

Mechanistic Studies on Aluminum-Catalyzed Ring-Opening Alternating Copolymerization of Maleic Anhydride with Epoxides: Ligand Effects and Quantitative Structure-Activity Relationship Model
 , , ,
, , , 
Abstract
:1. Introduction
2. Results and Discussion
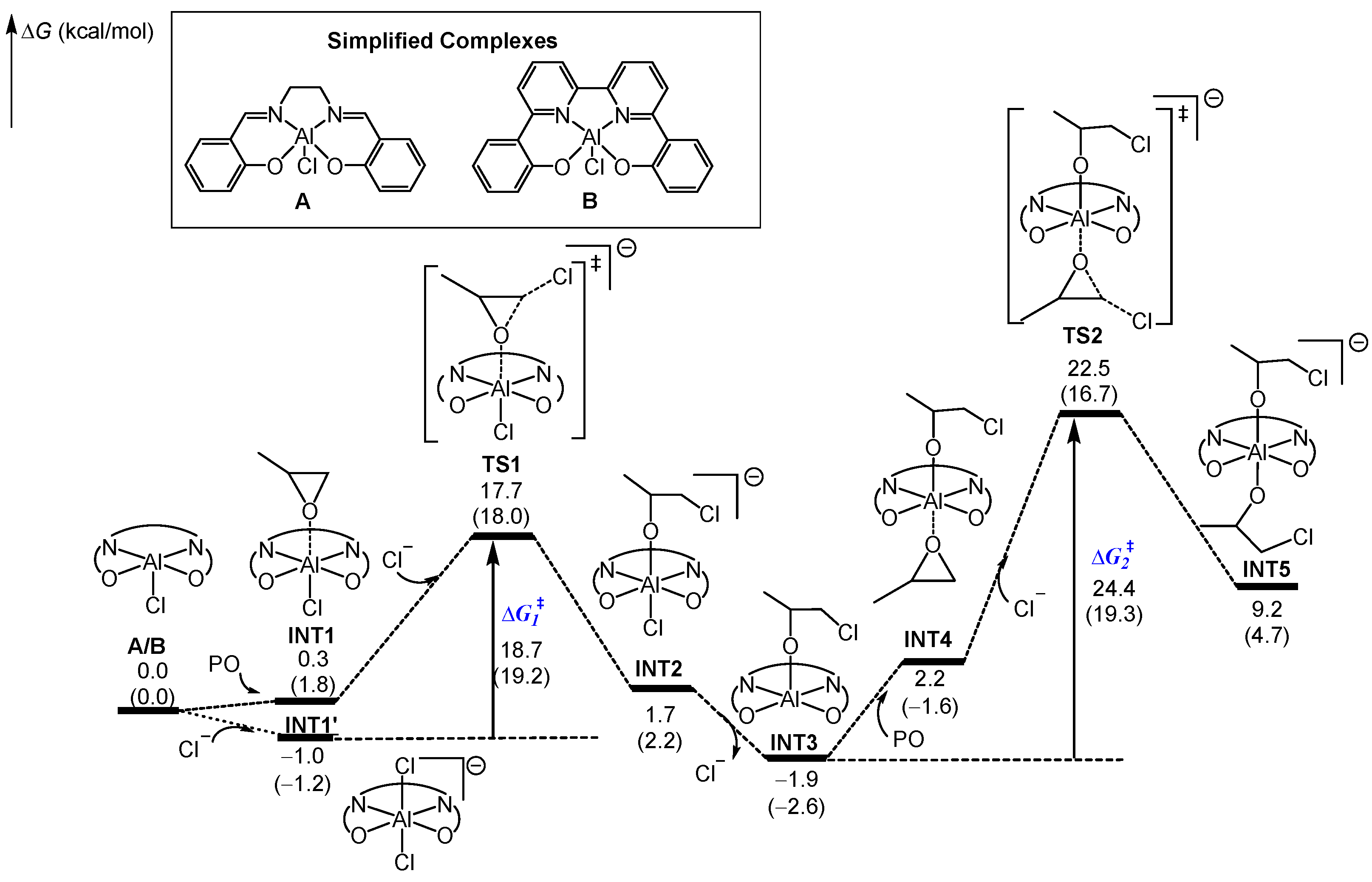
2.1. Chain Initiation Stage
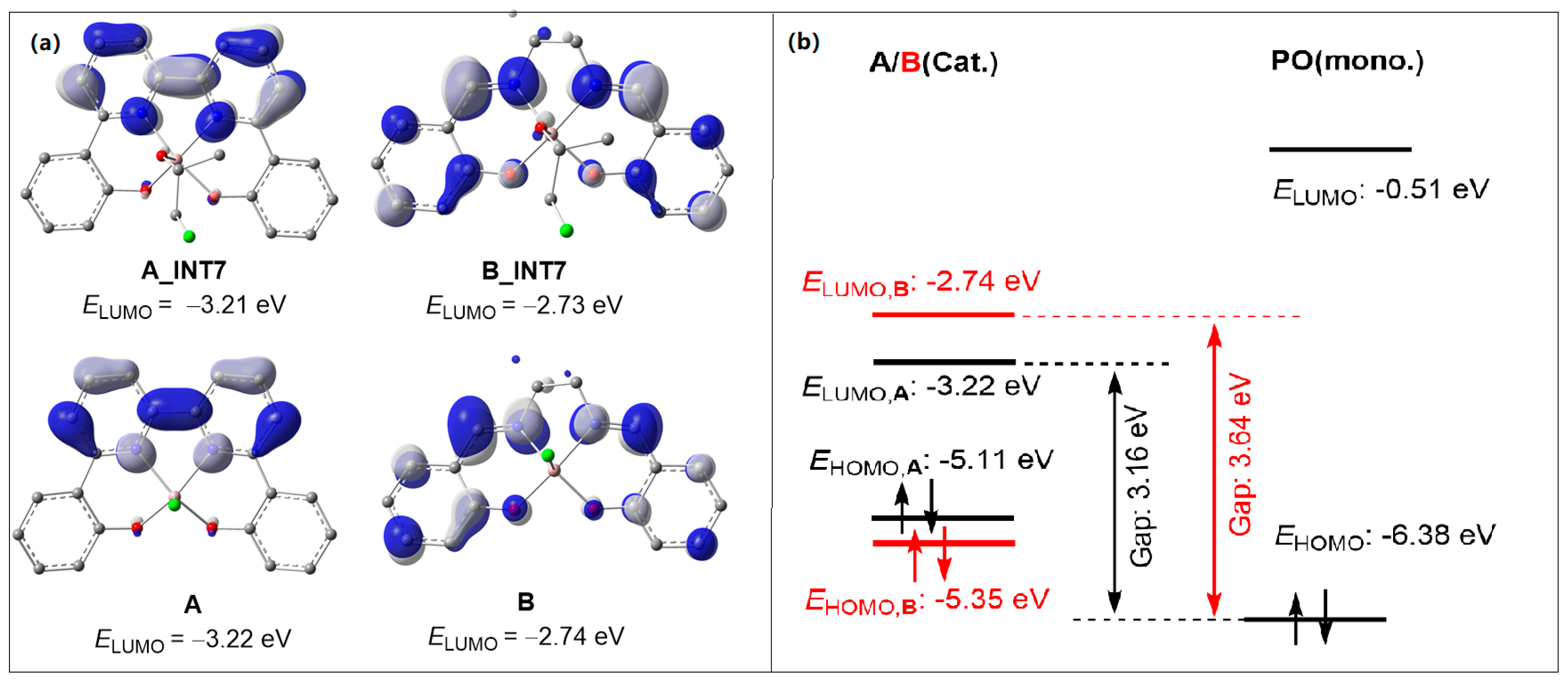
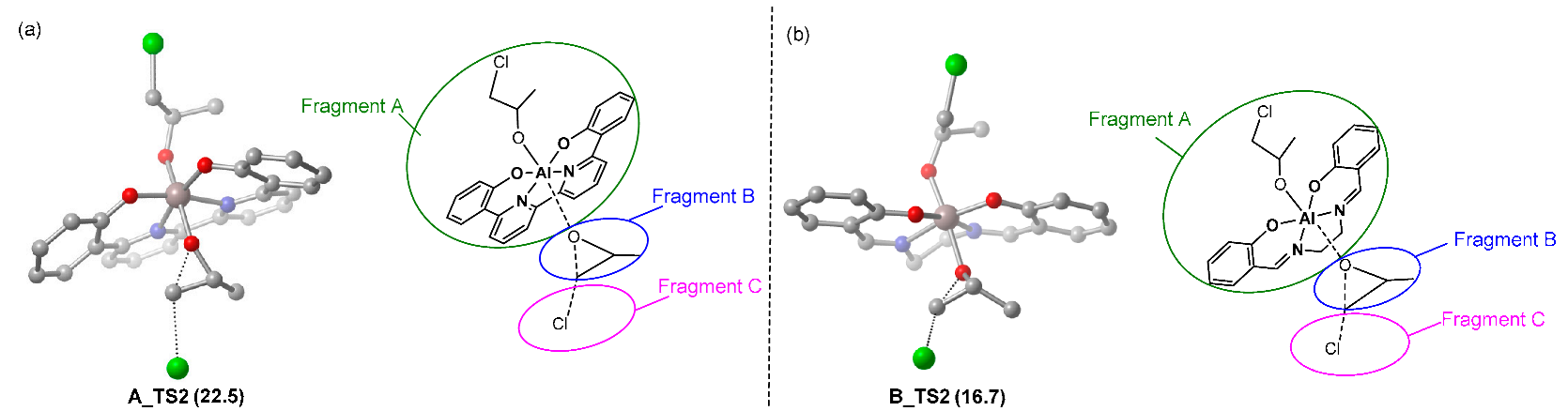
2.2. Chain Propagation Stage
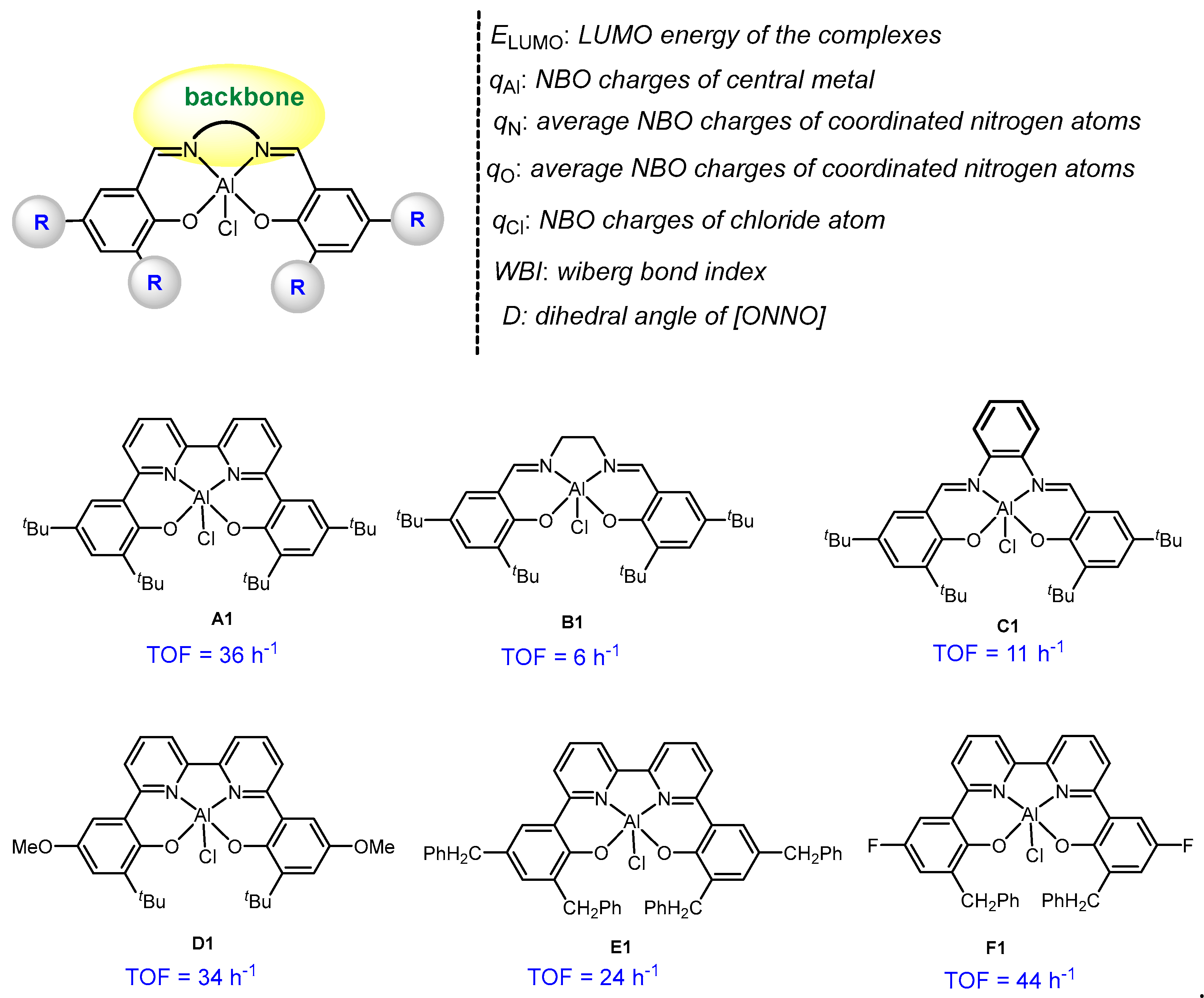
2.3. Multivariate Linear Regression Analysis
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Müller, R.J.; Kleeberg, I.; Deckwer, W.D. Biodegradation of polyesters containing aromatic constituents. J. Biotechnol. 2001, 86, 87. [Google Scholar] [CrossRef] [PubMed]
- Olson, D.A.; Gratton, S.E.A.; Desimone, J.M.; Sheares, V.V. Amorphous Linear Aliphatic Polyesters for the Facile Preparation of Tunable Rapidly Degrading Elastomeric Devices and Delivery Vectors. J. Am. Chem. Soc. 2006, 128, 13625. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.H.; Sheares, V.V. Amorphous Unsaturated Aliphatic Polyesters Derived from Dicarboxylic Monomers Synthesized by Diels-Alder Chemistry. Macromolecules 2007, 40, 4848. [Google Scholar] [CrossRef]
- Paul, S.; Zhu, Y.; Romain, C.; Brooks, R.; Saini, P.K.; Williams, C.K. Ring-opening copolymerization (ROCOP): Synthesis and properties of polyesters and polycarbonates. Chem. Commun. 2015, 51, 6459. [Google Scholar] [CrossRef] [PubMed]
