Abstract
Replacing expensive platinum oxygen reduction reaction (ORR) catalysts with atomically dispersed single-atom catalysts is an effective way to improve the energy conversion efficiency of fuel cells. Herein, a series of single-atom catalysts, TM-N2O2Cx (TM=Sc-Zn) with TM-N2O2 active units, were designed, and their catalytic performance for electrocatalytic O2 reduction was investigated based on density functional theory. The results show that TM-N2O2Cx exhibits excellent catalytic activity and stability in acidic media. The eight catalysts (TM=Sc, Ti, V, Cr, Mn, Fe, Co, and Ni) are all 4e− reaction paths, among which Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx follow dissociative mechanisms and the rest are consistent with associative mechanisms. In particular, Co-N2O2Cx and Ni-N2O2Cx enable a smooth reduction in O2 at small overpotentials (0.44 V and 0.49 V, respectively). Furthermore, a linear relationship between the adsorption free energies of the ORR oxygen-containing intermediates was evident, leading to the development of a volcano plot for the purpose of screening exceptional catalysts for ORR. This research will offer a novel strategy for the design and fabrication of exceptionally efficient non-precious metal catalysts on an atomic scale.
1. Introduction
Fuel cells (FCs) have garnered much attention as a technology for converting chemical energy into electrical energy because of their high energy conversion efficiency, high energy density, and non-polluting characteristics [1,2]. Compared with the anode reaction, the oxygen reduction reaction (ORR) at the cathode is kinetically slow, and in-depth research on the ORR process is of great significance for improving the overall performance of FCs [3,4,5]. Superior catalysts can be effective in solving the bottleneck problem of ORR. Among the many developed catalysts, Pt and its alloys are considered the best ORR catalysts for their high current density and low initial voltage. However, the scarcity of Pt resources, expensive price, poor durability, and easy deactivation limit its large-scale commercial application. Therefore, developing ORR electrocatalysts with a high catalytic activity, cheap availability, and low overpotential has recently been a hot research topic [6,7,8].
Single-atom catalysts (SACs) can achieve a high dispersion of metal atoms, which not only maximizes the utilization of metal atoms but also significantly improves the catalytic activity of the catalyst [9,10,11,12]. Since Pt1/FeOx single-atom catalysts were first reported by Zhang et al., a series of SACs have been notified and widely used electrocatalysis [13]. Guo et al. investigated the catalytic performance of a transition metal-anchored N-doped graphene single-atom catalyst (TMNx-GR) for electrocatalytic CO2 reduction based on density functional theory [14]. The results showed that TMNx-GR exhibited good catalytic activity, with NiN4-GR showing the best selectivity for the product CO. Chen’s group successfully constructed single-atom Cu-containing catalysts on N-doped carbon nanosheets and applied them to overall water splitting [15]. The results indicate that the oxygen evolution reaction at 200 mV and the hydrogen evolution reaction at 216 mV was achieved at a current density of 10 mA·cm−2, providing a new strategy for constructing non-precious metal catalysts. Wang et al. investigated the potential of transition metal-embedded g-C4N3 (TM@g-C4N3) single-atom catalysts for nitrogen reduction reactions (NRR) by a high-throughput screening system. Among the 30 candidate materials [4], V@g-C4N3 was identified as the most active NRR catalyst, with a limiting potential of −0.37 V. This work leads the way for the construction of efficient NRR single-atom catalysts using g-C4N3 as a novel carrier. Yi and co-workers prepared atomically dispersed Fe-Nx species (Fe loading up to 8.3 wt %) on porous porphyrin triazine-based frameworks (FeSAs/PTF) using a simple isothermal method [16]. FeSAs/PTF-600 has a high density of single-atom Fe-N4 active sites and a high layered porosity, and good electrical conductivity, resulting efficient activity, methanol resistance, and ORR superstability under alkaline and acidic conditions. Hunter et al. theoretically investigated the catalytic performance of N-doped graphene single-atom catalysts (M1M2@N6V4 and M1M2@N8V4, M = Co, Pt, Fe, Ni) for ORRs [17]. It was found that CoPt@N8V4 exhibited the most desirable ORR catalytic activity (ƞ = 0.30 V), and a basis for the screening of highly active ORR catalysts was proposed by volcano mapping.
