.; Patel, S.; Modi, P.; S Brahmkshatriya, P. Thiazole: A review on chemistry, synthesis and therapeutic importance of its derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Inza, S.; Aydillo, C.; Sanmartín, C.; Plano, D. Thiazole moiety: An interesting scaffold for developing new antitumoral compounds. In Heterocycles-Synthesis and Biological Activities; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Aly, A.A.; Bräse, S.; Hassan, A.A.; Mohamed, N.K.; Abd El-Haleem, L.E.; Nieger, M.; Morsy, N.M.; Abdelhafez, E.M. New paracyclophanylthiazoles with anti-leukemia activity: Design, synthesis, molecular docking, and mechanistic studies. Molecules 2020, 25, 3089. [Google Scholar] [CrossRef] [PubMed]
- Peng-Cheng, L.; Zhou, C.; Chen, J.; Liu, P.; Wang, K.; Mao, W.; Huan-Qiu, L.; Yang, Y.; Xiong, J.; Zhu, H. Design, synthesis and biological evaluation of thiazolidinone derivatives as potential egfr and her-2 kinase inhibitors. Bioorg. Med. Chem. Lett 2010, 18, 314–319. [Google Scholar] [CrossRef]
- Aziz, M.W.; Kamal, A.M.; Mohamed, K.O.; Elgendy, A.A. Design, synthesis and assessment of new series of quinazolinone derivatives as egfr inhibitors along with their cytotoxic evaluation against mcf7 and a549 cancer cell lines. Bioorganic Med. Chem. Lett. 2021, 41, 127987. [Google Scholar] [CrossRef]
- Walaa, S.; Mohamed, N.A.; Weam, S.; Nossier, E.S.; Mahmoud, K. Synthesis, molecular modeling studies and biological evaluation of novel pyrazole derivatives as antitumor and egfr inhibitors. Int. J. Pharm. Technol. 2016, 8, 25192–25209. [Google Scholar]
