
Constrained Phosphine Chalcogenide Selenoethers Supported by peri-Substitution

 , , , and
, , , and
Abstract
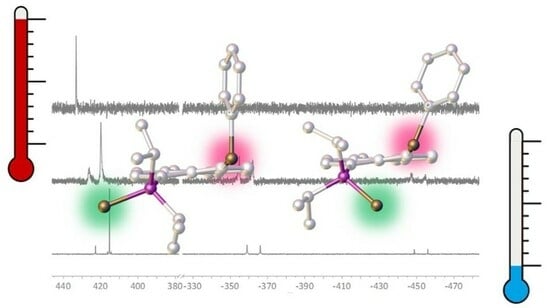
:
1. Dedication
2. Introduction
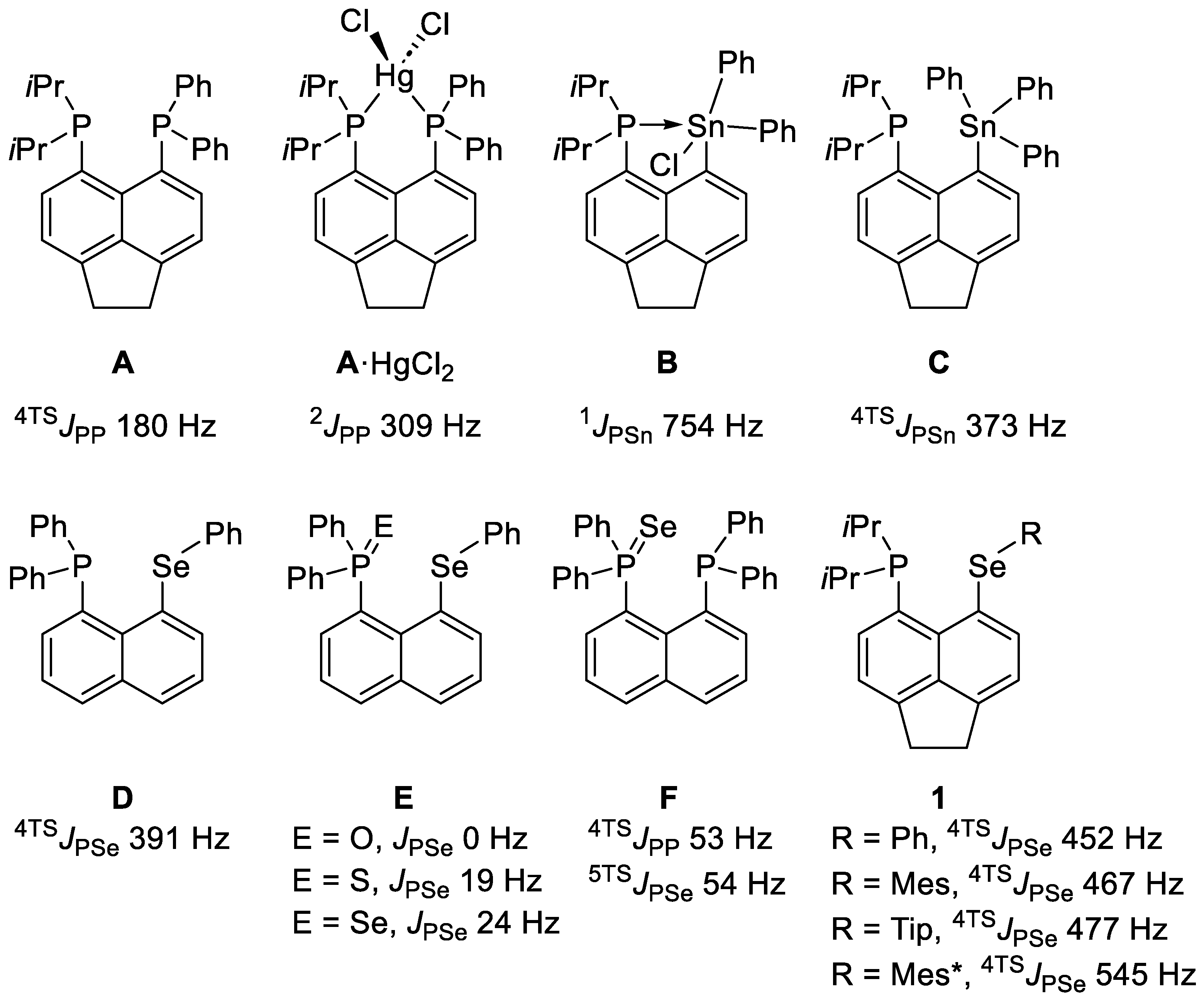
3. Results and Discussion
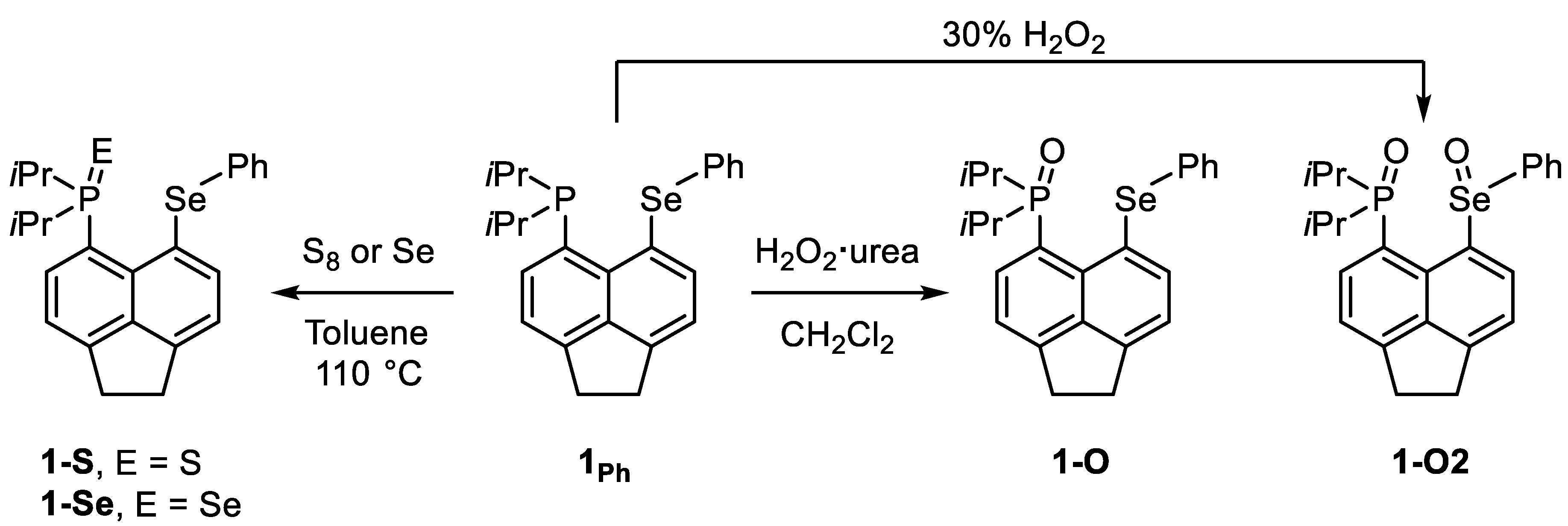
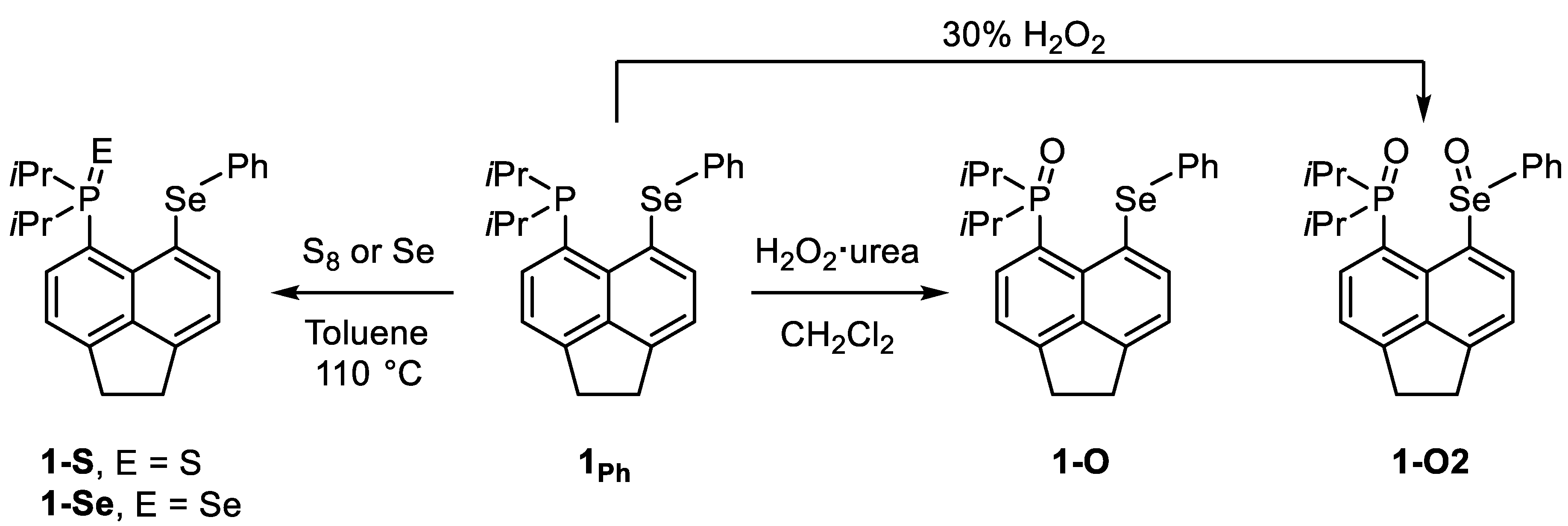
3.1. Synthesis
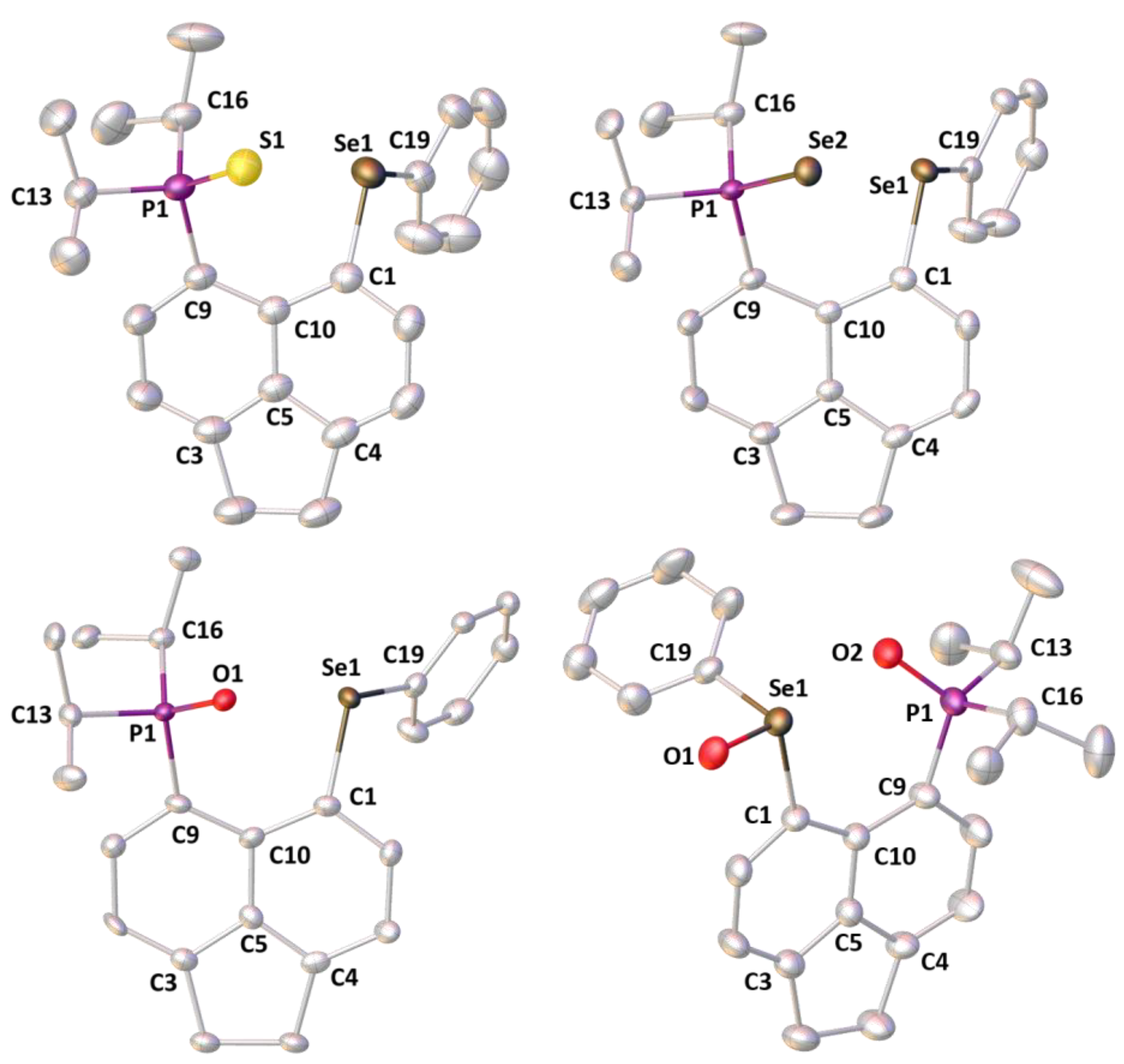
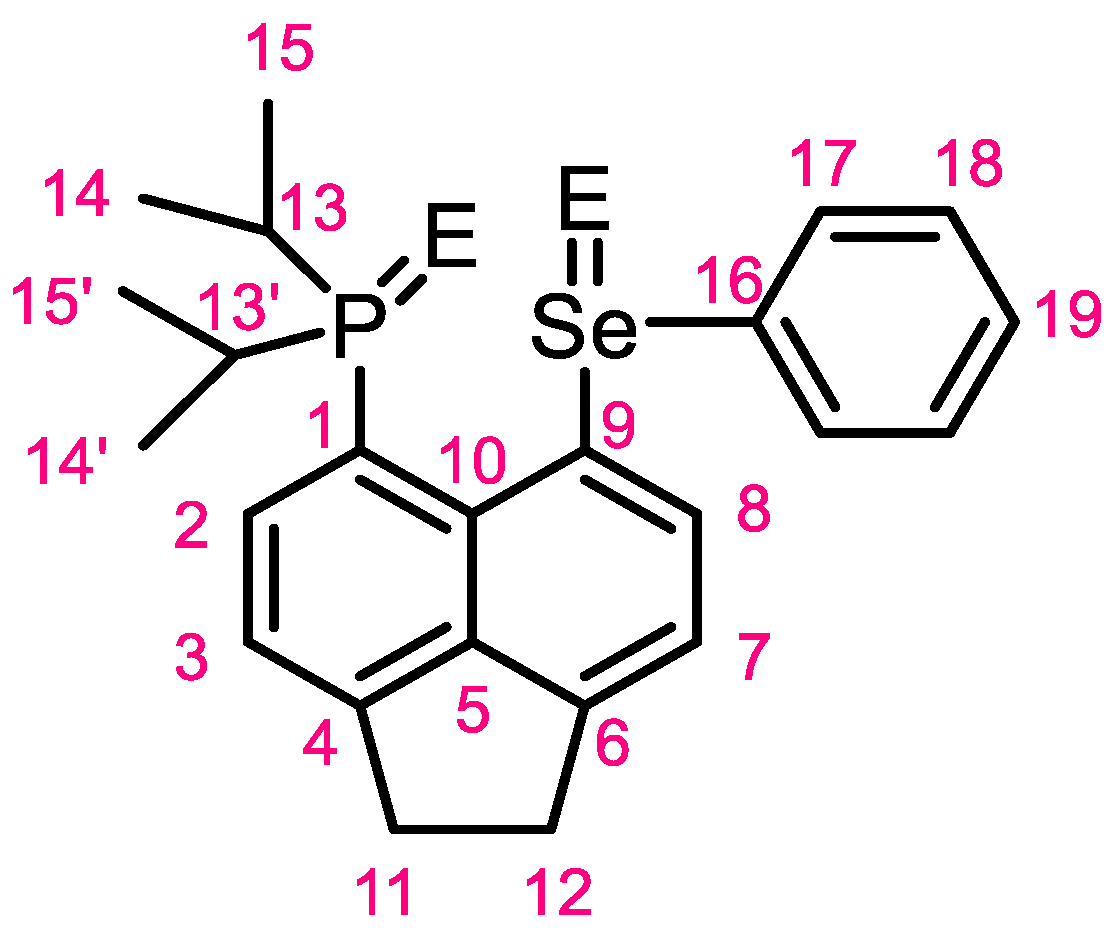
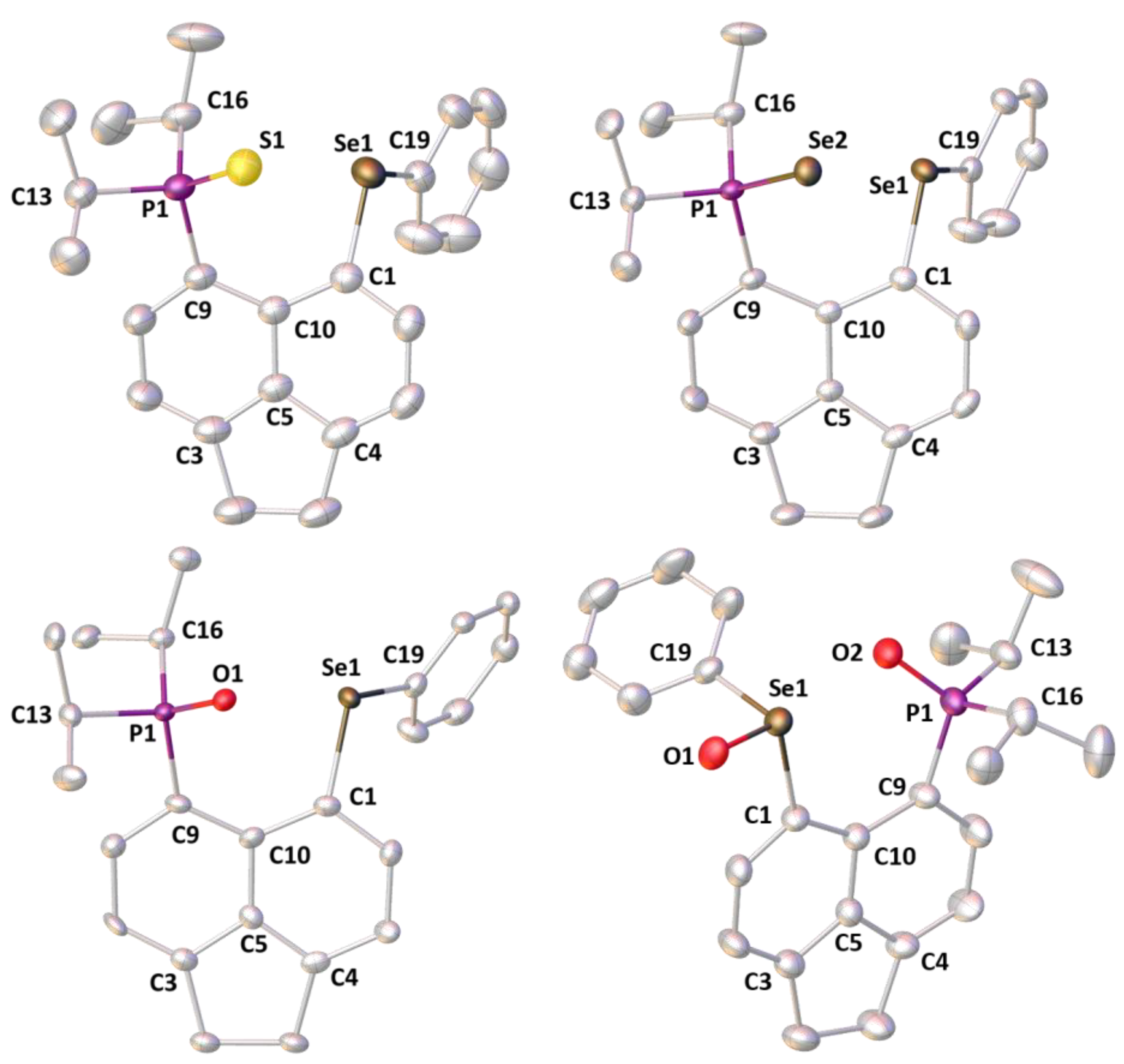
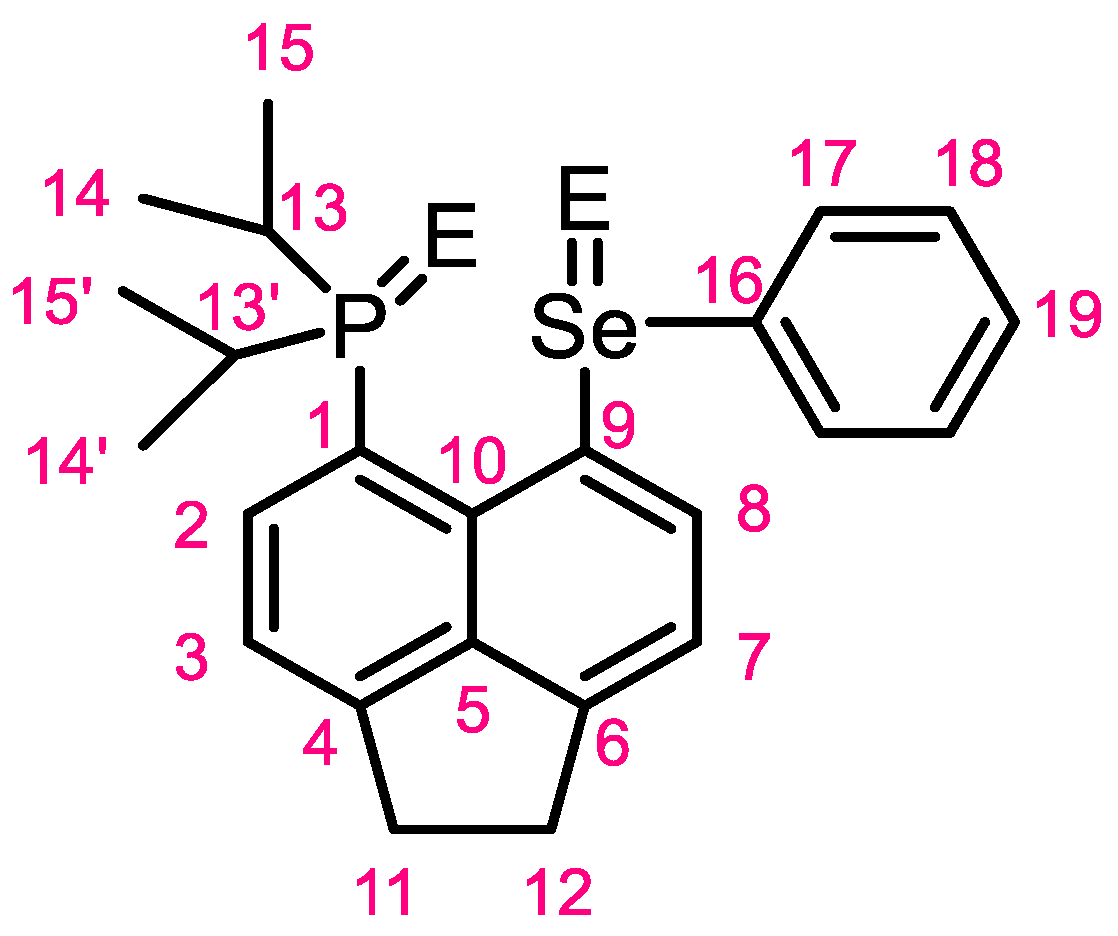
3.2. Crystallography
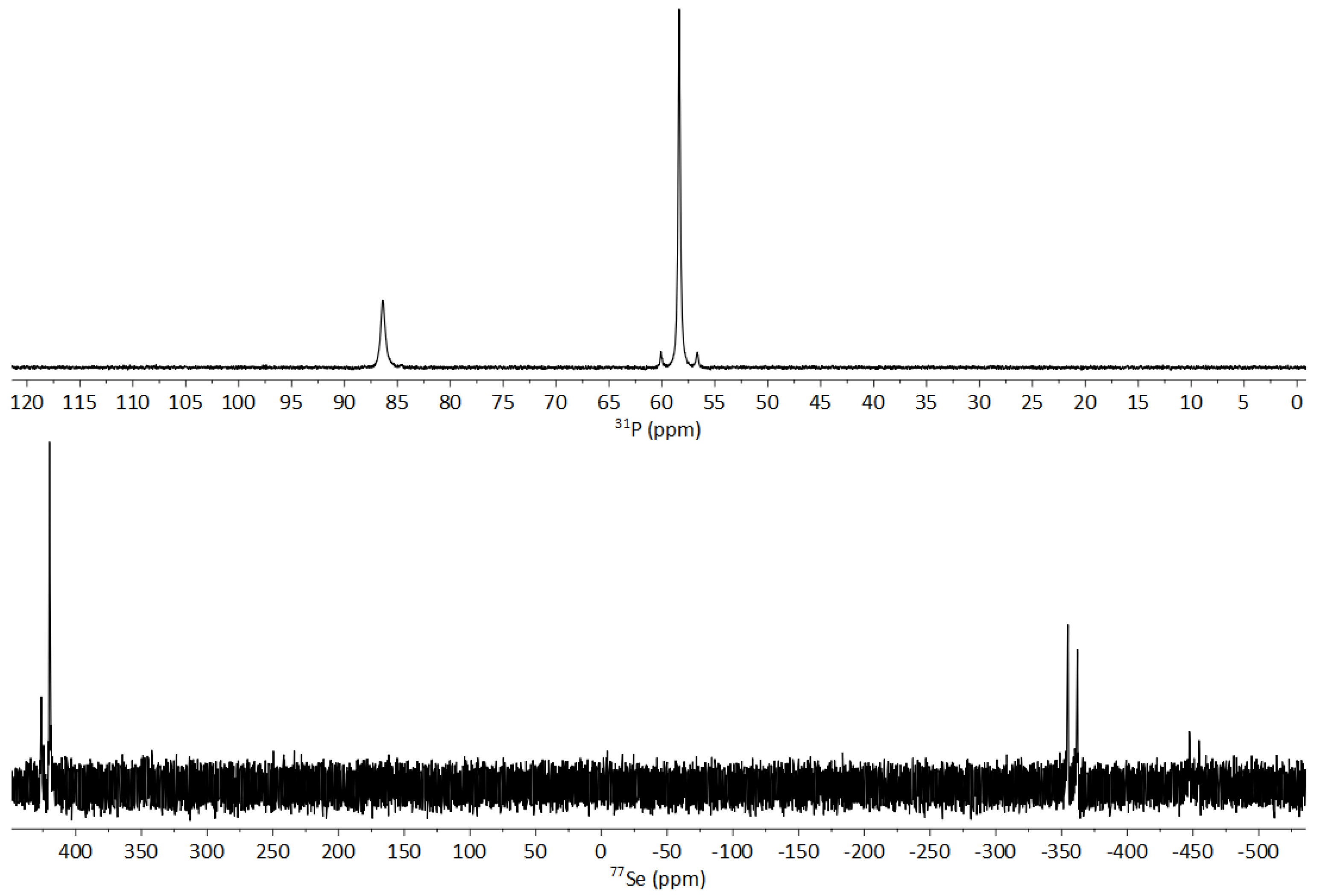
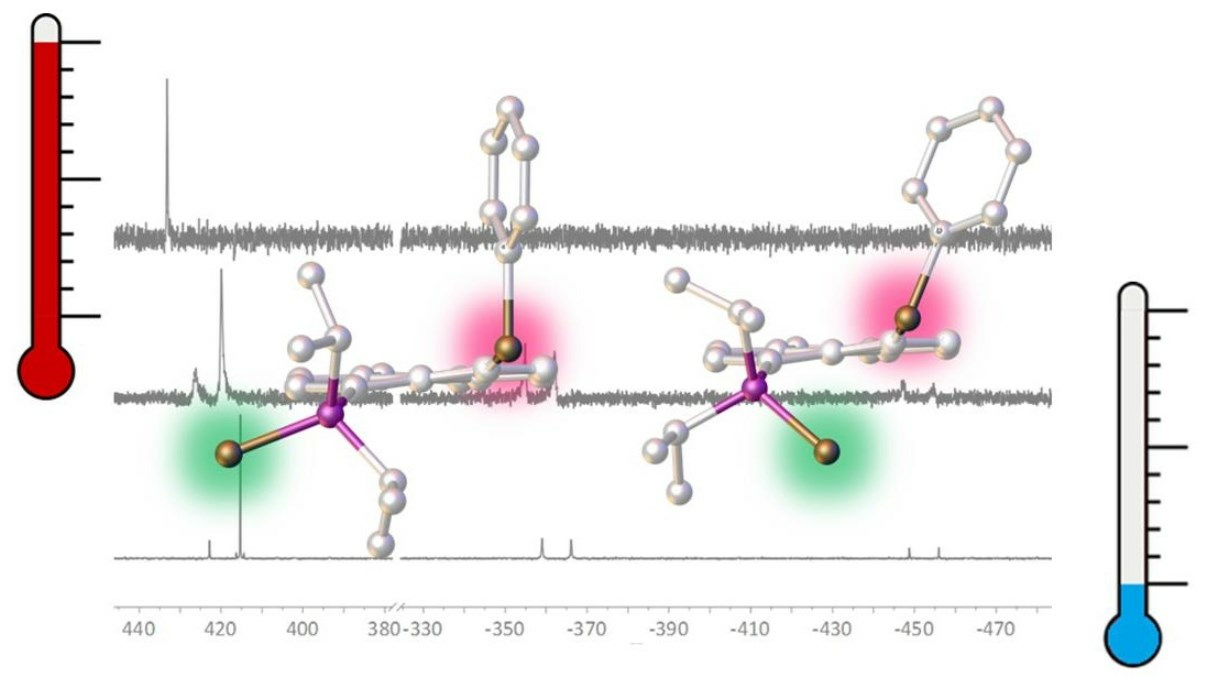
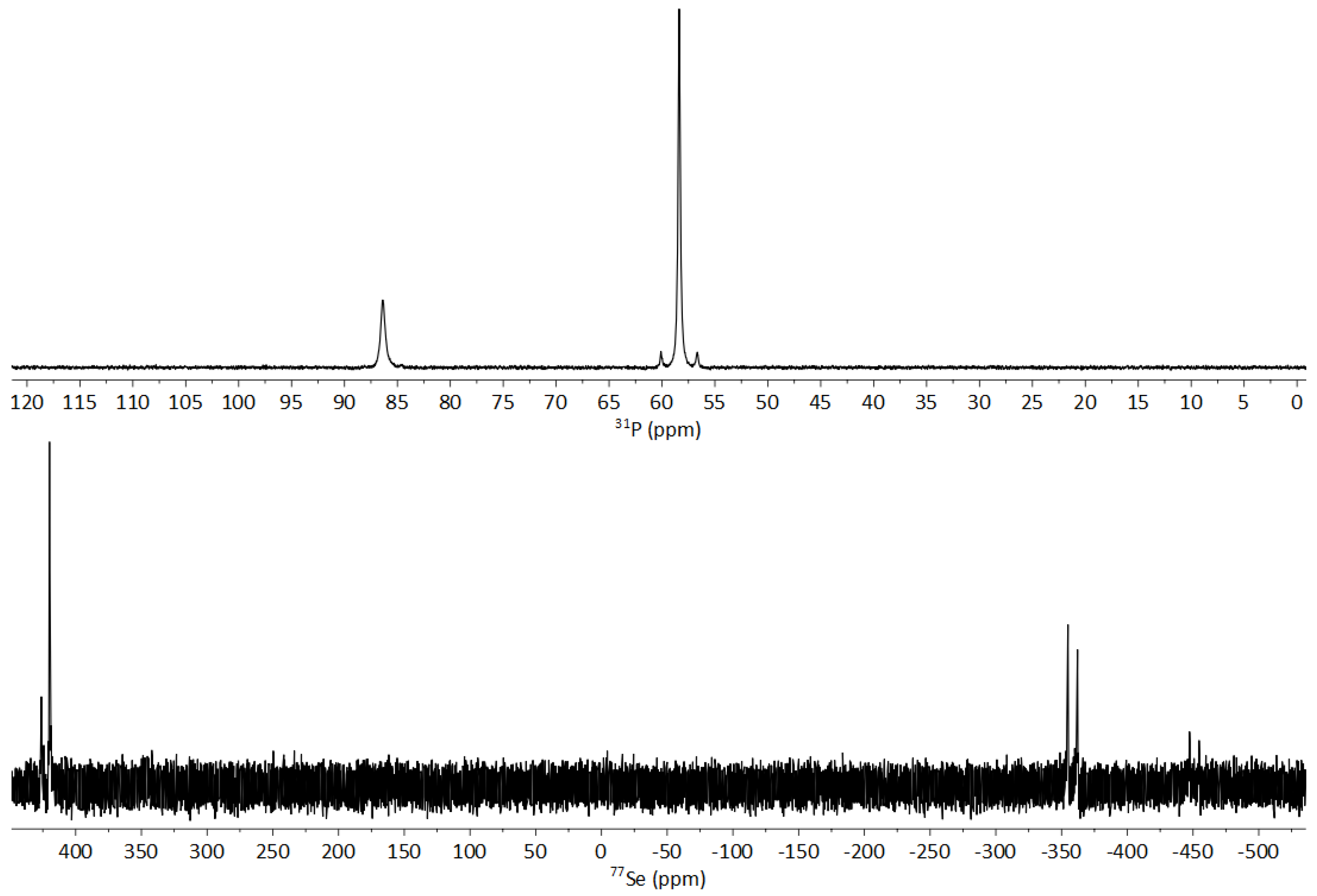
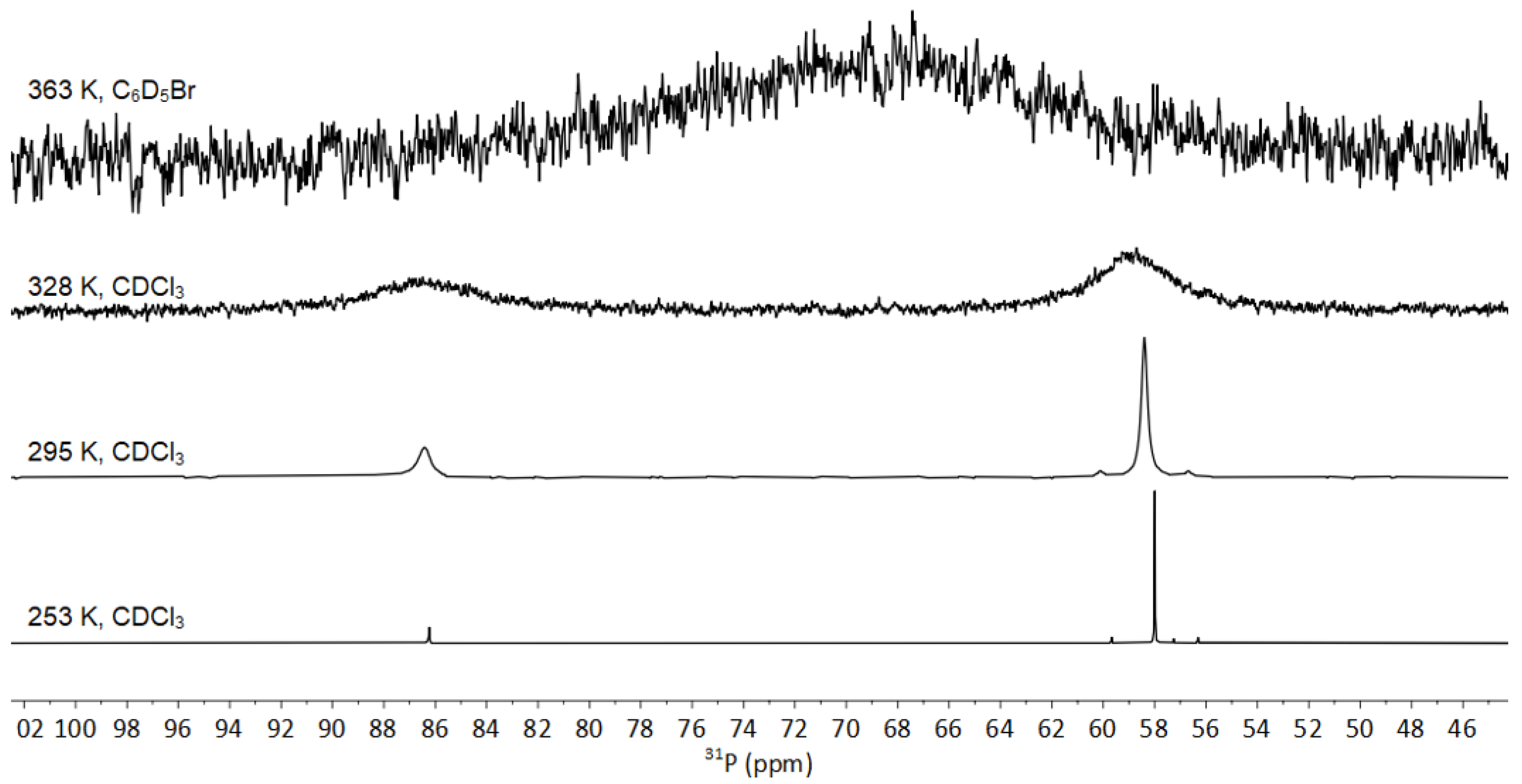
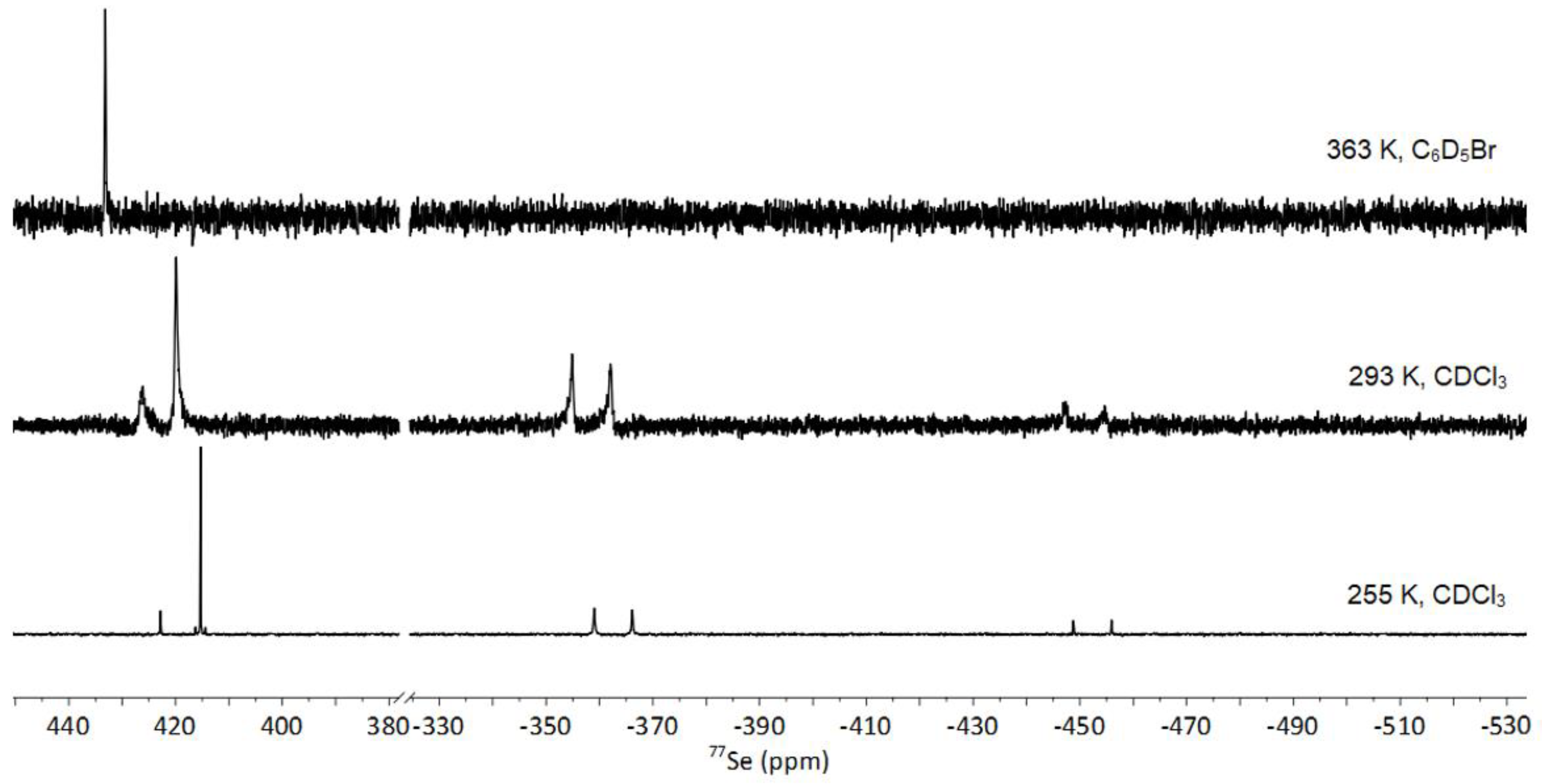
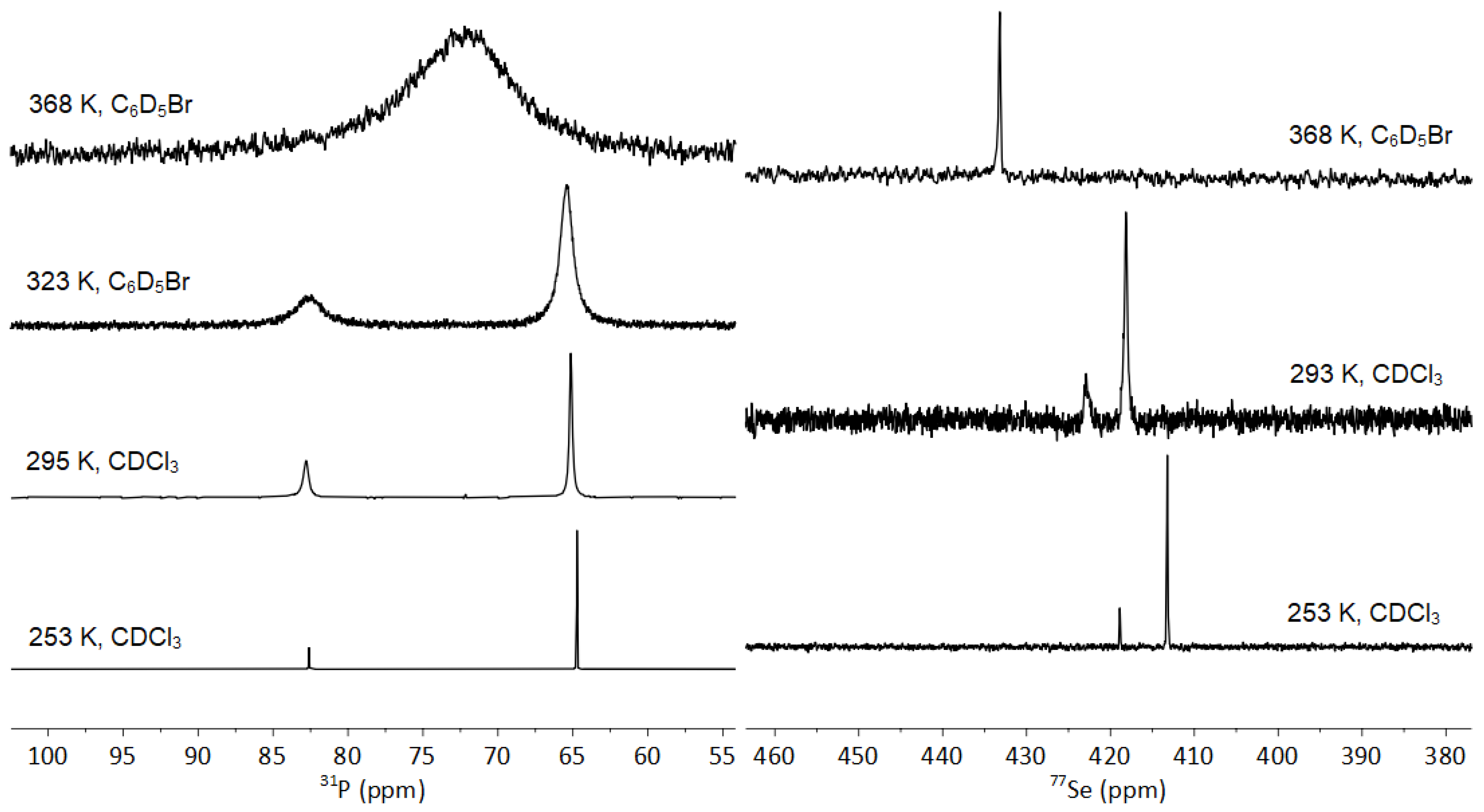
3.3. NMR Spectroscopy of 1-Se
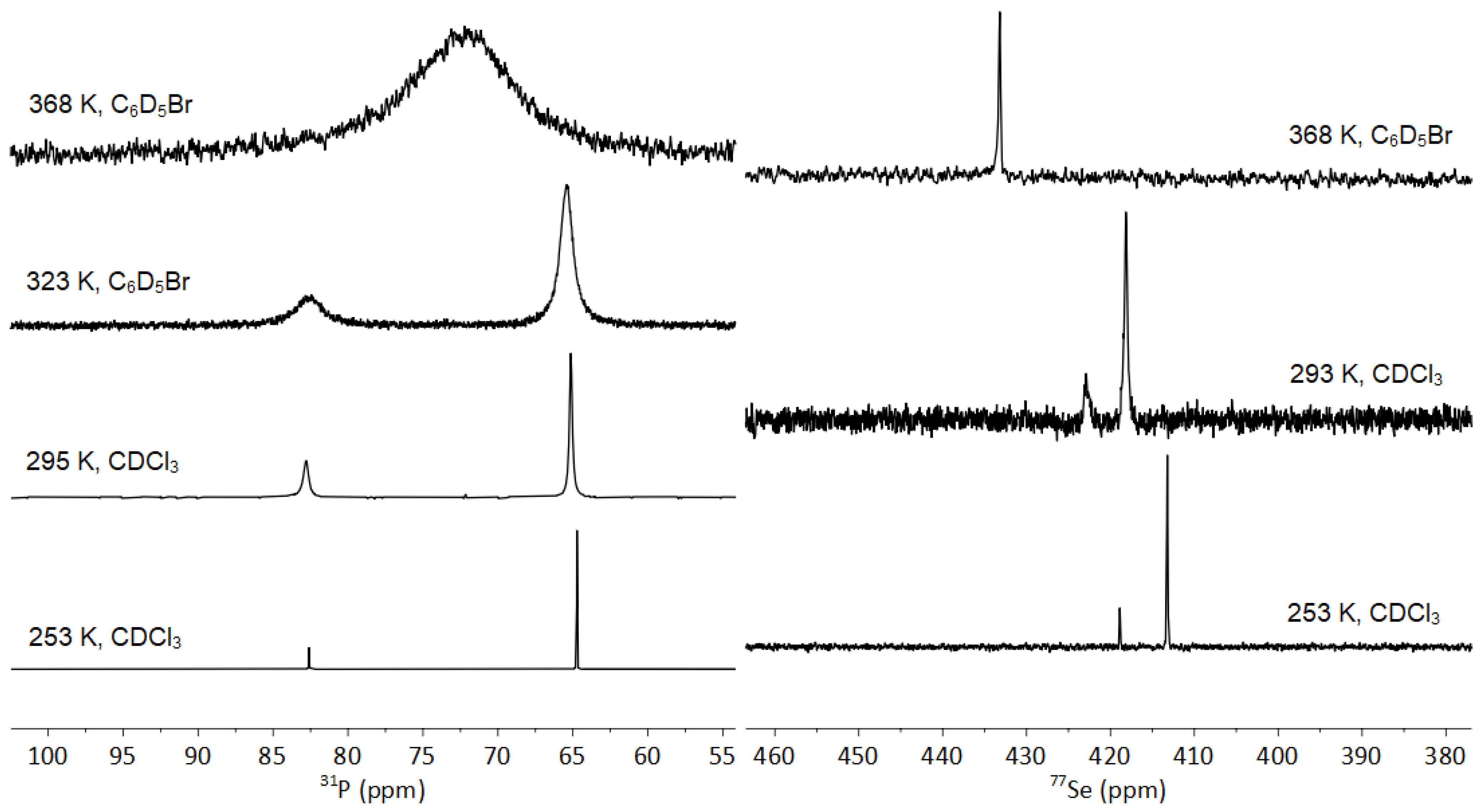
3.3.1. Fluxionality in Solution
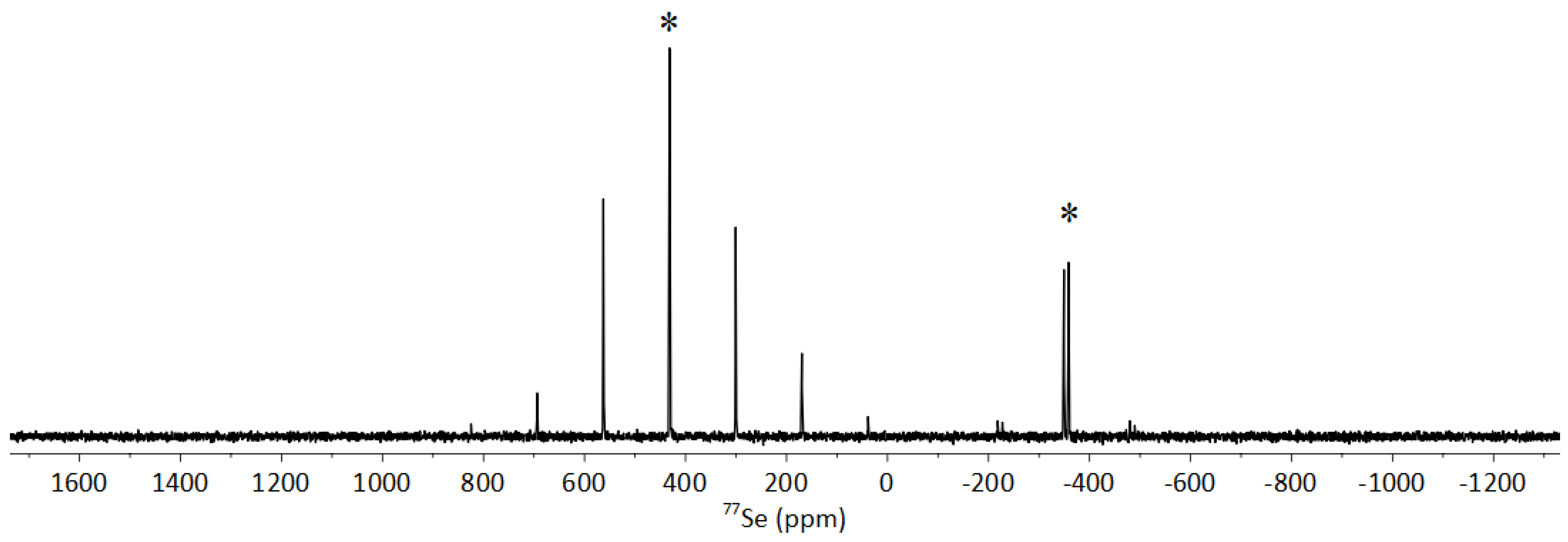
3.3.2. Solid-State NMR of 1-Se
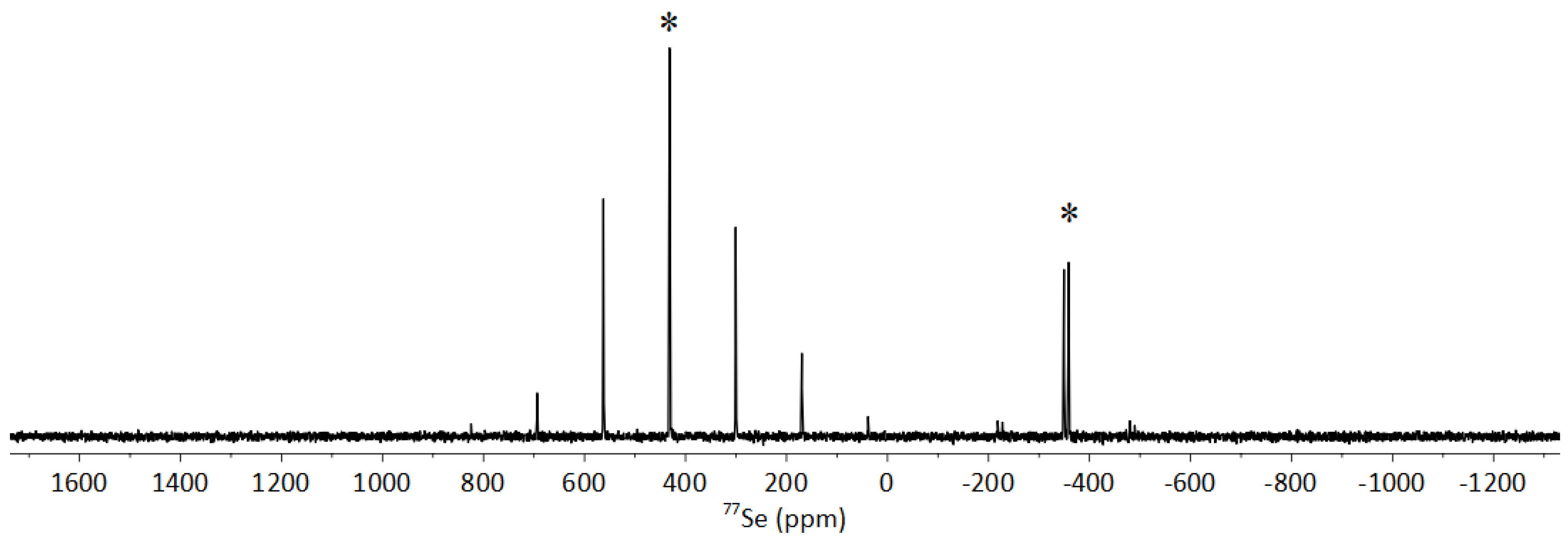
3.4. NMR Spectroscopy of 1-S and 1-O
3.5. NMR Spectroscopy of 1-O2
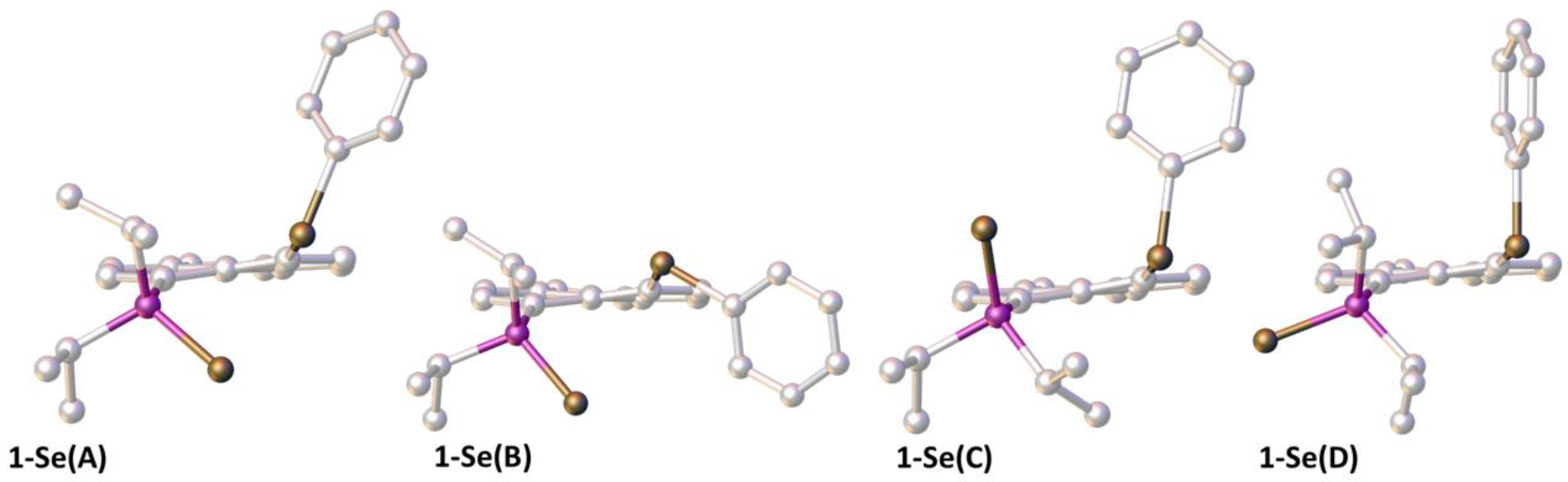
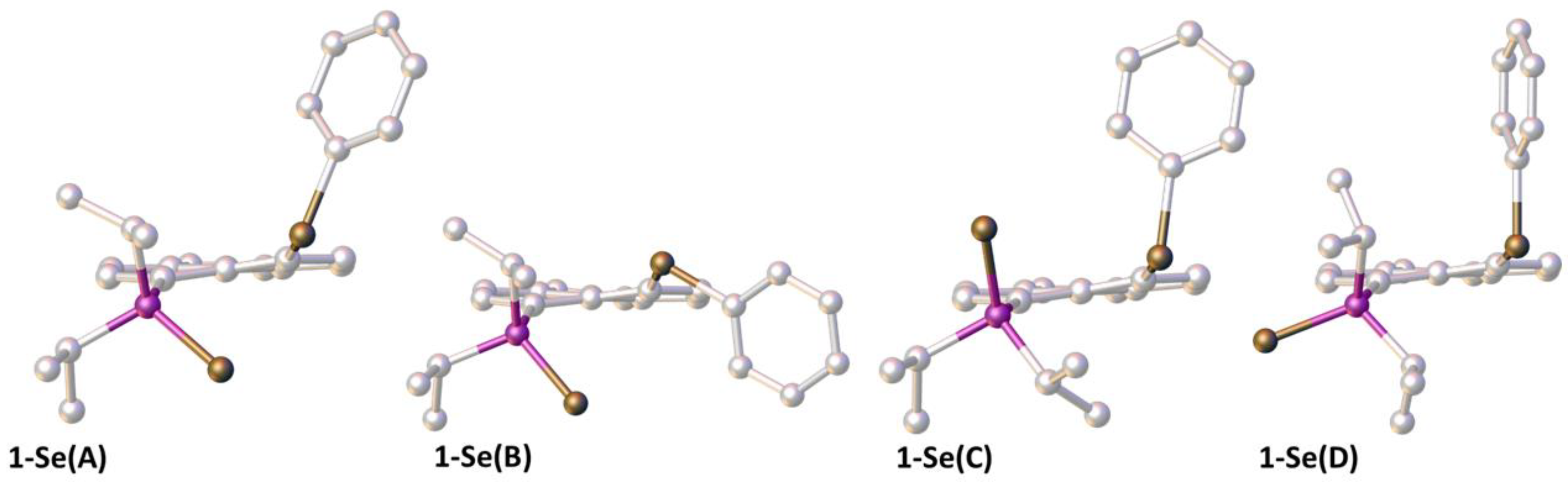
3.6. Computational Studies
4. Materials and Methods
4.1. General Considerations
4.2. Synthetic Procedures and Analytical Data
4.2.1. Synthesis of 1-O
4.2.2. Synthesis of 1-O2
4.2.3. Synthesis of 1-S
4.2.4. Synthesis of 1-Se
4.3. Crystallographic Details