- Longo, J.M.; Sanford, M.J.; Coates, G.W. Ring-Opening Copolymerization of Epoxides and Cyclic Anhydrides with Discrete Metal Complexes: Structure−Property Relationships. Chem. Rev. 2016, 116, 15167. [Google Scholar] [CrossRef]
- Stößer, T.; Chen, T.T.D.; Zhu, Y.; Williams, C.K. ‘Switch’ catalysis: From monomer mixtures to sequence-controlled block copolymers. Philos. Trans. Royal. Soc. A Math. Phys. Eng. Sci. 2017, 376, 20170066. [Google Scholar] [CrossRef]
- Lustoň, J.; Maňasek, Z. Copolymerization of Epoxides with Cyclic Anhydrides Catalyzed by Tertiary Amines in the Presence of Proton-Donating Compounds. J. Macromol. Sci. Part A Chem. 1979, 13, 853. [Google Scholar] [CrossRef]
- Lustoň, J.; Vašš, F. Anionic Copolymerization of Cyclic Ethers with Cyclic Anhydrides. Adv. Polym. Sci. 1984, 54, 91. [Google Scholar]
- Aida, T.; Inoue, S. Catalytic Reaction on Both Sides of a Metalloporphyrin Plane. Alternating Copolymerization of Phthalic Anhydride and Epoxypropane with an Aluminum Porphyrin-Quaternary Salt System. J. Am. Chem. Soc. 1985, 107, 1358. [Google Scholar] [CrossRef]
- Jeske, R.C.; DiCiccio, A.M.; Coates, G.W. Alternating Copolymerization of Epoxides and Cyclic Anhydrides: An Improved Route to Aliphatic Polyesters. J. Am. Chem. Soc. 2007, 129, 11330. [Google Scholar] [CrossRef]
- Jeske, R.C.; Rowley, J.M.; Coates, G.W. Pre-rate-determining selectivity in the terpolymerization of epoxides, cyclic Anhydrides, and CO2: A one-step route to diblock copolymers. Angew. Chem. Int. Ed. 2008, 47, 6041. [Google Scholar] [CrossRef] [PubMed]