Most of the reported ORR SACs are of metal–N4 coordination [18,19]. However, recent studies have found that single-atom catalysts with N and O co-coordination with metal atoms exhibit excellent catalytic activity, even better than metal–N4 coordination catalysts. Ge et al. investigated the catalytic performance of the oxygen reduction reaction (ORR) of M-N4-xOx (M=Fe, Co, and Ni; x = 1–4) in detail based on density functional theory [20]. The results show that Co-N3O1 and Ni-N2O2 exhibit the best catalytic activity, with overpotentials of 0.27 and 0.32 V, respectively, which is significantly better than the Pt catalysts. Electronic structure and density of states analyses revealed that one of the reasons for the higher activity of Co-N3O1 and Ni-N2O2 is their small energy gap. O doping can improve the electronic structure of the original catalyst, which can adjust the adsorption capacity of ORR intermediates. Dong et al. successfully synthesized a low-Mn-content single-atom catalyst (Mn-NO/CNs) with Mn-N2O2 sites, which exhibited good activity in the catalysis of CO2 [21]. The results show that the addition of oxygen atoms changed the coordination environment surrounding the Mn atoms, modifying the electronic structure of the catalyst and improving the catalytic efficiency of Mn-NO/CNs for CO2 reduction. Wei et al. designed seven two-dimensional (2D) metal–organic framework (MOF) materials with TMN2O2 ligand units and conducted theoretical investigations into their catalytic activities towards oxygen reduction and evolution reaction (ORR and OER). The findings demonstrate that CoN2O2 manifests superior catalytic performance, displaying low overpotential values of 0.33 V and 0.30 V for ORRs and OERs, respectively [22]. The above reports open up new avenues for the development of ORR catalysts.
Although TM-N2O2 active units have been reported as catalytic materials for ORR applications, it has not been explicitly addressed whether the catalytic activity of TM-N2O2 active units would be significantly influenced by ligand variations. Based on this, we devised a series of TM-N2O2 (TM=Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn) coordination-type single-atom catalysts (TM-N2O2Cx) by employing 4-hydroxy benzonitrile as the ligand and investigated their catalytic activities and reaction mechanisms for electrocatalytic O2 reduction in an acidic environment. The comprehensive analysis and discussion of the stability, reaction pathway, overpotentials, and electronic structure of TM-N2O2Cx elucidate that a majority of the catalysts exhibit remarkable catalytic activity. Furthermore, a comparative analysis of the work conducted by Wei et al [22]. revealed that changing the ligand does indeed exert an effect on the catalytic activity of the TM-N2O2 site, but without altering the trend of the volcano curve. These findings will serve as a stimulus for further experimental and theoretical explorations in ORRs.