4.4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chalmers, B.A.; Athukorala Arachcige, K.S.; Prentis, J.K.D.; Knight, F.R.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Sterically Encumbered Tin and Phosphorus peri-Substituted Acenaphthenes. Inorg. Chem. 2014, 53, 8795–8808. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Nejman, P.S.; Llewellyn, A.V.; Felaar, A.M.; Griffiths, B.L.; Portman, E.I.; Gordon, E.-J.L.; Fan, K.J.H.; Woollins, J.D.; Bühl, M.; et al. A Study of Through-Space and Through-Bond JPP Coupling in a Rigid Nonsymmetrical Bis(phosphine) and Its Metal Complexes. Inorg. Chem. 2018, 57, 3387–3398. [Google Scholar] [CrossRef]
- Knight, F.R.; Randall, R.A.M.; Roemmele, T.L.; Boeré, R.T.; Bode, B.E.; Crawford, L.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Electrochemically Informed Synthesis: Oxidation versus Coordination of 5,6-Bis(phenylchalcogeno)acenaphthenes. ChemPhysChem 2013, 14, 2199–3203. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, B.A.; Bühl, M.; Athukorala Arachcige, K.S.; Slawin, A.M.Z.; Kilian, P. A Strutural, Spectroscopic, and Computational Examination of the Dative Interaction in Constrained Phosphine-Stibines and Phosphine-Stiboranes. Chem. Eur. J. 2015, 21, 7520–7531. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, B.A.; Bühl, M.; Athukorala Arachcige, K.S.; Slawin, A.M.Z.; Kilian, P. Geometrically Enforced Donor-Facilitated De-hydrocoupling Leading to an Isolable Arsanylidine-Phosphorane. J. Am. Chem. Soc. 2014, 136, 6247–6250. [Google Scholar] [CrossRef] [PubMed]