- Gomaa, H.A.; El-Sherief, H.A.; Hussein, S.; Gouda, A.M.; Salem, O.I.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G. Novel 1, 2, 4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorganic Chem. 2020, 105, 104369. [Google Scholar] [CrossRef] [PubMed]
- Marzouk, A.A.; Abdel-Aziz, S.A.; Abdelrahman, K.S.; Wanas, A.S.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design and synthesis of new 1, 6-dihydropyrimidin-2-thio derivatives targeting vegfr-2: Molecular docking and antiproliferative evaluation. Bioorganic Chem. 2020, 102, 104090. [Google Scholar] [CrossRef] [PubMed]
- Mekheimer, R.A.; Allam, S.M.; Al-Sheikh, M.A.; Moustafa, M.S.; Al-Mousawi, S.M.; Mostafa, Y.A.; Youssif, B.G.; Gomaa, H.A.; Hayallah, A.M.; Abdelaziz, M. Discovery of new pyrimido [5, 4-c] quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and adme studies. Bioorganic Chem. 2022, 121, 105693. [Google Scholar] [CrossRef]
- Mahmoud, M.A.; Mohammed, A.F.; Salem, O.I.; Gomaa, H.A.; Youssif, B.G. New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the egfr tyrosine kinase. Arch. Der Pharm. 2022, 355, 2200009. [Google Scholar] [CrossRef]
- Hisham, M.; Hassan, H.A.; Gomaa, H.A.; Youssif, B.G.; Hayallah, A.M.; Abdel-Aziz, M. Structure-based design, synthesis and antiproliferative action of new quinazoline-4-one/chalcone hybrids as egfr inhibitors. J. Mol. Struct. 2022, 1254, 132422. [Google Scholar] [CrossRef]