2. Results and Discussion
2.1. The Structural Features of TM-N2O2Cx Single-Atom Catalysts
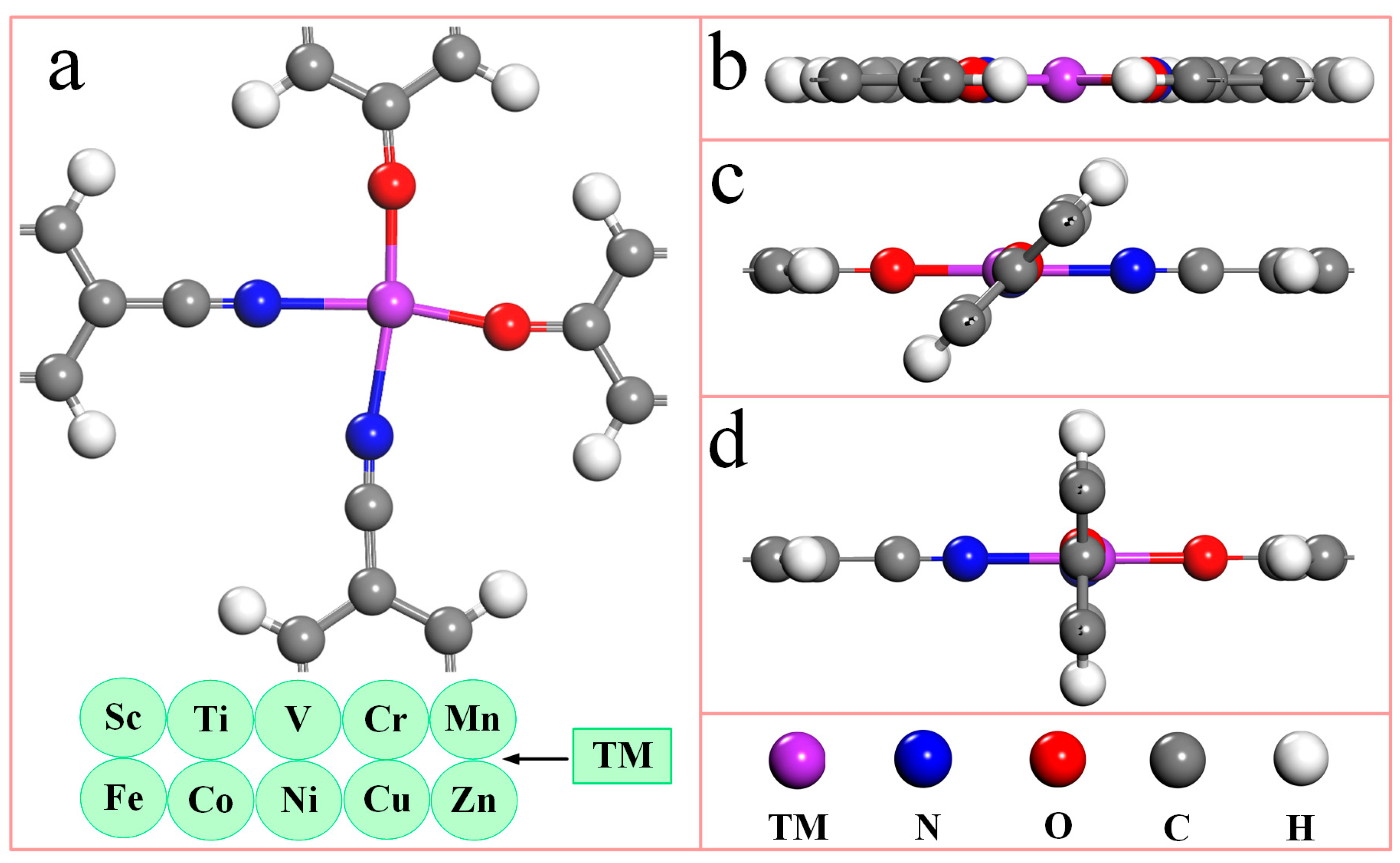
Figure 1 is the top (1a) and side views (1b-d) of the unit cell structure model of the TM-N2O2Cx single-atom catalyst. It can be seen from Figure 1a that the unit cell of the TM-N2O2Cx single-atom catalyst contains one transition metal atom, two nitrogen atoms, two oxygen atoms, fourteen carbon atoms, and eight hydrogen atoms, and each transition metal atom coordinates with two nitrogen atoms and two oxygen atoms simultaneously. Due to the different orientations of ligands in the bonding process, the TM-N2O2Cx single-atom catalyst has three initial configurations, as shown in Figure 1b–d, respectively. The structural optimization of ten SACs of the first transition metal series (Table S1) offering the stable configurations of Sc-N2O2Cx and Cu-N2O2Cx are demonstrated in Figure 1b,d, respectively. Structural optimization of ten single-atom catalysts (SACs) from the first transition metal series (refer to Table S1) reveals that stable configurations of Sc-N2O2Cx and Cu-N2O2Cx are illustrated in Figure 1b and 1d, respectively. In comparison, the regular structures of the remaining eight catalysts are shown in Figure 1c.