- Nejman, P.S.; Curzon, T.E.; Bühl, M.; McKay, D.; Woollins, J.D.; Ashbrook, S.E.; Cordes, D.B.; Slawin, A.M.Z.; Kilian, P. Phosphorus-Bismuth peri-Substituted Acenaphthenes: A Synthetic, Structural, and Computational Study. Inorg. Chem. 2020, 59, 5616–5625. [Google Scholar] [CrossRef]
- Nordheider, A.; Hupf, E.; Chalmers, B.A.; Knight, F.R.; Bühl, M.; Mebs, S.; Checinska, L.; Lork, E.; Camacho, P.S.; Ashbrook, S.E.; et al. Peri-Substituted Phosphorus–Tellurium Systems—An Experimental and Theoretical Investigation of the P···Te through-Space Interaction. Inorg. Chem. 2015, 54, 2435–2446. [Google Scholar] [CrossRef]
- Hupf, E.; Lork, E.; Mebs, S.; Checinska, L.; Beckmann, J. Probing Donor−Acceptor Interactions in peri-Substituted Diphenylphosphinoacenaphthyl−Element Dichlorides of Group 13 and 15 elements. Organometallics 2014, 33, 7247–7259. [Google Scholar] [CrossRef]
- Hupf, E.; Lork, E.; Mebs, S.; Beckmann, J. 6-Diphenylphosphinoacenaphth-5-yl-mercurials as Ligands for d10 Metals. Observation of Closed-Shell Interactions of the Type Hg(II)···M; M = Hg(II), Ag(I), Au(I). Inorg. Chem. 2015, 54, 1847–1859. [Google Scholar] [CrossRef]
- Furan, S.; Vogt, M.; Winkels, K.; Lork, E.; Mebs, S.; Hupf, E.; Beckmann, J. (6-Diphenylphosphinoacenaphth-5-yl)indium and -nickel Compounds: Synthesis, Structure, Transmetalation, and Cross-Coupling Reactions. Organometallics 2021, 40, 1284–1295. [Google Scholar] [CrossRef]
