

Integrative Analysis of Machine Learning and Molecule Docking Simulations for Ischemic Stroke Diagnosis and Therapy

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Biomarkers and Nomogram for Ischemic Stroke Diagnosis
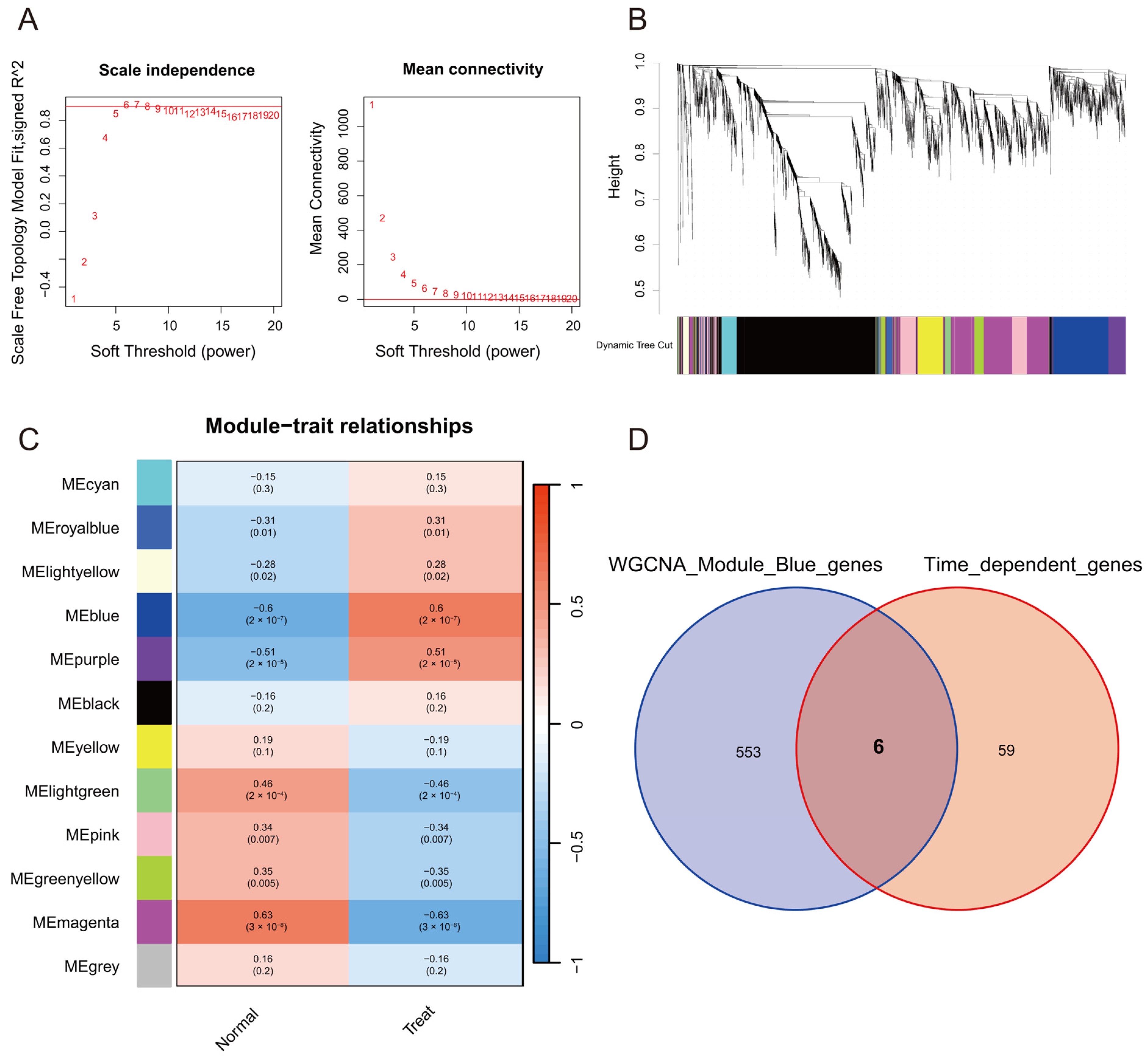
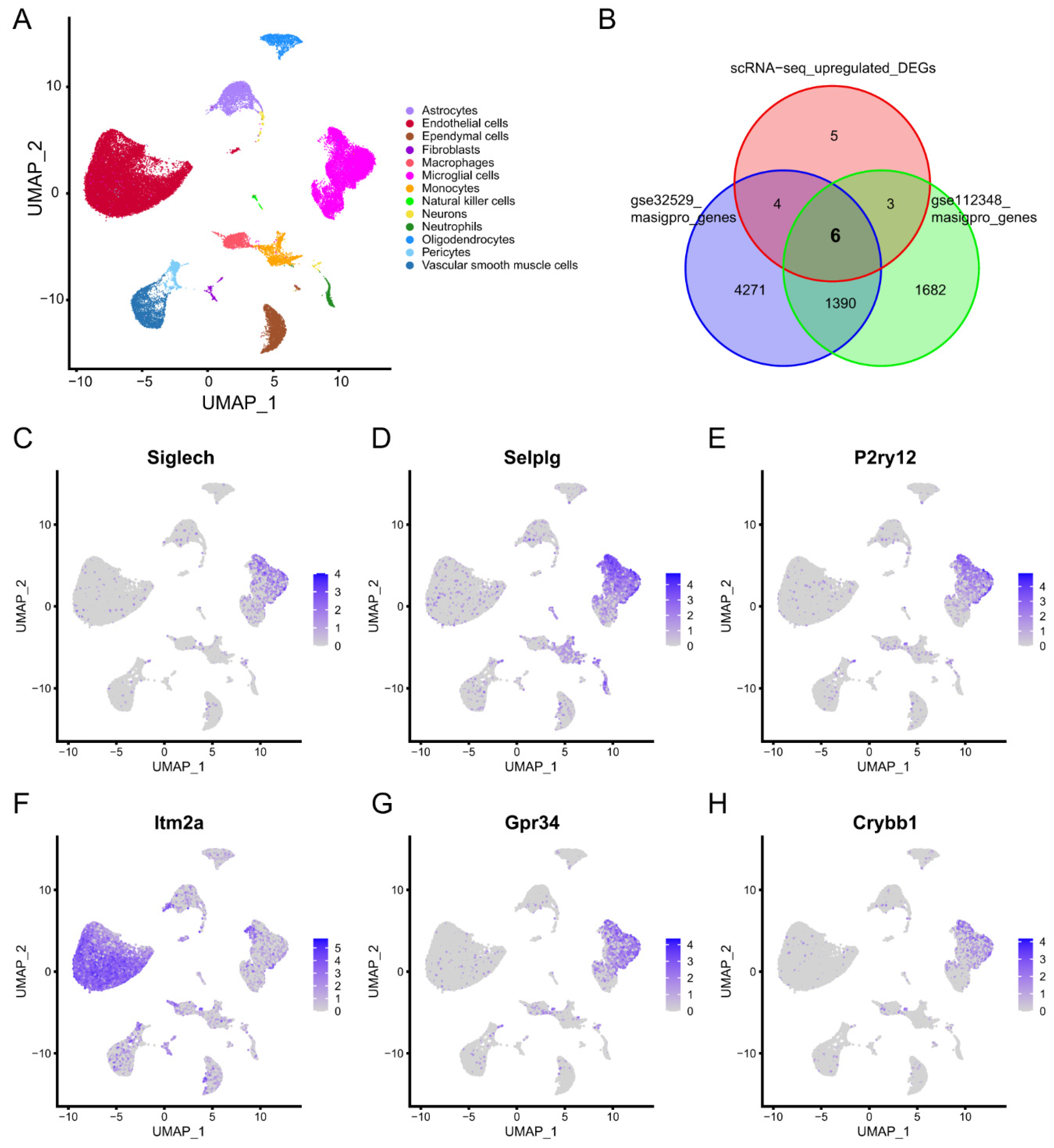
2.1.1. Weighted Correlation Network Analysis and Time-Dependent Genes in the Peripheral Blood of Ischemic Stroke
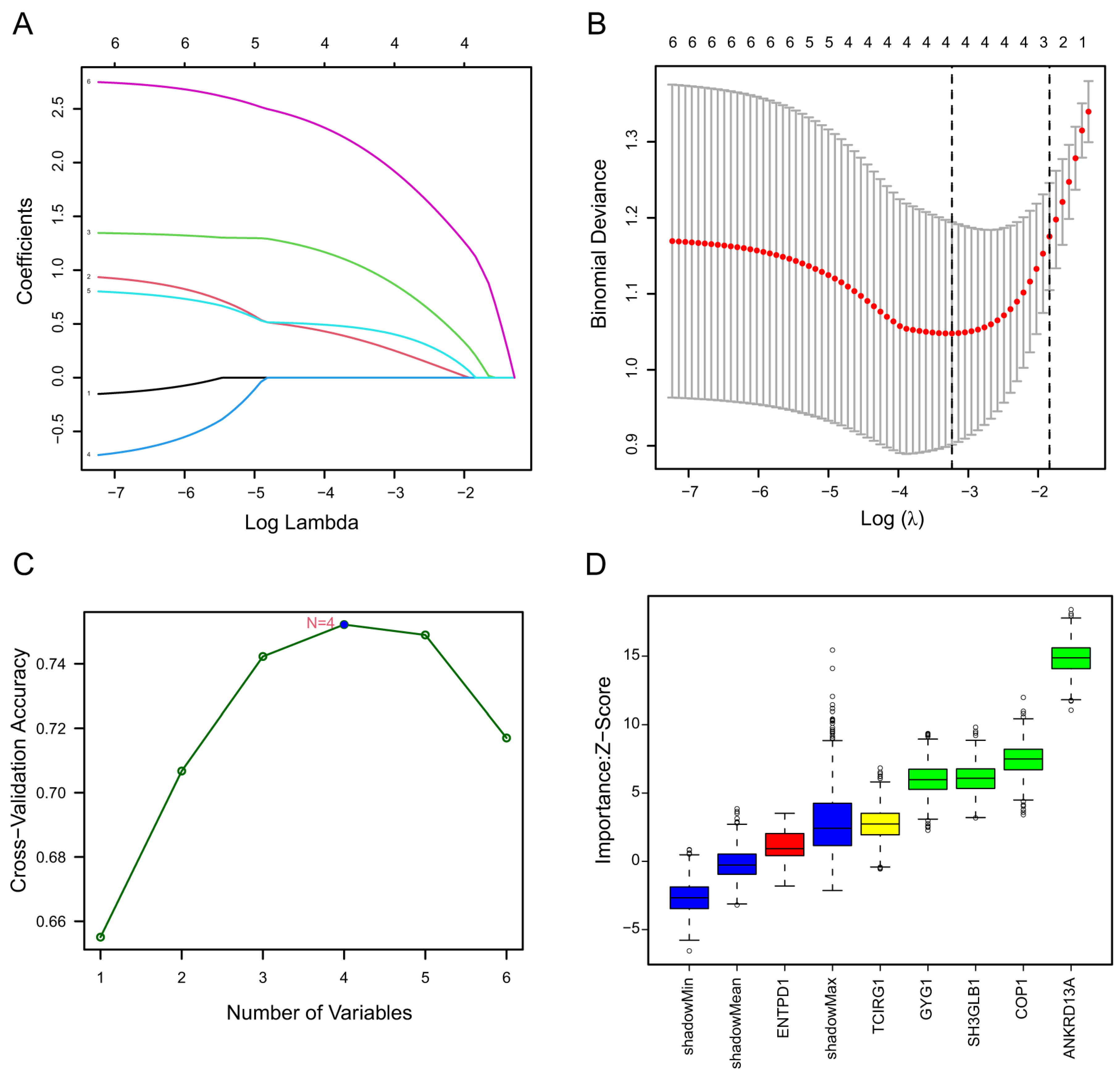
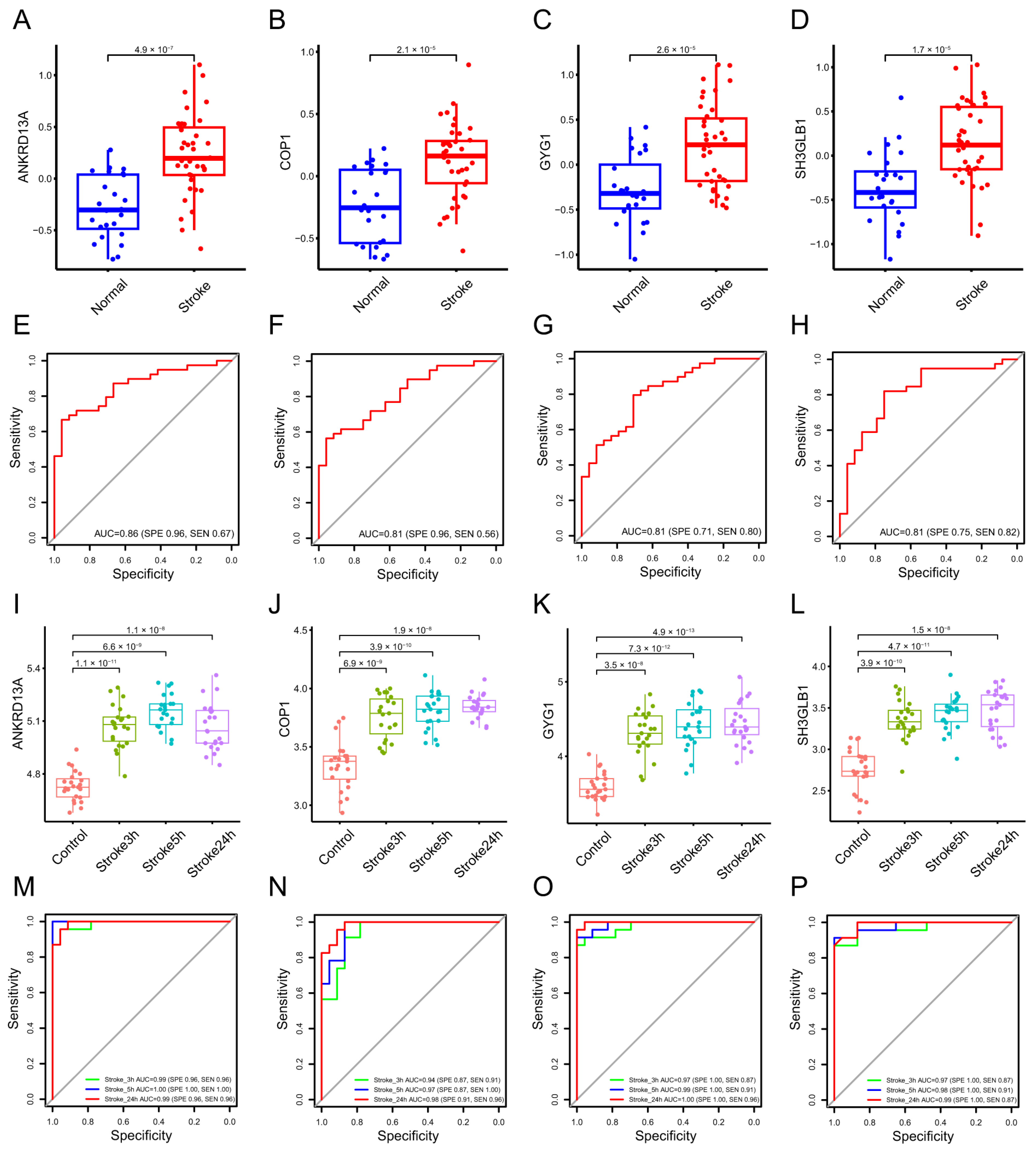
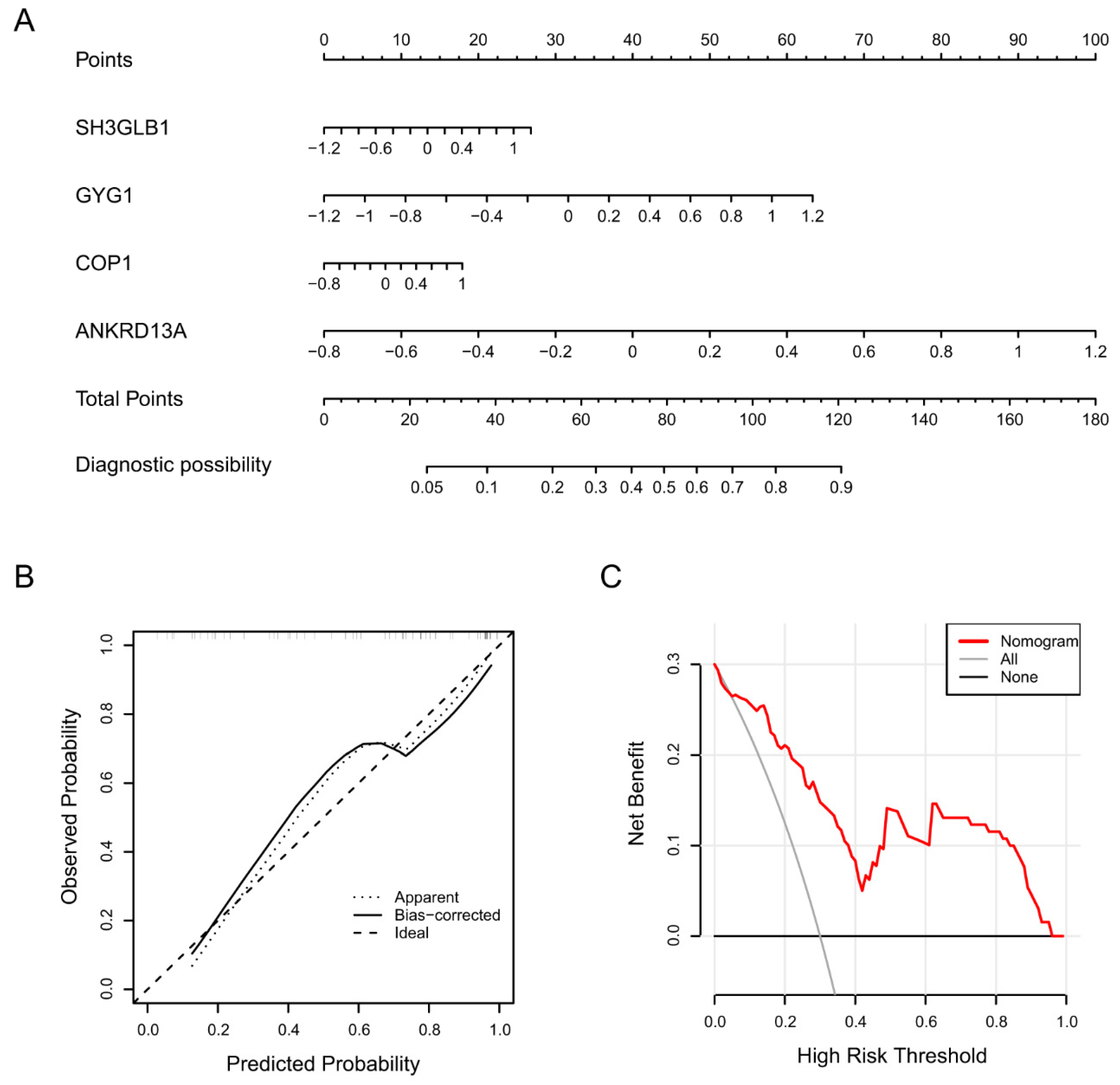
2.1.2. Identification of Diagnostic Biomarkers and Construction of Clinical Nomogram for Ischemic Stroke
2.2. The Role of Microglia Subclusters in Ischemic Stroke
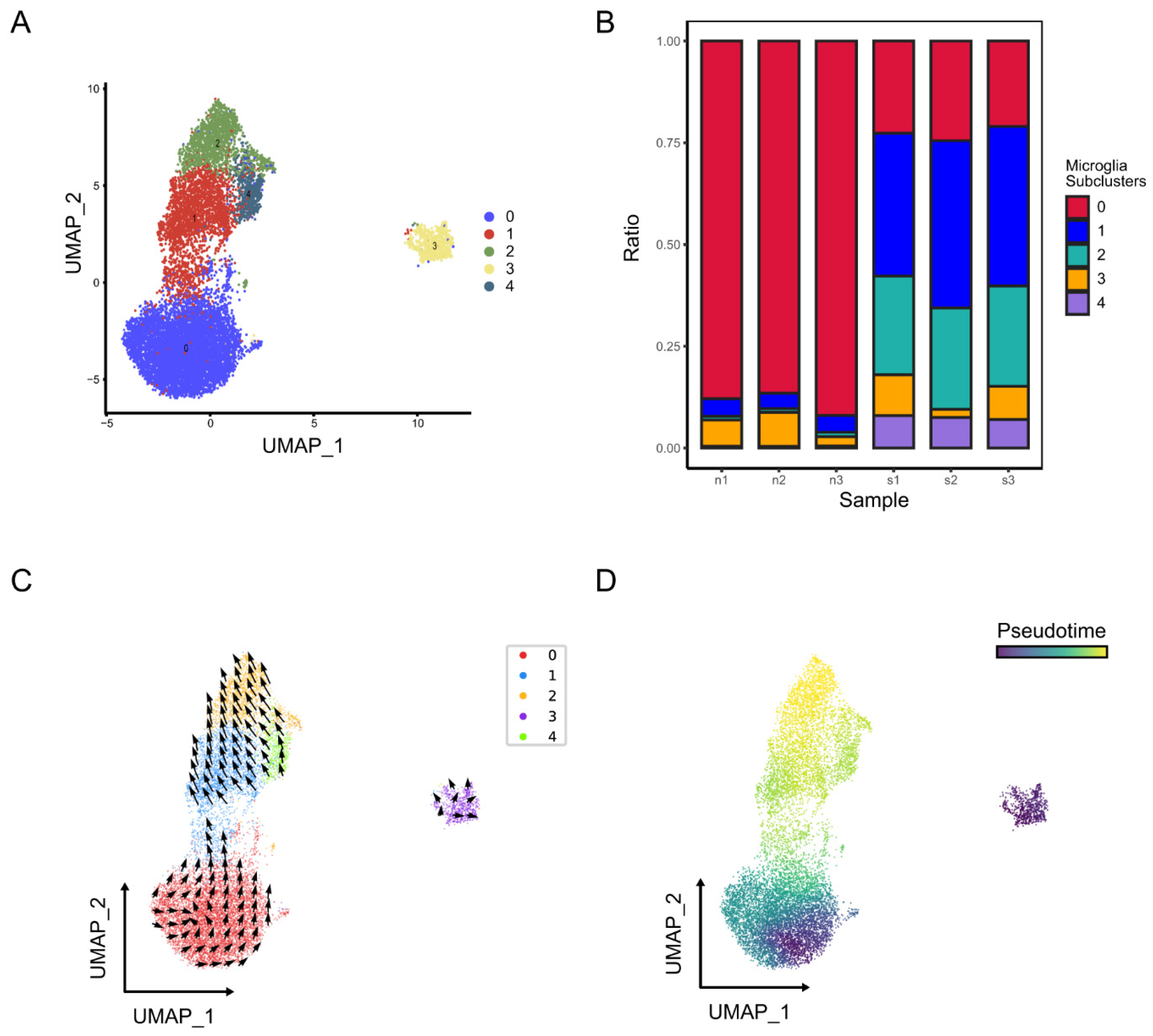
2.2.1. Identification of Microglia Subclusters and Their Dynamic Changes in Ischemic Stroke
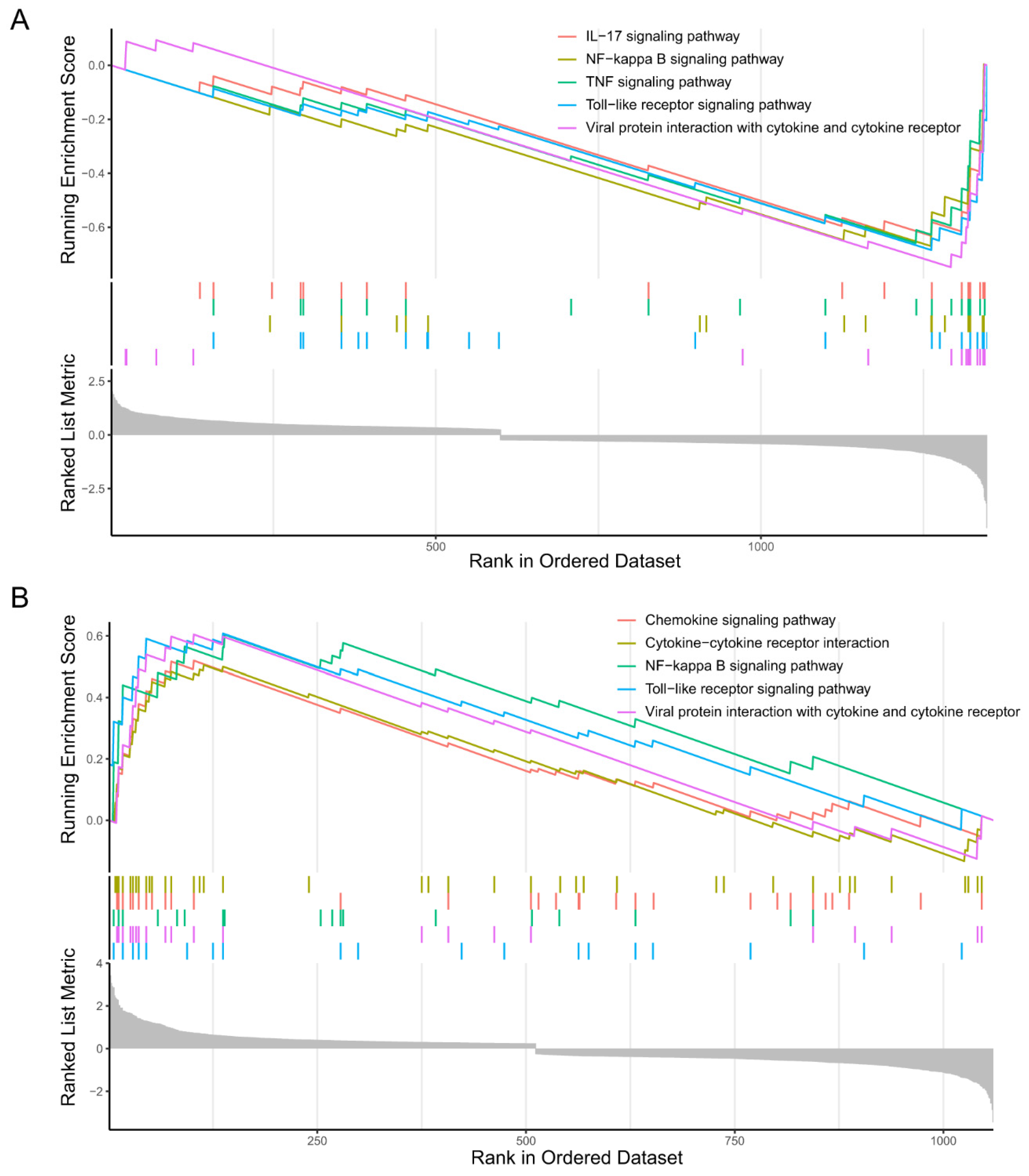
2.2.2. GSEA of the Microglia Subclusters 0 and 2
2.3. Molecule Docking Simulations for Ischemic Stroke Therapy
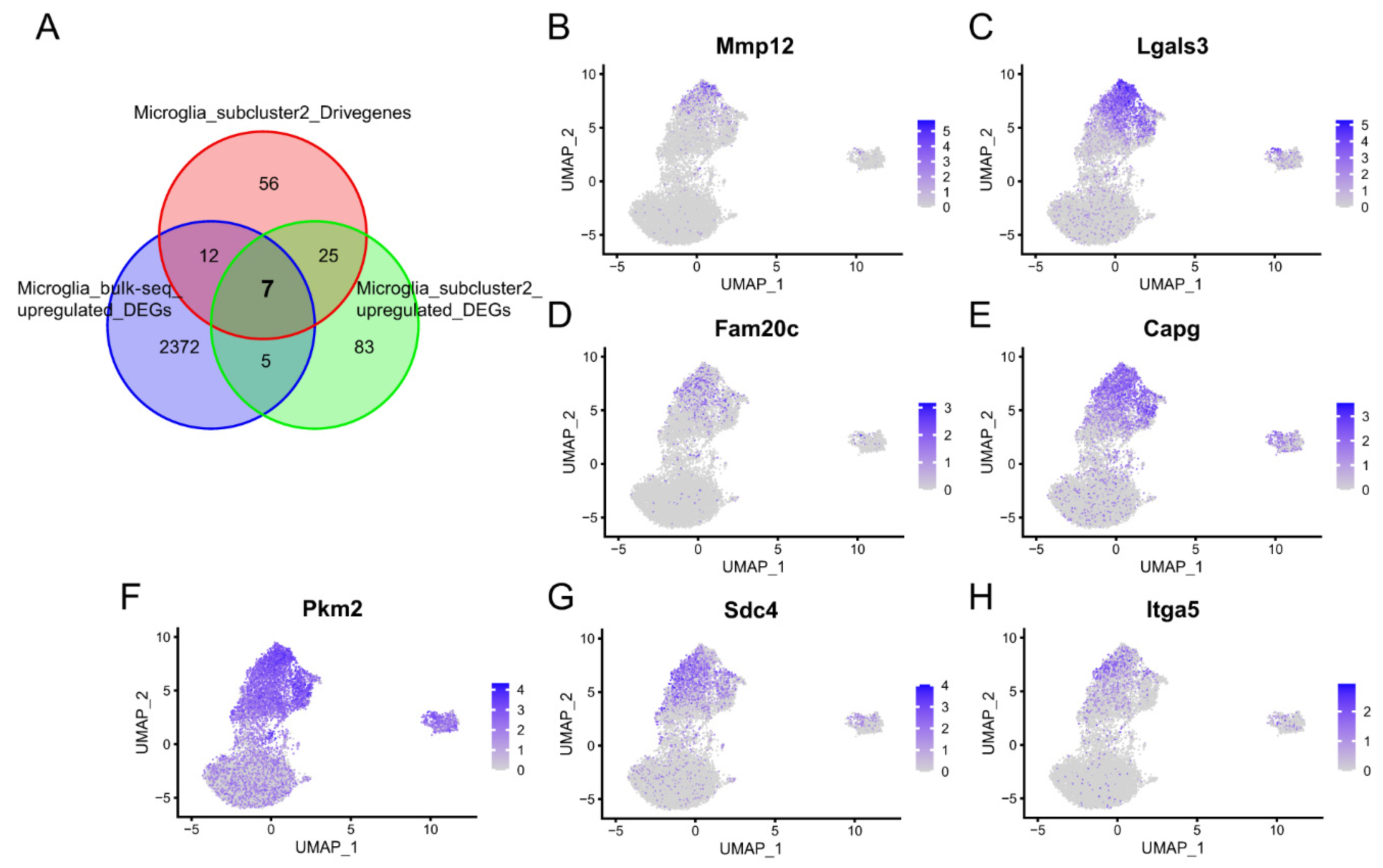
2.3.1. Key Genes of Microglia Subcluster 2
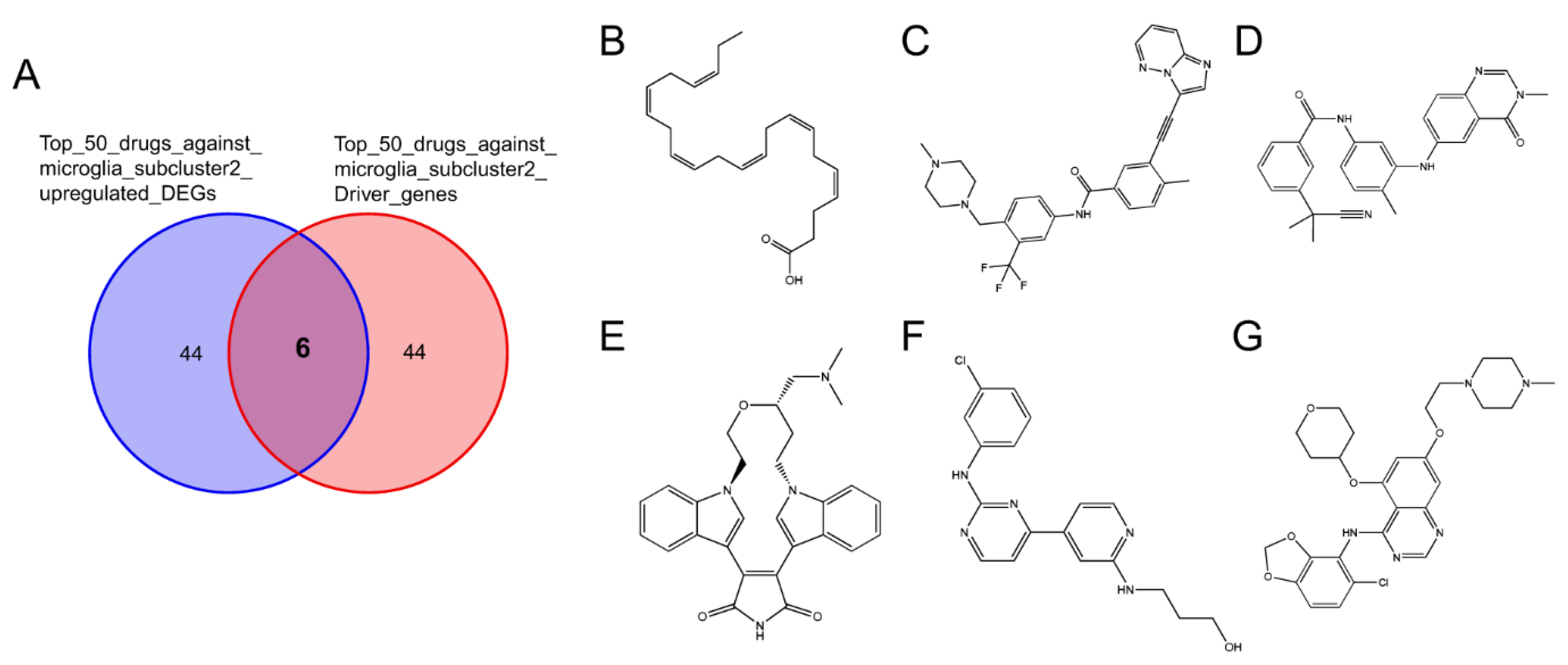
2.3.2. Connectivity Map Analysis Identified Potential Candidate Compounds Targeting Microglia Subcluster 2
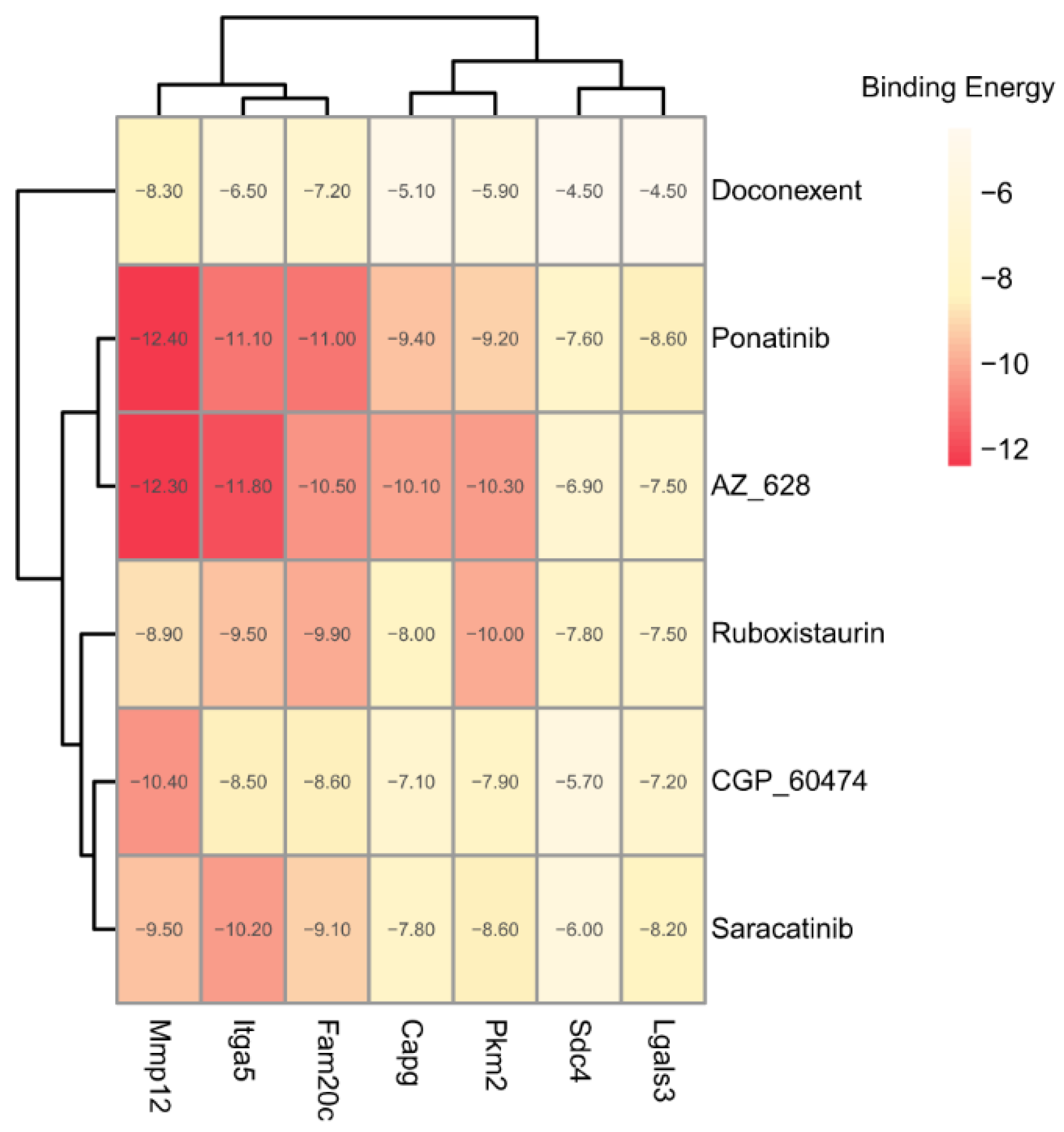
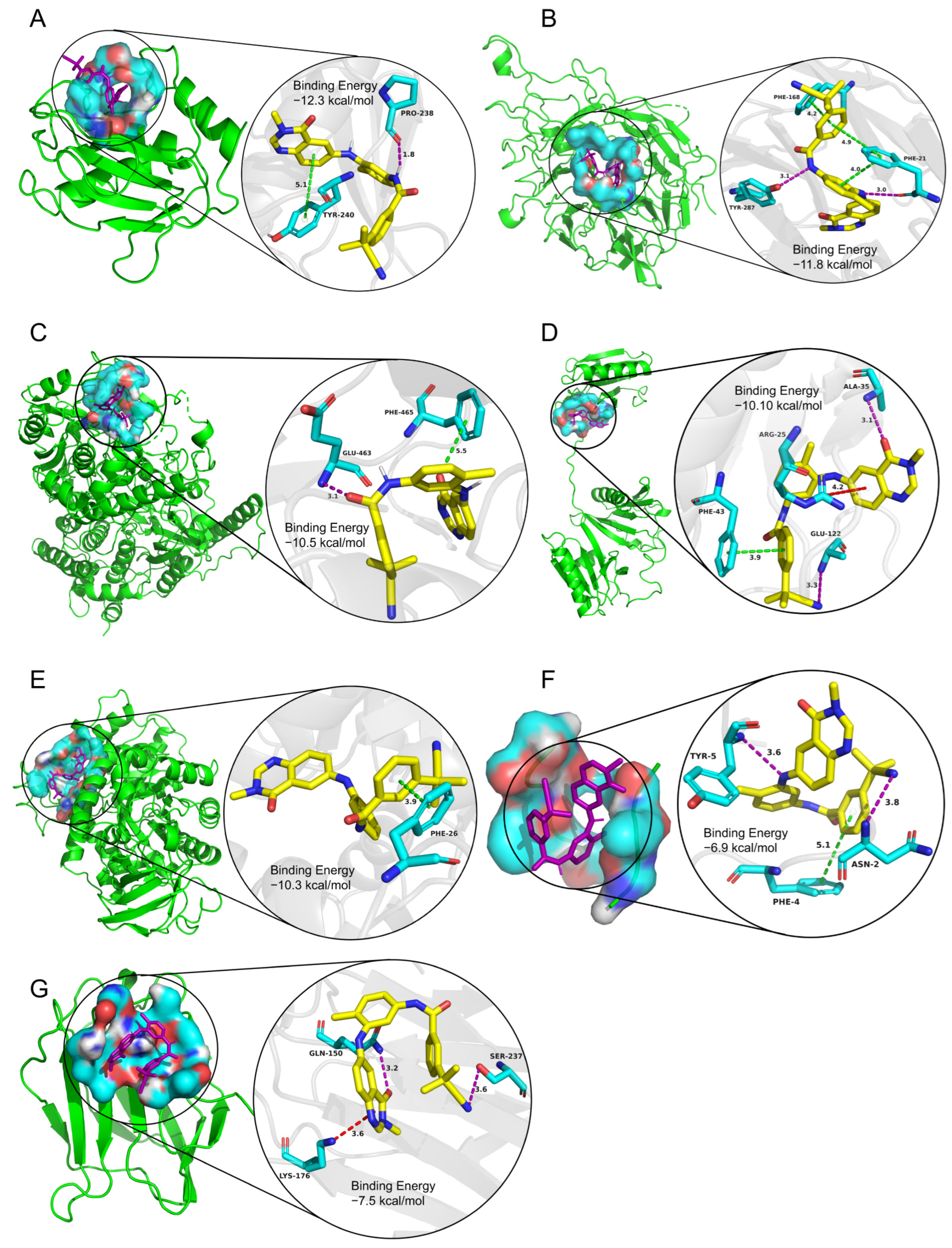
2.3.3. Potential Therapeutic Agents Predicted by Molecule Docking
2.4. Limitations
3. Materials and Methods
3.1. Data Collection and Settings
3.2. scRNA-Seq Data Analysis
3.3. MicroArray Data Processing
3.4. RNA-Seq Data Processing
3.5. Weighted Gene Coexpression Network Analysis
3.6. Time-Dependent Genes in the Progression of Stroke
3.7. Screening of Diagnostic Biomarkers for Stroke
3.8. Nomogram Construction and Evaluation
3.9. RNA Velocity and Pseudo Time Analysis
3.10. Gene Set Enrichment Analysis (GSEA)
3.11. Connectivity Map Analysis
3.12. Molecular Docking
3.13. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Collaborators, G.B.D.S. Global, Regional, and National Burden of Stroke and Its Risk Factors, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet Neurol. 2021, 20, 795–820. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.Y.; Kim, B.; Lee, S.R.; Cha, S.H.; Lee, D.S.; Lee, H.J. Prospects of Therapeutic Target and Directions for Ischemic Stroke. Pharmaceuticals 2021, 14, 321. [Google Scholar] [CrossRef]