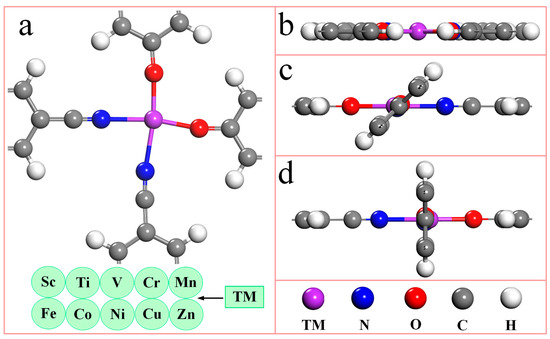
Figure 1.
The optimized geometric structures of TM-N2O2Cx single-atom catalysts. (a) Top view of unit cell. (b–d) Side views of unit cell. The purple, blue, red, gray, and white spheres represent TM, N, O, C, and H atoms, respectively. TM represents 10 metal atoms of the first transition metal series.
A Hirshfeld charges analysis shows that the metal atoms in the ten SACs all have partial positive charges [23], while the N and O atoms coordinated with them all have partial negative charges (Table S1), which indicates that there is charge transfer between the metal atoms and their neighboring atoms in the process of binding with the ligand so that the metal atoms can effectively bind with the ligand. Figure S1 is the projected partial density of states of the TM-N2O2Cx SACs. The green line is the 3d orbit of the metal atoms, while the blue and red lines are the 2p orbitals of the N and O atoms, respectively. The orbit’s degree of overlap can reflect the interatomic interaction’s strength. The better the overlap, the stronger the interaction; on the contrary, the weaker it is. Figure S1 shows that the 3d orbitals of the metal atoms and the N and O atoms in the ten poor catalysts overlap very well, proving that metal atoms have a strong coordination ability with N and O atoms in ligands. In addition, except for the metal atoms in Sc-N2O2Cx, Ni-N2O2Cx, and Zn-N2O2Cx, the metal atoms in the other SACs have magnetism and the Mn atom possesses the highest magnetic moment of 4.504 μB, which may also affect the catalytic activity of the catalysts.