- Huijser, S.; HosseiniNejad, E.; Sablong, R.; de Jong, C.; Koning, C.E.; Duchateau, R. Ring-Opening Co- and Terpolymerization of an Alicyclic Oxirane with Carboxylic Acid Anhydrides and CO2 in the Presence of Chromium Porphyrinato and Salen Catalysts. Macromolecules 2011, 44, 1132. [Google Scholar] [CrossRef]
- Robert, C.; de Montigny, F.; Thomas, C.M. Tandem synthesis of alternating polyesters from renewable resources. Nat. Commun. 2011, 2, 586. [Google Scholar] [CrossRef]
- DiCiccio, A.M.; Coates, G.W. Ring-Opening Copolymerization of Maleic Anhydride with Epoxides: A Chain-Growth Approach to Unsaturated Polyesters. J. Am. Chem. Soc. 2011, 133, 10724. [Google Scholar] [CrossRef] [PubMed]
- Hosseini Nejad, E.; van Melis, C.G.W.; Vermeer, T.J.; Koning, C.E.; Duchateau, R. Alternating Ring-Opening Polymerization of Cyclohexene Oxide and Anhydrides: Effect of Catalyst, Cocatalyst, and Anhydride Structure. Macromolecules 2012, 45, 1770. [Google Scholar] [CrossRef]
- Liu, F.P.; Li, J.; Liu, Y.; Ren, W.M.; Lu, X.B. Alternating Copolymerization of trans-Internal Epoxides and Cyclic Anhydrides Mediated by Dinuclear Chromium Catalyst Systems. Macromolecules 2019, 52, 5652. [Google Scholar] [CrossRef]
- Winkler, M.; Romain, C.; Meier, M.A.R.; Williams, C.K. Renewable polycarbonates and polyesters from 1,4-cyclohexadiene. Green Chem. 2015, 17, 300. [Google Scholar] [CrossRef]
- Jeon, J.Y.; Eo, S.C.; Varghese, J.K.; Lee, B.Y. Copolymerization and terpolymerization of carbon dioxide/propylene oxide/phthalic anhydride using a (salen)Co(III) complex tethering four quaternary ammonium salts. Beilstein J. Org. Chem. 2014, 10, 1787. [Google Scholar] [CrossRef]