- Peyravian, N.; Dikici, E.; Deo, S.; Toborek, M.; Daunert, S. Opioid Antagonists as Potential Therapeutics for Ischemic Stroke. Prog. Neurobiol. 2019, 182, 101679. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.N.; Gonzales, R.; Feng, Y.; Wood, K.; Chai, Z.; Dong, J.F.; La Cava, A.; Shi, F.D.; Liu, Q. Brain Ischemia Induces Diversified Neuroantigen-Specific T-Cell Responses That Exacerbate Brain Injury. Stroke 2018, 49, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Bourget, C.; Adams, K.V.; Morshead, C.M. Reduced Microglia Activation Following Metformin Administration or Microglia Ablation Is Sufficient to Prevent Functional Deficits in a Mouse Model of Neonatal Stroke. J. Neuroinflamm. 2022, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jin, M.Z.; Yang, Z.Y.; Jin, W.L. Microglia in Neurodegenerative Diseases. Neural Regen. Res. 2021, 16, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of ‘Homeostatic’ Microglia and Patterns of Their Activation in Active Multiple Sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2y12 Receptor Regulates Microglial Activation by Extracellular Nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New Tools for Studying Microglia in the Mouse and Human Cns. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Cao, Z.; Harvey, S.S.; Chiang, T.; Foltz, A.G.; Lee, A.G.; Cheng, M.Y.; Steinberg, G.K. Unique Subtype of Microglia in Degenerative Thalamus after Cortical Stroke. Stroke 2021, 52, 687–698. [Google Scholar] [CrossRef]
- Wicks, E.E.; Ran, K.R.; Kim, J.E.; Xu, R.; Lee, R.P.; Jackson, C.M. The Translational Potential of Microglia and Monocyte-Derived Macrophages in Ischemic Stroke. Front. Immunol. 2022, 13, 897022. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Shao, Y.; Zhang, X.; Xiang, J.; Zhang, R.; Sun, Z.; Mei, S.; Zhou, J.; Zhang, J.; Shi, L. A Unique Type of Highly-Activated Microglia Evoking Brain Inflammation Via Mif/Cd74 Signaling Axis in Aged Mice. Aging Dis. 2021, 12, 2125–2139. [Google Scholar] [CrossRef] [PubMed]