- Kordts, N.; Künzler, S.; Rathjen, S.; Sieling, T.; Großekappenberg, H.; Schmidtmann, M.; Müller, T. Silyl Chalconium Ions: Synthesis, Structure and Application in Hydrodefluorination Reactions. Chem. Eur. J. 2017, 23, 10068–10079. [Google Scholar] [CrossRef] [PubMed]
- Hierso, J.C. Indirect Nonbonded Nuclear Spin−Spin Coupling: A Guide for the Recognition and Understanding of “Through-Space” NMR J Constants in Small Organic, Organometallics, and Coordination Compounds. Chem. Rev. 2014, 114, 4838–4867. [Google Scholar] [CrossRef] [PubMed]
- Athukorala Arachcige, K.S.; Camacho, P.S.; Ray, M.J.; Chalmers, B.A.; Knight, F.R.; Ashbrook, S.E.; Bühl, M.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Sterically Restricted Tin Phosphines, Stabilised by Weak Intramolecular Donor-Acceptor Interactions. Organometallics 2014, 33, 2424–2433. [Google Scholar] [CrossRef]
- Hupf, E.; Lork, E.; Mebs, S.; Beckmann, J. Intramolecularly Coordinated (6-(Diphenylphosphino)acenapth-5-yl)stannanes. Repulsion vs Attraction of P- and Sn-Containing Substituents in the peri Positions. Organometallics 2014, 33, 2409–2423. [Google Scholar] [CrossRef]
- Knight, F.R.; Fuller, A.L.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Sterically Crowded peri-Substituted Naphthalene Phosphines and their PV Derivatives. Chem. Eur. J. 2010, 16, 7617–7634. [Google Scholar] [CrossRef] [PubMed]