- Duan, Z.; Wang, X.; Gao, Q.; Zhang, L.; Liu, B.; Kim, I. Highly active bifunctional cobalt-salen complexes for the synthesis of poly(ester-block-carbonate) copolymerviaterpolymerization of carbon dioxide, propylene oxide, and norbornene anhydride isomer: Roles of anhydride conformation consideration. J. Polym. Sci. A Polym. Chem. 2014, 52, 789. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Z.; Zhang, X.; Lü, X. Alternating Ring-Opening Copolymerization of Cyclohexene Oxide and Maleic Anhydride with Diallyl-Modified Manganese(III)–Salen Catalysts. Aust. J. Chem. 2016, 69, 47. [Google Scholar] [CrossRef]
- Mundil, R.; Hošt’álek, Z.; Šeděnková, I.; Merna, J. Alternating ring-opening copolymerization of cyclohexene oxide with phthalic anhydride catalyzed by iron(III) salen complexes. Macromol. Res. 2015, 23, 161. [Google Scholar] [CrossRef]
- Chen, X.L.; Wang, B.; Pan, L.; Li, Y.S. Synthesis of Unsaturated (Co)polyesters from Ring-Opening Copolymerization by Aluminum Bipyridine Bisphenolate Complexes with Improved Protonic Impurities Tolerance. Macromolecules 2022, 55, 3502. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G.; McCullough, L. DFT and QSAR Studies of Ethylene Polymerization by Zirconocene Catalysts. ACS Catal. 2019, 9, 9339. [Google Scholar] [CrossRef]
- Yang, W.; Ma, Z.; Yi, J.; Ahmed, S.; Sun, W.H. Catalytic performance of bis(imino)pyridine Fe/Co complexes toward ethylene polymerization by 2D-/3D-QSPR modeling. J. Comput. Chem. 2019, 40, 1374. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Wang, J. Quantum chemical descriptors based QSAR modeling of neodymium carboxylate catalysts for coordination polymerization of isoprene. Chin. J. Chem. Eng. 2020, 28, 1805. [Google Scholar] [CrossRef]