- Zia, S.; Rawji, K.S.; Michaels, N.J.; Burr, M.; Kerr, B.J.; Healy, L.M.; Plemel, J.R. Microglia Diversity in Health and Multiple Sclerosis. Front. Immunol. 2020, 11, 588021. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ru, W.; Adolacion, J.R.; Spurgat, M.S.; Liu, X.; Yuan, S.; Liang, R.X.; Dong, J.; Potter, A.S.; Potter, S.S.; et al. Single-Cell Rna-Seq Analysis Reveals Compartment-Specific Heterogeneity and Plasticity of Microglia. iScience 2021, 24, 102186. [Google Scholar] [CrossRef]
- Benakis, C.; Simats, A.; Tritschler, S.; Heindl, S.; Besson-Girard, S.; Llovera, G.; Pinkham, K.; Kolz, A.; Ricci, A.; Theis, F.J.; et al. T Cells Modulate the Microglial Response to Brain Ischemia. eLife 2022, 11, e8203. [Google Scholar] [CrossRef]
- Xu, H.; Qin, W.; Hu, X.; Mu, S.; Zhu, J.; Lu, W.; Luo, Y. Lentivirus-Mediated Overexpression of Otulin Ameliorates Microglia Activation and Neuroinflammation by Depressing the Activation of the Nf-Kappab Signaling Pathway in Cerebral Ischemia/Reperfusion Rats. J. Neuroinflamm. 2018, 15, 83. [Google Scholar] [CrossRef]
- Duan, R.N.; Yang, C.L.; Du, T.; Liu, A.; Wang, A.R.; Sun, W.J.; Li, X.; Li, J.X.; Yan, C.Z.; Liu, Q.J. Smek1 Deficiency Exacerbates Experimental Autoimmune Encephalomyelitis by Activating Proinflammatory Microglia and Suppressing the Ido1-Ahr Pathway. J. Neuroinflamm. 2021, 18, 145. [Google Scholar] [CrossRef]
- Yan, J.; Zhang, Y.; Wang, L.; Li, Z.; Tang, S.; Wang, Y.; Gu, N.; Sun, X.; Li, L. Trem2 Activation Alleviates Neural Damage Via Akt/Creb/Bdnf Signalling after Traumatic Brain Injury in Mice. J. Neuroinflamm. 2022, 19, 289. [Google Scholar] [CrossRef]
- Chen, J.; Cheong, C.; Lan, L.; Zhou, X.; Liu, J.; Lyu, A.; Cheung, W.K.; Zhang, L. Deepdrim: A Deep Neural Network to Reconstruct Cell-Type-Specific Gene Regulatory Network Using Single-Cell Rna-Seq Data. Brief. Bioinform. 2021, 22, bbab325. [Google Scholar] [CrossRef]