2.2. The Structural Stability of TM-N2O2Cx
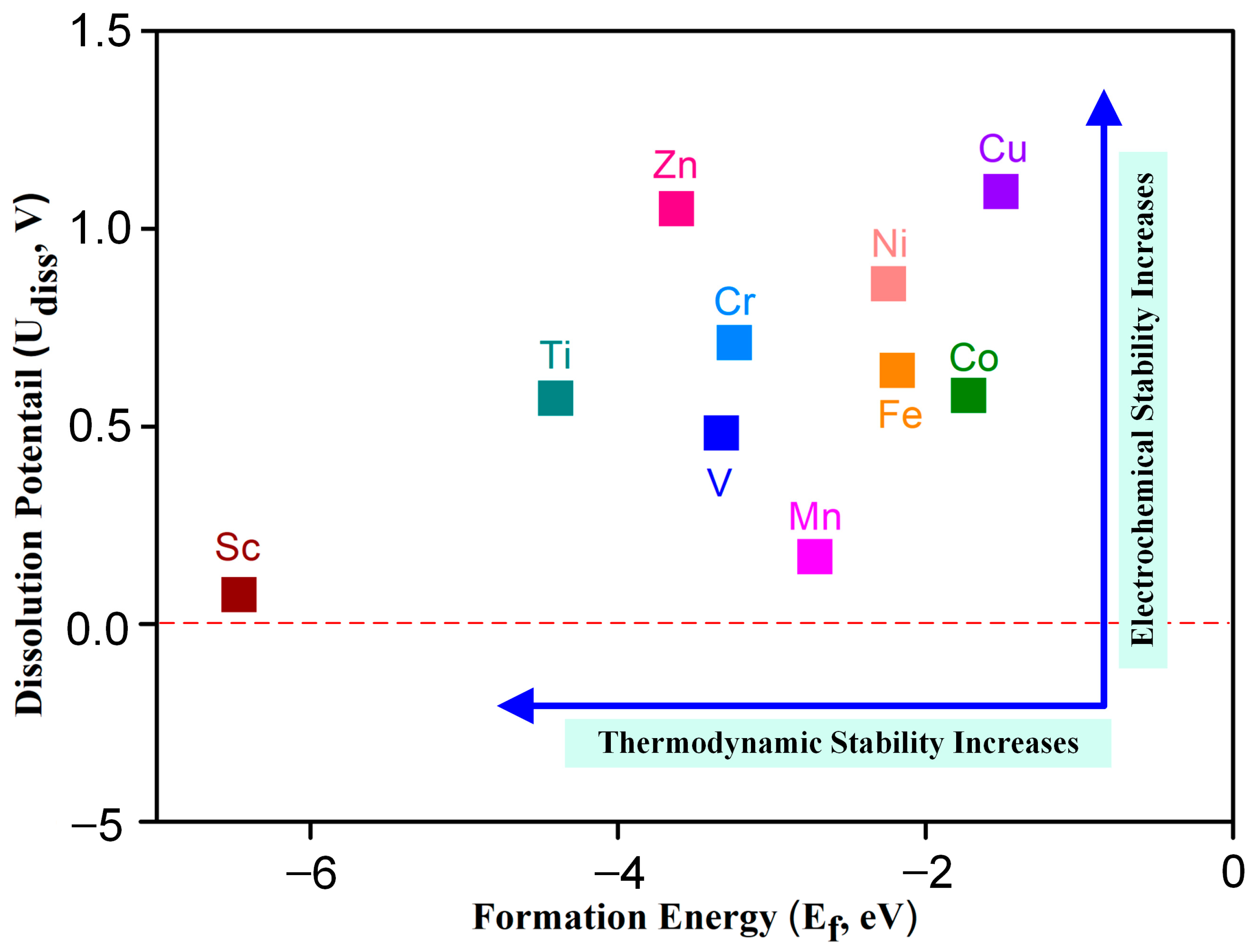
As the structural stability of the catalyst plays an essential role in maintaining the catalytic activity of the motivation, the thermodynamic and electrochemical stability of TM-N2O2Cx were studied according to the formation energy (Ef) and dissolution potential (Udiss) [24], respectively. Ef and Udiss can be obtained from the formulas Ef = ETM-N2O2Cx − ETM − EN2O2Cx (1) and Udiss = U°diss − Ef/ne (2) (detailed data are shown in Table S2), where ETM-N2O2Cx, ETM, and EN2O2Cx are the total energies of TM-N2O2Cx, TM, and N2O2Cx, respectively. U°diss and n are the standard dissolution potential and the number of electrons involved in the dissolution of transition metals, respectively. The negative value of formation energy indicates that it is an exothermic reaction in the process of a metal atom combining with a ligand to form a single-atom catalyst; so, the more negative the value of formation energy is, the stronger the thermodynamic stability of the catalyst is and vice versa. Figure 2 shows that the formation energies of ten kinds of SACs are negative, indicating that TM-N2O2Cx SACs offer good thermodynamic stability.