- Zhao, Y.; Lu, H.; Luo, G.; Kang, X.; Hou, Z.; Luo, Y. Origin of stereoselectivity and multidimensional quantitative analysis of ligand effects on yttrium-catalysed polymerization of 2-vinylpyridine. Catal. Sci. Technol. 2019, 9, 6227. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Poland, R.R.; Escobedo, C. Kinetic Studies of the Alternating Copolymerization of Cyclic Acid Anhydrides and Epoxides, and the Terpolymerization of Cyclic Acid Anhydrides, Epoxides, and CO2 Catalyzed by (salen)CrIIICl. Macromolecules 2012, 45, 2242. [Google Scholar] [CrossRef]
- Fieser, M.E.; Sanford, M.J.; Mitchell, L.A.; Dunbar, C.R.; Mandal, M.; Van Zee, N.J.; Urness, D.M.; Cramer, C.J.; Coates, G.W.; Tolman, W.B. Mechanistic Insights into the Alternating Copolymerization of Epoxides and Cyclic Anhydrides Using a (Salph)AlCl and Iminium Salt Catalytic System. J. Am. Chem. Soc. 2017, 139, 15222. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070. [Google Scholar] [CrossRef]
- Kitaura, K.; Morokuma, K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Y.; Houk, K.N. Bioorthogonal cycloadditions: Computational analysis with the distortion/interaction model and predictions of reactivities. Acc. Chem. Res. 2017, 50, 2297. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2007, 120, 215. [Google Scholar]