- Martin, K.E.; Garcia, A.J. Macrophage Phenotypes in Tissue Repair and the Foreign Body Response: Implications for Biomaterial-Based Regenerative Medicine Strategies. Acta Biomater. 2021, 133, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.; Zafar, H. Inference of Cell State Transitions and Cell Fate Plasticity from Single-Cell with Margaret. Nucleic Acids Res. 2022, 50, e86. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, P.; Chen, W.; Ye, L.; Brannan, K.W.; Le, N.T.; Abe, J.I.; Cooke, J.P.; Wang, G. A Relay Velocity Model Infers Cell-Dependent Rna Velocity. Nat. Biotechnol. 2023. [Google Scholar] [CrossRef]
- Shao, X.; Lu, X.; Liao, J.; Chen, H.; Fan, X. New Avenues for Systematically Inferring Cell-Cell Communication: Through Single-Cell Transcriptomics Data. Protein Cell 2020, 11, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Agu, P.C.; Afiukwa, C.A.; Orji, O.U.; Ezeh, E.M.; Ofoke, I.H.; Ogbu, C.O.; Ugwuja, E.I.; Aja, P.M. Molecular Docking as a Tool for the Discovery of Molecular Targets of Nutraceuticals in Diseases Management. Sci. Rep. 2023, 13, 13398. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef]
- Xiong, T.; Lv, X.S.; Wu, G.J.; Guo, Y.X.; Liu, C.; Hou, F.X.; Wang, J.K.; Fu, Y.F.; Liu, F.Q. Single-Cell Sequencing Analysis and Multiple Machine Learning Methods Identified G0s2 and Hpse as Novel Biomarkers for Abdominal Aortic Aneurysm. Front. Immunol. 2022, 13, 907309. [Google Scholar] [CrossRef]
- Zheng, P.F.; Chen, L.Z.; Liu, P.; Pan, H.W.; Fan, W.J.; Liu, Z.Y. Identification of Immune-Related Key Genes in the Peripheral Blood of Ischaemic Stroke Patients Using a Weighted Gene Coexpression Network Analysis and Machine Learning. J. Transl. Med. 2022, 20, 361. [Google Scholar] [CrossRef]
- Sarafidis, M.; Lambrou, G.I.; Zoumpourlis, V.; Koutsouris, D. An Integrated Bioinformatics Analysis Towards the Identification of Diagnostic, Prognostic, and Predictive Key Biomarkers for Urinary Bladder Cancer. Cancers 2022, 14, 3358. [Google Scholar] [CrossRef]
- Liu, L.; Liu, J.; Wang, Y.; Wang, D.; Wang, Y. Substantial Improvement of Stroke Care in China. Stroke 2018, 49, 3085–3091. [Google Scholar] [CrossRef]
- Lv, J.; Hu, W.; Yang, Z.; Li, T.; Jiang, S.; Ma, Z.; Chen, F.; Yang, Y. Focusing on Claudin-5: A Promising Candidate in the Regulation of Bbb to Treat Ischemic Stroke. Prog. Neurobiol. 2018, 161, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Wita, K.; Kulach, A.; Wilkosz, K.; Wybraniec, M.; Wojakowski, W.; Kuczmik, W.; Lelek, M.; Tomalski, W.; Ochala, A.; Uchwat, U.; et al. Mechanical Thrombectomy in Acute Ischemic Stroke-the Role of Interventional Cardiologists: A Prospective Single-Center Study. Cardiovasc. Interv. 2022, 15, 550–558. [Google Scholar] [CrossRef]