- Youssif, B.G.; Gouda, A.M.; Moustafa, A.H.; Abdelhamid, A.A.; Gomaa, H.A.; Kamal, I.; Marzouk, A.A. Design and synthesis of new triarylimidazole derivatives as dual inhibitors of brafv600e/p38α with potential antiproliferative activity. J. Mol. Struct. 2022, 1253, 132218. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Bokhtia, R.M.; Al-Mahmoudy, A.M.; Taher, E.S.; AlAwadh, M.A.; Elagawany, M.; Abdel-Aal, E.H.; Panda, S.; Gouda, A.M.; Asfour, H.Z. Design, synthesis and biological evaluation of novel 5-((substituted quinolin-3-yl/1-naphthyl) methylene)-3-substituted imidazolidin-2, 4-dione as hiv-1 fusion inhibitors. Bioorganic Chem. 2020, 99, 103782. [Google Scholar] [CrossRef]
- Shaykoon, M.S.; Marzouk, A.A.; Soltan, O.M.; Wanas, A.S.; Radwan, M.M.; Gouda, A.M.; Youssif, B.G.; Abdel-Aziz, M. Design, synthesis and antitrypanosomal activity of heteroaryl-based 1, 2, 4-triazole and 1, 3, 4-oxadiazole derivatives. Bioorganic Chem. 2020, 100, 103933. [Google Scholar] [CrossRef]
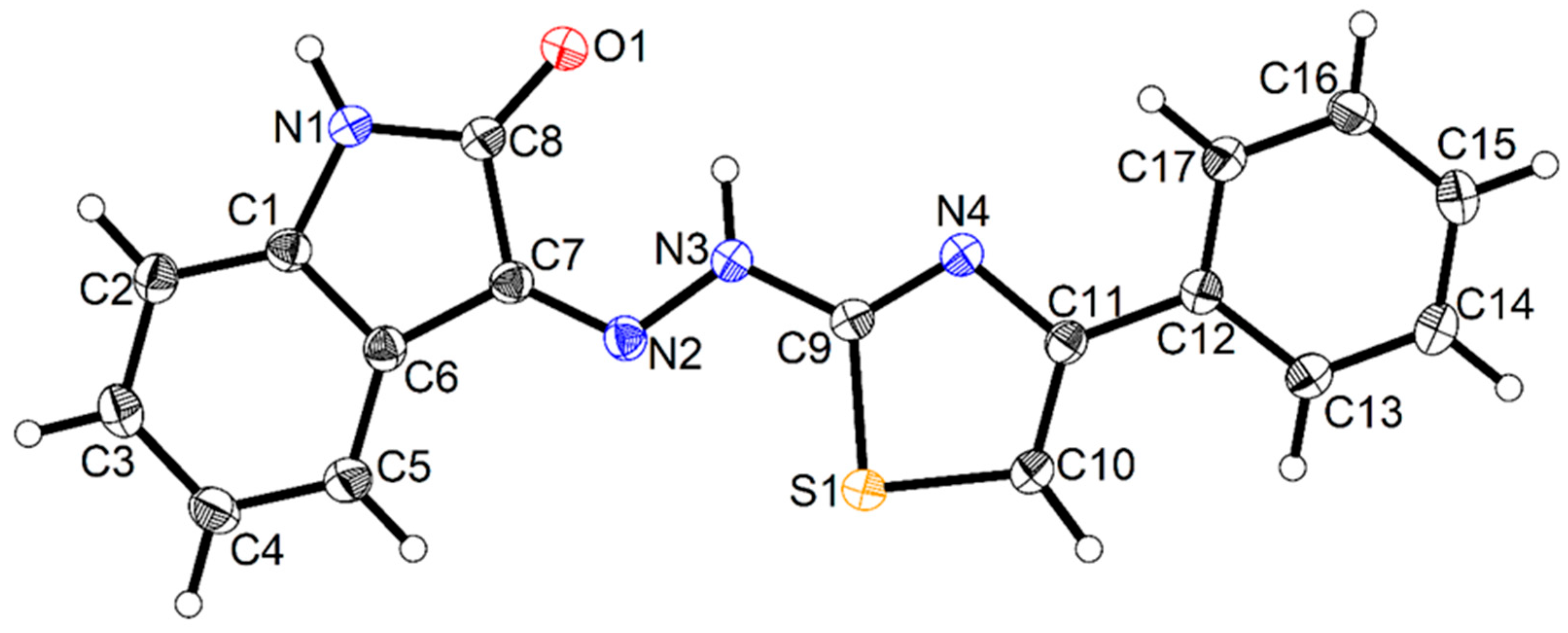
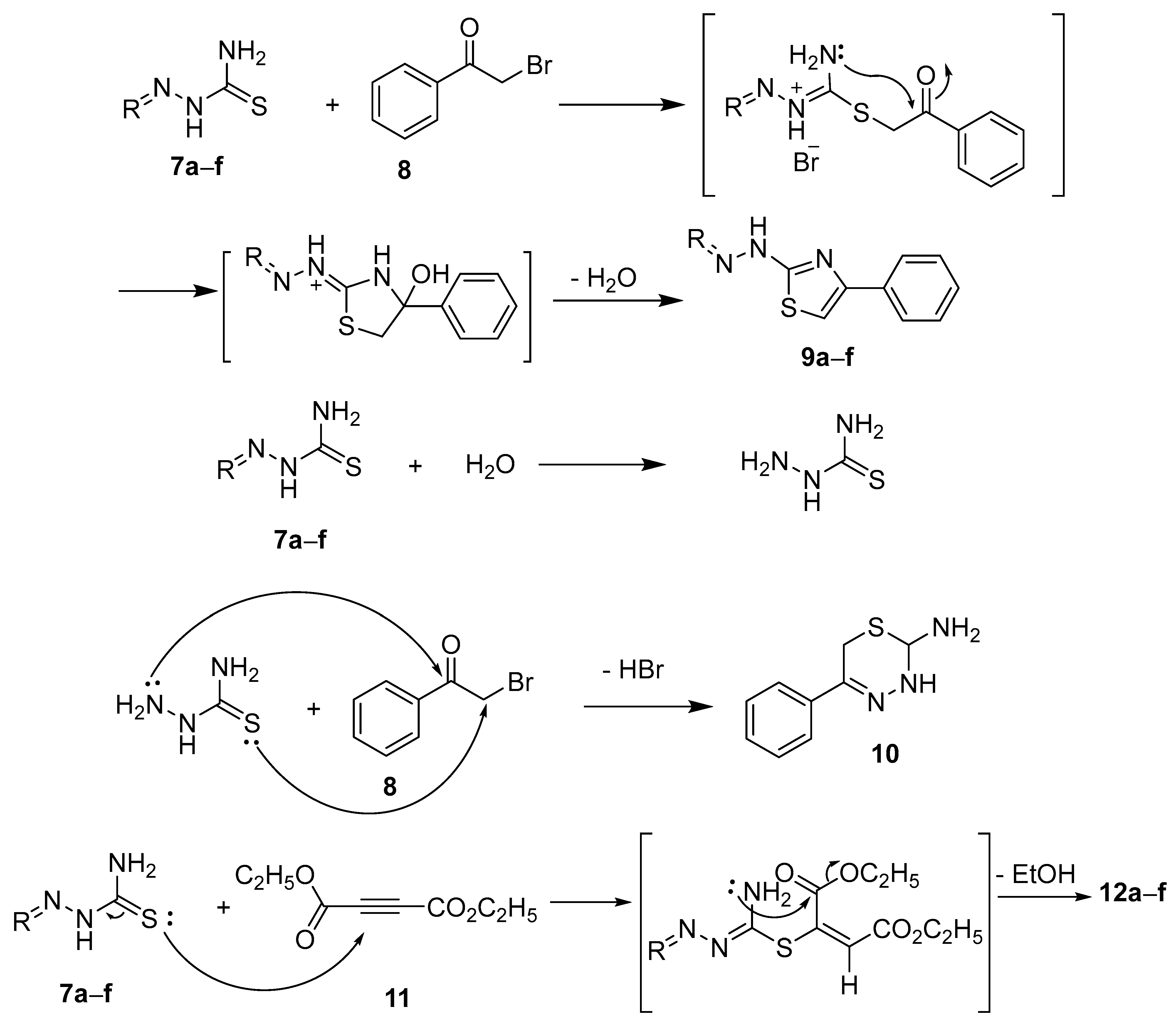
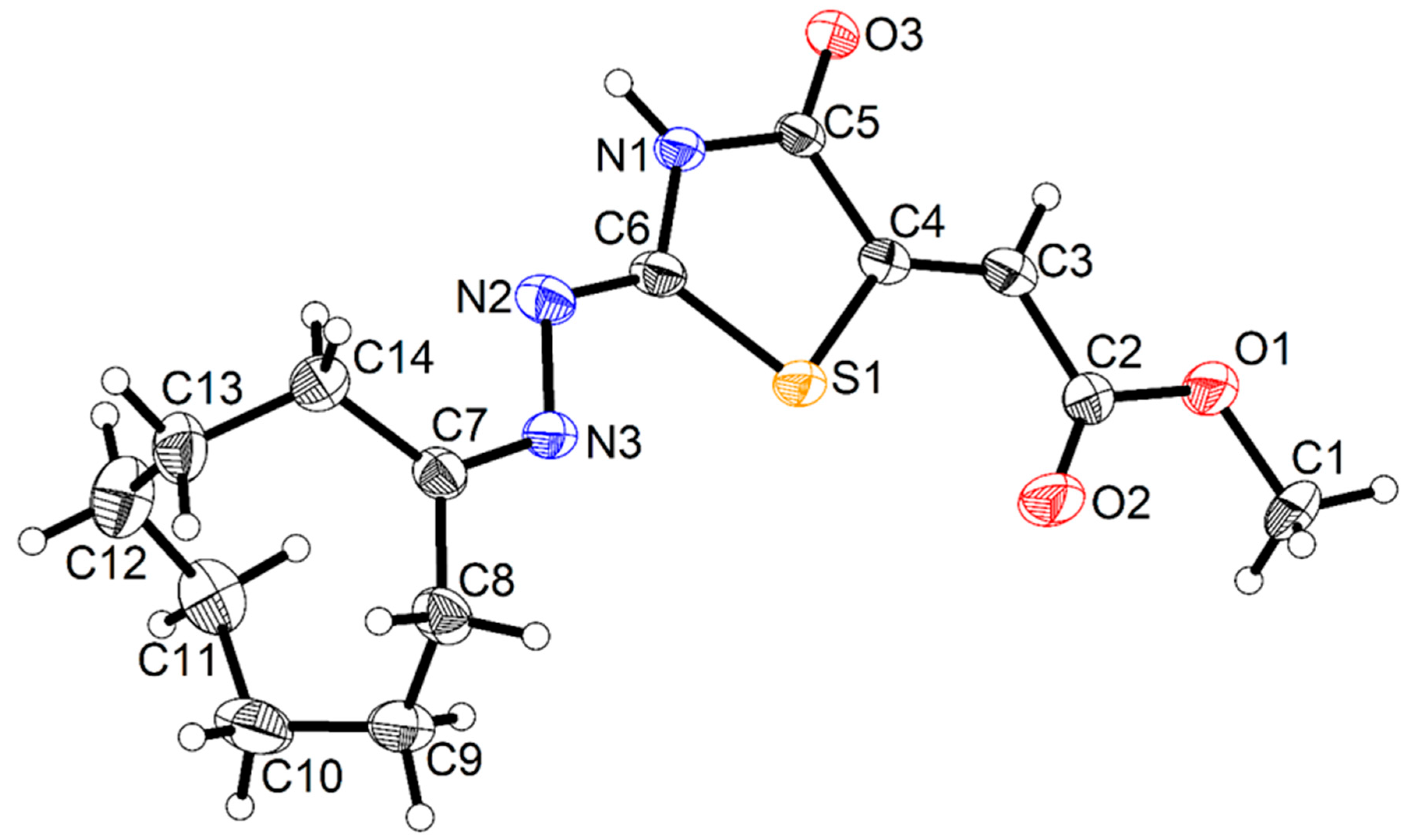
- Hassan, A.A.; Mohamed, S.K.; Mohamed, N.K.; El-Shaieb, K.M.; Abdel-Aziz, A.T.; Mague, J.T.; Akkurt, M. The behavior of (cyclic-alkylidene) hydrazinecarbothioamides in cyclization with dimethyl acetylenedicarboxylate. J. Heterocycl. Chem. 2017, 54, 2043–2053. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (nM) | ||||
|---|---|---|---|---|---|---|
| A-549 | MCF-7 | Panc-1 | HT-29 | Average (GI50) | ||
| 9a | 90 | 76 ± 6 | 80 ± 7 | 78 ± 6 | 78 ± 6 | 78 |
| 9b | 89 | 62 ± 5 | 66 ± 5 | 64 ± 5 | 62 ± 5 | 64 |
| 9c | 92 | 34 ± 3 | 37 ± 3 | 35± 3 | 34 ± 3 | 35 |
| 9d | 90 | 57 ± 5 | 61 ± 5 | 59± 5 | 58 ± 5 | 59 |
| 9e | 93 | 48 ± 4 | 52 ± 4 | 50 ± 4 | 50 ± 4 | 50 |