- Malkina, O.L.; Hierso, J.-C.; Malkin, V.G. Distinguishing “Through-Space” from “Through-Bonds” Contribution in Indirect Nuclear Spin−Spin Coupling; General Approaches Applied to Complex JPP and JPSe Scalar Couplings. J. Am. Chem. Soc. 2022, 144, 10768–10784. [Google Scholar] [CrossRef]
- Zhang, L.; Christie, F.A.; Tarcza, A.E.; Lancaster, H.G.; Taylor, L.J.; Bühl, M.; Malkinia, O.L.; Woollins, J.D.; Carpenter-Warren, C.L.; Cordes, D.B.; et al. Phosphine and Selenoether peri-Substituted Acenaphthenes and Their Transition-Metal Complexes: Structural and NMR Investigations. Inorg. Chem. 2023, 62, 16084–16100. [Google Scholar] [CrossRef]
- Chernick, C.L.; Skinner, H.A. 285. Thermochemistry of organophosphorus compounds. Part II. Triethyl phoshate, tripropylphosphine oxide, and tributylphosphine oxide. J. Chem. Soc. 1956, 1401–1405. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Meguerian, G. Reactions of Elemental Sulfur. I. The Uncatalysed Teaction of Sulfur with Triarylphosphines. J. Am. Chem. Soc. 1956, 78, 3701–3715. [Google Scholar] [CrossRef]
- Capps, K.B.; Wixmerten, B.; Bauer, A.; Hoff, C.D. Thermochemistry of Sulfur Atom Transfer. Enthalpies of Reaction of Phosphines with Sulfur, Selenium, and Tellurium, and of Desulfurization of Triphenylarsenic Sulfide, Triphenylantimony Sulfide, and Benzyl Trisulfide. Inorg. Chem. 1998, 37, 2861–2864. [Google Scholar] [CrossRef]
- Cinderalla, A.P.; Vulovic, B.; Watson, D.A. Palladium-Catalysed Cross-Coupling of Silyl Electrophiles with Akylzinc Halides: A Silyl-Negishi Reaction. J. Am. Chem. Soc. 2017, 139, 7741–7744. [Google Scholar] [CrossRef] [PubMed]