- Liang, L.; Yu, J.; Li, J.; Li, N.; Liu, J.; Xiu, L.; Zeng, J.; Wang, T.; Wu, L. Integration of Scrna-Seq and Bulk Rna-Seq to Analyse the Heterogeneity of Ovarian Cancer Immune Cells and Establish a Molecular Risk Model. Front. Oncol. 2021, 11, 711020. [Google Scholar] [CrossRef] [PubMed]
- Statnikov, A.; Wang, L.; Aliferis, C.F. A Comprehensive Comparison of Random Forests and Support Vector Machines for Microarray-Based Cancer Classification. BMC Bioinform. 2008, 9, 319. [Google Scholar] [CrossRef]
- Ding, X.; Qin, J.; Huang, F.; Feng, F.; Luo, L. The Combination of Machine Learning and Untargeted Metabolomics Identifies the Lipid Metabolism -Related Gene Ch25h as a Potential Biomarker in Asthma. Inflamm. Res. 2023, 72, 1099–1119. [Google Scholar] [CrossRef] [PubMed]
- Won, M.; Park, K.A.; Kim, S.; Ju, E.; Ko, Y.; Yoo, H.; Ro, H.; Lee, J.; Oh, J.; Lee, E.G.; et al. Ankrd13a Controls Early Cell-Death Checkpoint by Interacting with Rip1 Independent of Nf-Kappab. Cell Death Differ. 2022, 29, 1152–1163. [Google Scholar] [CrossRef]
- Stamova-kiossepacheva, B.; Jickling, G.C.; Sharp, F. Methods of Distinguishing Ischemic Stroke from Intracerebral Hemorrhage. U.S. Patent 11,525,161, 13 December 2022. [Google Scholar]
- Zhang, L.; Liu, X.; Sheng, H.; Liu, S.; Li, Y.; Zhao, J.Q.; Warner, D.S.; Paschen, W.; Yang, W. Neuron-Specific Sumo Knockdown Suppresses Global Gene Expression Response and Worsens Functional Outcome after Transient Forebrain Ischemia in Mice. Neuroscience 2017, 343, 190–212. [Google Scholar] [CrossRef]
- Wu, J.; Wu, Z.; He, A.; Zhang, T.; Zhang, P.; Jin, J.; Li, S.; Li, G.; Li, X.; Liang, S.; et al. Genome-Wide Screen and Validation of Microglia Pro-Inflammatory Mediators in Stroke. Aging Dis. 2021, 12, 786–800. [Google Scholar] [CrossRef]
- He, C.; Wang, T.; Han, Y.; Zuo, C.; Wang, G. E3 Ubiquitin Ligase Cop1 Confers Neuroprotection in Cerebral Ischemia/Reperfusion Injury Via Regulation of Transcription Factor C/Ebpbeta in Microglia. Int. J. Biol. Macromol. 2022, 222, 1789–1800. [Google Scholar] [CrossRef]