- Fukui, K. The Path of chemical reactions-The IRC approach. Acc. Chem. Res. 1981, 14, 363. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of equilibrium geometries and transition structures. J. Comput. Chem. 1982, 3, 214. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theoret. Chim. Acta 1973, 28, 213. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378. [Google Scholar] [CrossRef]
- Zhang, D.D.; Feng, X.; Gnanou, Y.; Huang, K.W. Theoretical Mechanistic Investigation into Metal-Free Alternating Copolymerization of CO2 and Epoxides: The Key Role of Triethylborane. Macromolecules 2018, 51, 5600. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B Condens. Matter Mater. Phys. 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H.A. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Legault, C.Y. Cylview; Version 1.0 b; Universitéde Sherbrooke: Sherbrooke, QC, Canada, 2009; Available online: https://www.cylview.org/ (accessed on 23 October 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Catalyst | t (h) | Conv. (%) | TOF (h−1) | Ester Linkage (%) | Mn,thero (kDa) | Mn,sec (kDa) | PDI |
| A1/PPNCl | 2 | 71 | 36 | >99 | 5.6 | 4.6 | 1.28 |
| B1/PPNCl | 2 | 12 | 6.0 | >99 | 0.9 | n.d. | n.d. |
 | |||||||
|---|---|---|---|---|---|---|---|
| TS2 | ΔG‡ | ΔEint(A-B-C) | ΔEdist(A) | ΔEdist(B) | ΔEdist(C) | ΔEdist | ΔE‡ |
| A_TS2 | 24.4 | −33.9 | 17.9 | 20.0 | 0.0 | 37.9 | 3.9 |
| B_TS2 | 19.3 | −34.1 | 15.0 | 18.6 | 0.0 | 33.6 | −0.5 |
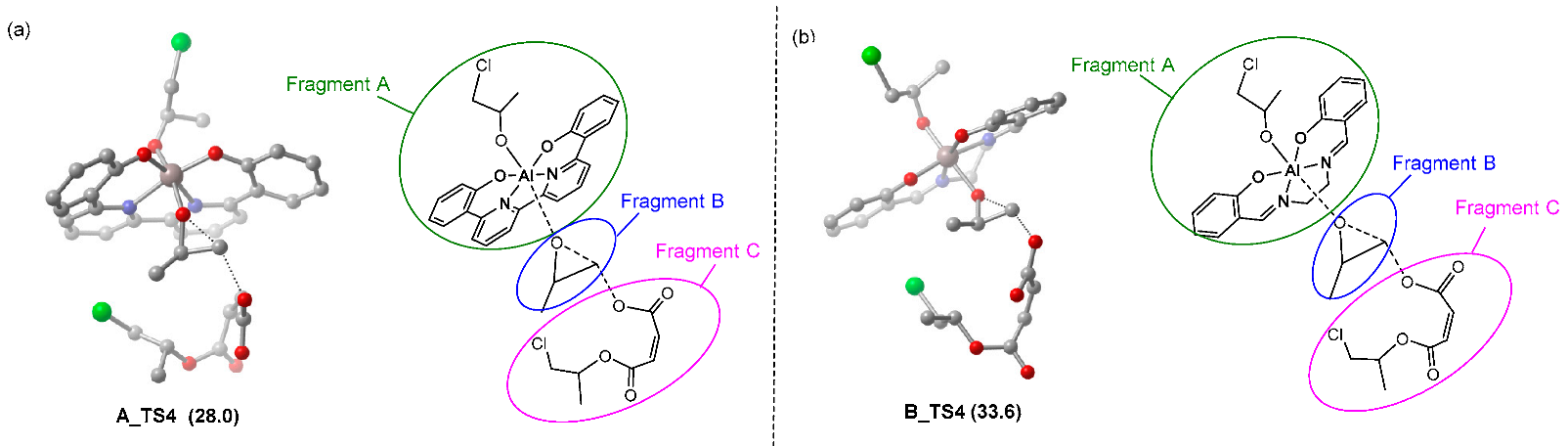 | |||||||
|---|---|---|---|---|---|---|---|
| TS4 | ΔG‡ | ΔEint(A-B-C) | ΔEdist(A) | ΔEdist(B) | ΔEdist(C) | ΔEdist | ΔE‡ |