- Kilian, P.; Milton, H.L.; Slawin, A.M.Z.; Woollins, J.D. Chlorides, Oxochlorides, and Oxoacids of 1,8-Diphosphanaphthalene: A System with Imposed Close P···P Interaction. Inorg. Chem. 2004, 43, 2252–2260. [Google Scholar] [CrossRef] [PubMed]
- Eventova, V.A.; Belov, K.V.; Efimov, S.V.; Khodov, I.A. Conformational Screening of Arbidol Solvates: Investigation via 2D NOESY. Pharmaceutics 2023, 15, 226. [Google Scholar] [CrossRef]
- Pyykko, P.; Atsumi, M. Molecular Single-Bond Covalent Radii for Elements 1–118. Chem. Eur. J. 2009, 15, 186−197. [Google Scholar] [CrossRef] [PubMed]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent ver der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- CrystalStructure 4.3.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5642–5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a function of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Binning, R.C.; Curtiss, L.A. Compact contracted basis sets for third-row atoms: Ga–Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Kreig, H. A consistent and accurate ab initio parametrization of density function dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. Exchange-hole dipole moment and the dispersion interaction. J. Chem. Phys. 2005, 122, 154104. [Google Scholar] [CrossRef]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
- Kutzelnigg, W.; Fleischer, U.; Schindler, M. The IGLO-Method: Ab Initio Calculation and Interpretation of NMR Chemical Shifts and Magnetic Susceptibilities. In NMR Basic Principles and Progress; Springer: Berlin/Heidelberg, Germany, 1990; Volume 23. [Google Scholar] [CrossRef]
- Albright, T.A.; Freeman, W.J.; Schweizer, E.E. Nuclear Magnetic Resonance Studies. IV. The Carbon and Phosphorus Nuclear Magnetic Resonance of Phosphine Oxides and Related Compounds. J. Org. Chem. 1975, 40, 3437–3441. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1-O | 1-S | 1-Se | 1-O2 [b] |
|---|---|---|---|---|
| peri-region bond distances | ||||
| P1···Se1 | 3.322(2) | 3.4863(5) | 3.5012(7) | 3.578(1) [3.610(1)] |
| P1-E | 1.491(6) | 1.9657(5) | 2.1219(7) | 1.487(4) [1.492(4)] |
| Se1···E | 2.825(6) | 3.2272(5) | 3.2829(6) | 2.646(3) [2.625(3)] |
| Se1-O1 | − | − | − | 1.669(3) [1.671(4)] |
| peri-region bond angles | ||||
| C9-P1-E | 110.7(4) | 112.39(5) | 112.40(8) | 112.7(2) [113.1(2)] |
| P1-E-Se1 | 95.7(3) | 80.36(2) | 77.47(2) | 117.1(2) [118.5(2)] |
| E-Se1-C19 | 165.4(3) | 164.53(5) | 166.29(8) | 84.6(2) [86.4(2)] |
| O2-Se1-O1 | − | − | − | 169.3(1) [169.2(2)] |
| C1-Se1-C19 | 99.3(3) | 97.01(6) | 96.8(1) | 97.7(2) [95.4(2)] |
| Splay [a] | 18.5 | 19.9 | 20.2 | 28.1 [29.1] |
| dihedral angles | ||||
| C9-C10-C5-C4 | 174.8(8) | 173.7(1) | 173.2(2) | 178.3(5) [177.4(5)] |
| P1-C9···C1-Se1 | 22.2(5) | 31.87(8) | 32.0(1) | 1.8(3) [1.7(3)] |
| out-of-plane displacements | ||||
| P1 | 0.508 | 0.605 | 0.593 | 0.065 [0.007] |