- Visuttijai, K.; Hedberg-Oldfors, C.; Thomsen, C.; Glamuzina, E.; Kornblum, C.; Tasca, G.; Hernandez-Lain, A.; Sandstedt, J.; Dellgren, G.; Roach, P.; et al. Glycogenin Is Dispensable for Glycogen Synthesis in Human Muscle, and Glycogenin Deficiency Causes Polyglucosan Storage. J. Clin. Endocrinol. Metab. 2020, 105, 557–566. [Google Scholar] [CrossRef]
- Cai, Y.; Guo, H.; Fan, Z.; Zhang, X.; Wu, D.; Tang, W.; Gu, T.; Wang, S.; Yin, A.; Tao, L.; et al. Glycogenolysis Is Crucial for Astrocytic Glycogen Accumulation and Brain Damage after Reperfusion in Ischemic Stroke. iScience 2020, 23, 101136. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, Y.; Hou, W.; Cai, Y. Brain Glycogen: An Angel or a Devil for Ischemic Stroke? Neurosci. Bull. 2023, 39, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.B.; Uo, T.; Kinoshita, C.; Sopher, B.L.; Lee, R.J.; Murphy, S.P.; Kinoshita, Y.; Garden, G.A.; Wang, H.G.; Morrison, R.S. Bax Interacting Factor-1 Promotes Survival and Mitochondrial Elongation in Neurons. J. Neurosci. 2014, 34, 2674–2683. [Google Scholar] [CrossRef]
- Xie, S.T.; Chen, A.X.; Song, B.; Fan, J.; Li, W.; Xing, Z.; Peng, S.Y.; Zhang, Q.P.; Dong, L.; Yan, C.; et al. Suppression of Microglial Activation and Monocyte Infiltration Ameliorates Cerebellar Hemorrhage Induced-Brain Injury and Ataxia. Brain Behav. Immun. 2020, 89, 400–413. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Z.; Wang, L.; Zhang, L.; Liu, Y.; Cao, J.; Fu, Q.; Liu, Y.; Li, H.; Lou, J.; et al. Cottonseed Oil Alleviates Ischemic Stroke Injury by Inhibiting the Inflammatory Activation of Microglia and Astrocyte. J. Neuroinflamm. 2020, 17, 270. [Google Scholar] [CrossRef]
- Ganbold, T.; Bao, Q.; Zandan, J.; Hasi, A.; Baigude, H. Modulation of Microglia Polarization through Silencing of Nf-Kappab P65 by Functionalized Curdlan Nanoparticle-Mediated Rnai. ACS Appl. Mater. Interfaces 2020, 12, 11363–11374. [Google Scholar] [CrossRef]