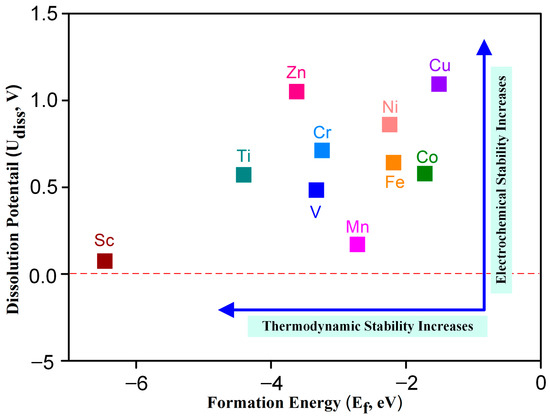
Figure 2.
Dissolution potential and formation energy of transition metal atoms in TM-N2O2Cx, the dash line represents a potential of 0 V.
During the process of electrocatalysis, if the dissolution potential of metal atoms in the single-atom catalyst is relatively low, it becomes easier for these metal atoms to undergo oxidation and subsequently dissolve into the solvent. This phenomenon ultimately leads to a substantial decrease in the overall catalytic activity exhibited by the catalyst. Consequently, in order to enhance the stability of the catalyst, it is essential for the metal atoms within it to possess a higher dissolution potential. The dissolution potentials of metal atoms for all ten catalysts investigated in Figure 2 were found to be greater than 0 V. Notably, these values surpass the standard dissolution potentials of the respective metals (refer to Table S2), thereby indicating the exceptional electrochemical stability displayed by the ten TM-N2O2Cx single-atom catalysts that were examined in this study.
2.3. O2 Adsorption
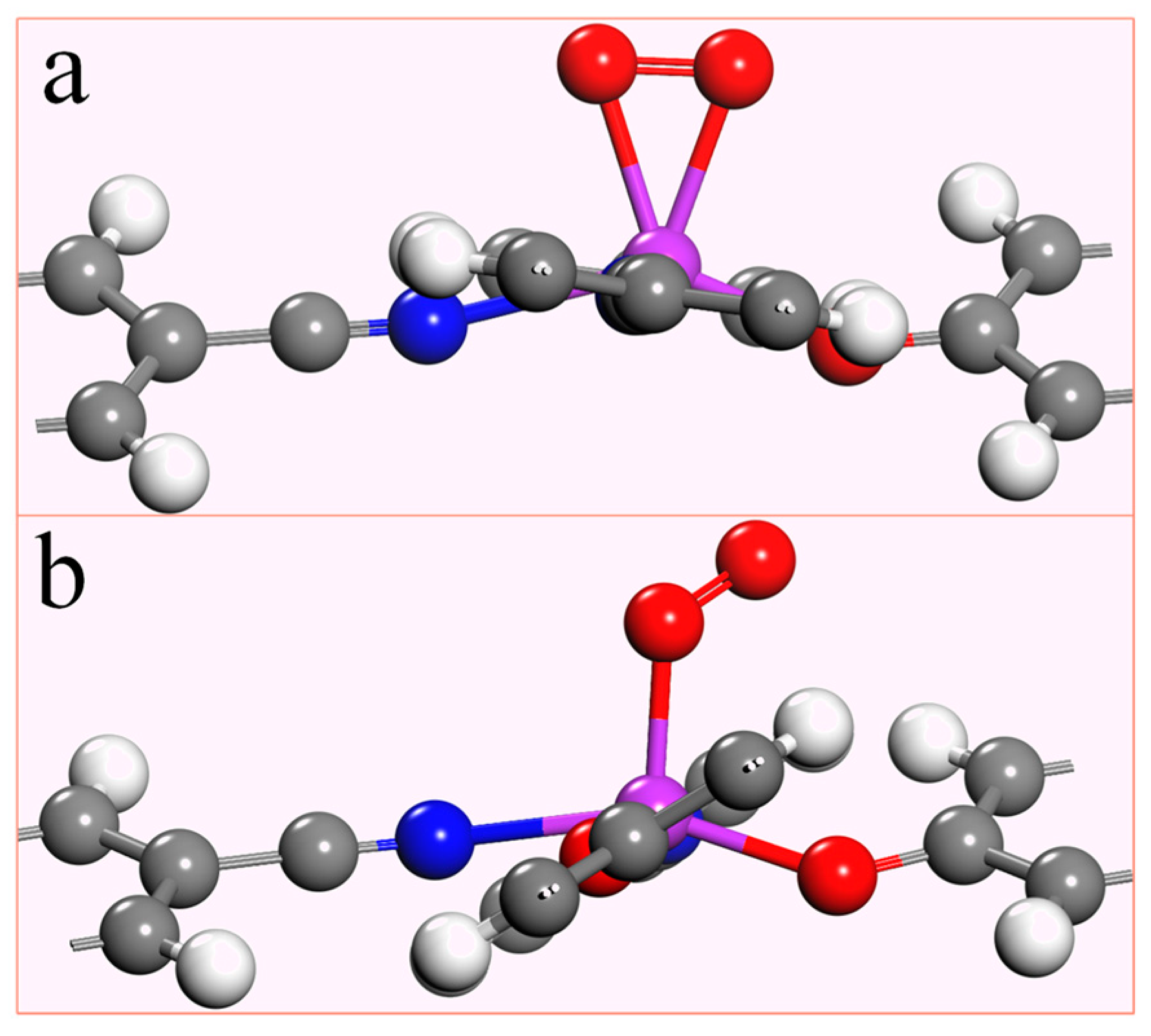
Since the electrocatalytic oxygen reduction reaction occurs in an aqueous solution, the O2 molecule must be effectively adsorbed by the catalyst for the response to happen smoothly. To investigate the adsorption