| Se1 | −0.393 | −0.700 | −0.725 | 0.006 [0.128] |
| Molecule | ΔE | ΔH298 | ΔG298 | δ(P) | δ(Se = P) | δ(SePh) |
|---|---|---|---|---|---|---|
| 1-Se(A) | 0 | 0 | 0 | 61.7 58.0 b | −389.9 −362.6 b | 385.1 415.3 b |
| 1-Se(B) | 19.0 | 19.7 | 23.5 | |||
| 1-Se(C) | 32.8 | 33.8 | 34.3 | |||
| 1-Se(D) | −2.6 | −2.4 | −2.4 | 89.1 86.2 b | −555.7 −452.4 b | 433.8 422.8 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarcza, A.E.; Slawin, A.M.Z.; Carpenter-Warren, C.L.; Bühl, M.; Kilian, P.; Chalmers, B.A. Constrained Phosphine Chalcogenide Selenoethers Supported by peri-Substitution. Molecules 2023, 28, 7297. https://doi.org/10.3390/molecules28217297
Tarcza AE, Slawin AMZ, Carpenter-Warren CL, Bühl M, Kilian P, Chalmers BA. Constrained Phosphine Chalcogenide Selenoethers Supported by peri-Substitution. Molecules. 2023; 28(21):7297. https://doi.org/10.3390/molecules28217297
Chicago/Turabian StyleTarcza, Anna E., Alexandra M. Z. Slawin, Cameron L. Carpenter-Warren, Michael Bühl, Petr Kilian, and Brian A. Chalmers. 2023. "Constrained Phosphine Chalcogenide Selenoethers Supported by peri-Substitution" Molecules 28, no. 21: 7297. https://doi.org/10.3390/molecules28217297
APA StyleTarcza, A. E., Slawin, A. M. Z., Carpenter-Warren, C. L., Bühl, M., Kilian, P., & Chalmers, B. A. (2023). Constrained Phosphine Chalcogenide Selenoethers Supported by peri-Substitution. Molecules, 28(21), 7297. https://doi.org/10.3390/molecules28217297