- Xue, Y.; Nie, D.; Wang, L.J.; Qiu, H.C.; Ma, L.; Dong, M.X.; Tu, W.J.; Zhao, J. Microglial Polarization: Novel Therapeutic Strategy against Ischemic Stroke. Aging Dis. 2021, 12, 466–479. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The Biphasic Function of Microglia in Ischemic Stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Awad, K.S.; Elinoff, J.M.; Wang, S.; Gairhe, S.; Ferreyra, G.A.; Cai, R.; Sun, J.; Solomon, M.A.; Danner, R.L. Raf/Erk Drives the Proliferative and Invasive Phenotype of Bmpr2-Silenced Pulmonary Artery Endothelial Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L187–L201. [Google Scholar] [CrossRef]
- Montagut, C.; Sharma, S.V.; Shioda, T.; McDermott, U.; Ulman, M.; Ulkus, L.E.; Dias-Santagata, D.; Stubbs, H.; Lee, D.Y.; Singh, A.; et al. Elevated Craf as a Potential Mechanism of Acquired Resistance to Braf Inhibition in Melanoma. Cancer Res. 2008, 68, 4853–4861. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Qiu, J.; Ye, J.; Jiang, T.; Zhang, W.; Zheng, X.; Zhu, Z.; Chen, L.; Wang, Z.; Mi, S.; et al. Az-628 Delays Osteoarthritis Progression Via Inhibiting the Tnf-Alpha-Induced Chondrocyte Necroptosis and Regulating Osteoclast Formation. Int. Immunopharmacol. 2022, 111, 109085. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.L.; Conley, Y.; Ding, J.; Dillman, A.; Warach, S.; Singleton, A.; Matarin, M. Genomic Biomarkers and Cellular Pathways of Ischemic Stroke by Rna Gene Expression Profiling. Neurology 2010, 75, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Stamova, B.; Jickling, G.C.; Ander, B.P.; Zhan, X.; Liu, D.; Turner, R.; Ho, C.; Khoury, J.C.; Bushnell, C.; Pancioli, A.; et al. Gene Expression in Peripheral Immune Cells Following Cardioembolic Stroke Is Sexually Dimorphic. PLoS ONE 2014, 9, e102550. [Google Scholar] [CrossRef] [PubMed]
- Stevens, S.L.; Leung, P.Y.; Vartanian, K.B.; Gopalan, B.; Yang, T.; Simon, R.P.; Stenzel-Poore, M.P. Multiple Preconditioning Paradigms Converge on Interferon Regulatory Factor-Dependent Signaling to Promote Tolerance to Ischemic Brain Injury. J. Neurosci. 2011, 31, 8456–8463. [Google Scholar] [CrossRef]
- Bhattarai, S.; Aly, A.; Garcia, K.; Ruiz, D.; Pontarelli, F.; Dharap, A. Deep Sequencing Reveals Uncharted Isoform Heterogeneity of the Protein-Coding Transcriptome in Cerebral Ischemia. Mol. Neurobiol. 2019, 56, 1035–1043. [Google Scholar] [CrossRef]