| A_TS4 | 33.3 | −46.1 | 18.0 | 23.3 | 1.0 | 42.4 | −3.7 |
| B_TS4 | 42.5 | −39.5 | 14.8 | 26.1 | 0.2 | 41.2 | 1.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Li, H.; Mehmood, A.; Chi, K.; Shi, D.; Wang, Z.; Wang, B.; Li, Y.; Luo, Y. Mechanistic Studies on Aluminum-Catalyzed Ring-Opening Alternating Copolymerization of Maleic Anhydride with Epoxides: Ligand Effects and Quantitative Structure-Activity Relationship Model. Molecules 2023, 28, 7279. https://doi.org/10.3390/molecules28217279
Xu X, Li H, Mehmood A, Chi K, Shi D, Wang Z, Wang B, Li Y, Luo Y. Mechanistic Studies on Aluminum-Catalyzed Ring-Opening Alternating Copolymerization of Maleic Anhydride with Epoxides: Ligand Effects and Quantitative Structure-Activity Relationship Model. Molecules. 2023; 28(21):7279. https://doi.org/10.3390/molecules28217279
Chicago/Turabian StyleXu, Xiaowei, Hao Li, Andleeb Mehmood, Kebin Chi, Dejun Shi, Zhuozheng Wang, Bin Wang, Yuesheng Li, and Yi Luo. 2023. "Mechanistic Studies on Aluminum-Catalyzed Ring-Opening Alternating Copolymerization of Maleic Anhydride with Epoxides: Ligand Effects and Quantitative Structure-Activity Relationship Model" Molecules 28, no. 21: 7279. https://doi.org/10.3390/molecules28217279
APA StyleXu, X., Li, H., Mehmood, A., Chi, K., Shi, D., Wang, Z., Wang, B., Li, Y., & Luo, Y. (2023). Mechanistic Studies on Aluminum-Catalyzed Ring-Opening Alternating Copolymerization of Maleic Anhydride with Epoxides: Ligand Effects and Quantitative Structure-Activity Relationship Model. Molecules, 28(21), 7279. https://doi.org/10.3390/molecules28217279