| 9f | 92 | 42 ± 3 | 46 ± 4 | 42 ± 3 | 45 ± 4 | 44 |
| 12a | 91 | 82 ± 7 | 88 ± 8 | 82 ± 7 | 84 ± 7 | 84 |
| 12b | 89 | 65 ± 5 | 70 ± 6 | 66 ±5 | 66 ± 5 | 67 |
| 12c | 94 | 53 ± 4 | 57 ± 4 | 54 ± 4 | 55 ± 5 | 55 |
| 12d | 90 | 36 ± 3 | 40 ± 3 | 36 ± 3 | 38 ± 3 | 38 |
| 12e | 91 | 40 ± 3 | 44 ± 3 | 40 ± 3 | 42 ± 3 | 42 |
| 12f | 89 | 45 ± 4 | 50 ± 4 | 46 ± 4 | 46 ± 4 | 47 |
| Erlotinib | ND | 30 ± 3 | 40 ± 3 | 30 ± 3 | 30 ± 3 | 33 |
| Compd. | EGFR Inhibition IC50 ± SEM (nM) | BRAFV600E Inhibition IC50 ± SEM (nM) |
|---|---|---|
| 9c | 86 ± 5 | 94 ± 6 |
| 9f | 97 ± 6 | 117 ± 8 |
| 12d | 89 ± 5 | 98 ± 6 |
| 12e | 91 ± 6 | 105 ± 7 |
| 12f | 100 ± 6 | 125 ± 9 |
| Erlotinib | 80 ± 5 | 60 ± 5 |
| 9c | 9f | 12d | 12e | 12f | Erlotinib | |
|---|---|---|---|---|---|---|
| EGFR (PDB ID: 1M17) | ||||||
| S (kcal/mol) | −6.97 | −6.09 | −6.69 | −6.20 | −6.01 | −7.38 |
| RMSD (Å) | 1.57 | 1.96 | 1.77 | 0.91 | 1.48 | 0.75 |
| Amino acids residues binding interactions and their bond length (Å) | 2Asp 831 (3.06, 3.90) c, Cys 773 (4.34) b | Cys 773 (4.25) b | Asp 831 (3.14) c | 2Lys 721 (3.14, 2.96) a, Gly 695 (4.02, 4.54) b, Val 702 (4.12) b | Asp 831 (3.39) a, HOH 10 (2.86) a, 2 Val 702 (4.11, 4.52) b | Met 769 (3.26) a, HOH 10 (3.11) a, Lys 721 (4.55) b |
| 9c | 9f | 12d | 12e | 12f | Erlotinib | |
|---|---|---|---|---|---|---|
| BRAFV600E (PDB ID: 5JRQ) | ||||||
| S (kcal/mol) | −7.90 | −7.03 | −7.75 | −7.33 | −6.74 | −8.04 |
| RMSD (Å) | 0.95 | 1.66 | 1.52 | 1.44 | 1.08 | 1.33 |