capacity of the TM-N2O2Cx single-atom catalyst for O2, the adsorption energy (taking O2 adsorption as an example) was obtained by the formula: Eads = ETM-N2O2Cx-O2 − ETM-N2O2Cx − EO2 (3). ETM-N2O2Cx-O2 is the total energy of the adsorbed O2 and TM-N2O2Cx, while ETM-N2O2Cx and EO2 are the total energy of TM-N2O2Cx and the single O2 molecule. If the adsorption energy is negative, indicating that O2 can be effectively adsorbed, and the more negative the value, the stronger the adsorption is. There are two forms of O2 adsorption on the catalyst, namely end-on configuration and side-on configuration (Figure 3). The most stable adsorption states and adsorption energies of O2 on ten single-atom catalysts of the first transition metal series are listed in Table S3, which shows that the stable adsorption states of O2 on Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx are side-on configuration (Figure 3a). At the same time, the remaining seven are end-on configuration (Figure 3b). Additionally, the adsorption energies of the ten stable states are in the range of −4.22 ~ −0.39 eV, which indicates that O2 can be effectively adsorbed on the catalyst to facilitate the reduction reaction.
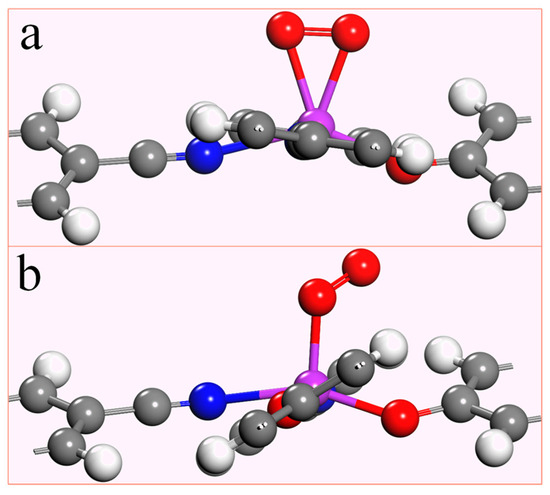
Figure 3.
Two different adsorption states of O2 on TM-N2O2Cx,(a) side-on configuration, (b) end-on configuration.