- Zheng, K.; Lin, L.; Jiang, W.; Chen, L.; Zhang, X.; Zhang, Q.; Ren, Y.; Hao, J. Single-Cell Rna-Seq Reveals the Transcriptional Landscape in Ischemic Stroke. J. Cereb. Blood Flow Metab. 2022, 42, 56–73. [Google Scholar] [CrossRef]
- Spitzer, D.; Guerit, S.; Puetz, T.; Khel, M.I.; Armbrust, M.; Dunst, M.; Macas, J.; Zinke, J.; Devraj, G.; Jia, X.; et al. Profiling the Neurovascular Unit Unveils Detrimental Effects of Osteopontin on the Blood-Brain Barrier in Acute Ischemic Stroke. Acta Neuropathol. 2022, 144, 305–337. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset ID | Control | Stroke | Tissue | Species | Data Type | Reference |
|---|---|---|---|---|---|---|
| GSE16561 | 24 | 39 | Peripheral blood | human | Microarray | [54] |
| GSE58294 | 23 | 23 | Peripheral blood | human | Microarray | [55] |
| GSE32529 | 6 | 8 | Cerebral cortex | mouse | RNA-Seq | [56] |
| GSE112348 | 3 | 9 | Cerebral cortex | mouse | RNA-Seq | [57] |
| GSE174574 | 3 | 3 | brain | mouse | scRNA-Seq | [58] |
| PRJNA687414 | 6 | 6 | microglia cells | mouse | RNA-Seq | [59] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.; Zaidi, S.A.A.; He, L.; Zhang, S.; Zhou, G. Integrative Analysis of Machine Learning and Molecule Docking Simulations for Ischemic Stroke Diagnosis and Therapy. Molecules 2023, 28, 7704. https://doi.org/10.3390/molecules28237704
Song J, Zaidi SAA, He L, Zhang S, Zhou G. Integrative Analysis of Machine Learning and Molecule Docking Simulations for Ischemic Stroke Diagnosis and Therapy. Molecules. 2023; 28(23):7704. https://doi.org/10.3390/molecules28237704
Chicago/Turabian StyleSong, Jingwei, Syed Aqib Ali Zaidi, Liangge He, Shuai Zhang, and Guangqian Zhou. 2023. "Integrative Analysis of Machine Learning and Molecule Docking Simulations for Ischemic Stroke Diagnosis and Therapy" Molecules 28, no. 23: 7704. https://doi.org/10.3390/molecules28237704
APA StyleSong, J., Zaidi, S. A. A., He, L., Zhang, S., & Zhou, G. (2023). Integrative Analysis of Machine Learning and Molecule Docking Simulations for Ischemic Stroke Diagnosis and Therapy. Molecules, 28(23), 7704. https://doi.org/10.3390/molecules28237704