| Amino acids residues binding interactions and their bond length (Å) | Gln 530 (4.45) c, Cys 532 (4.11) c | Cys 532 (3.95) c | Lys 483 (2.87) a, Asp 594 (2.65) a | Lys 483 (3.45) a, Asp 594 (2.68) a | Lys 483 (3.34) a, Asp 594 (2.98) a, Phe 583 (3.93) b | Phe 583 (3.98) b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, A.A.; Mohamed, N.K.; Aly, A.A.; Ramadan, M.; Gomaa, H.A.M.; Abdel-Aziz, A.T.; Youssif, B.G.M.; Bräse, S.; Fuhr, O. Synthesis and Antiproliferative Potential of Thiazole and 4-Thiazolidinone Containing Motifs as Dual Inhibitors of EGFR and BRAFV600E. Molecules 2023, 28, 7951. https://doi.org/10.3390/molecules28247951
Hassan AA, Mohamed NK, Aly AA, Ramadan M, Gomaa HAM, Abdel-Aziz AT, Youssif BGM, Bräse S, Fuhr O. Synthesis and Antiproliferative Potential of Thiazole and 4-Thiazolidinone Containing Motifs as Dual Inhibitors of EGFR and BRAFV600E. Molecules. 2023; 28(24):7951. https://doi.org/10.3390/molecules28247951
Chicago/Turabian StyleHassan, Alaa A., Nasr K. Mohamed, Ashraf A. Aly, Mohamed Ramadan, Hesham A. M. Gomaa, Ahmed T. Abdel-Aziz, Bahaa G. M. Youssif, Stefan Bräse, and Olaf Fuhr. 2023. "Synthesis and Antiproliferative Potential of Thiazole and 4-Thiazolidinone Containing Motifs as Dual Inhibitors of EGFR and BRAFV600E" Molecules 28, no. 24: 7951. https://doi.org/10.3390/molecules28247951
APA StyleHassan, A. A., Mohamed, N. K., Aly, A. A., Ramadan, M., Gomaa, H. A. M., Abdel-Aziz, A. T., Youssif, B. G. M., Bräse, S., & Fuhr, O. (2023). Synthesis and Antiproliferative Potential of Thiazole and 4-Thiazolidinone Containing Motifs as Dual Inhibitors of EGFR and BRAFV600E. Molecules, 28(24), 7951. https://doi.org/10.3390/molecules28247951