2.4. ORR Catalytic Performance
2.4.1. Selectivity of Reaction (2e− or 4e−)
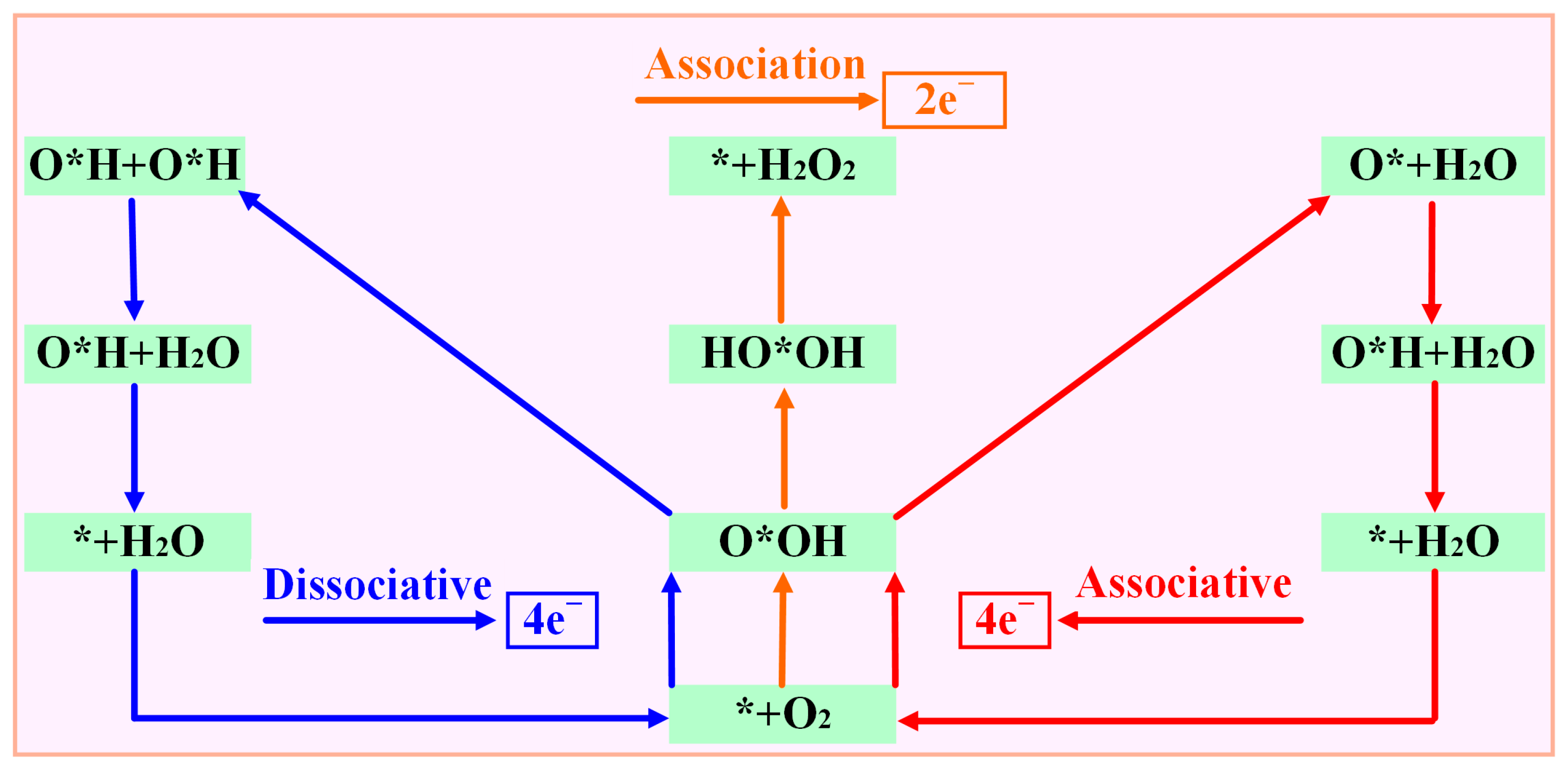
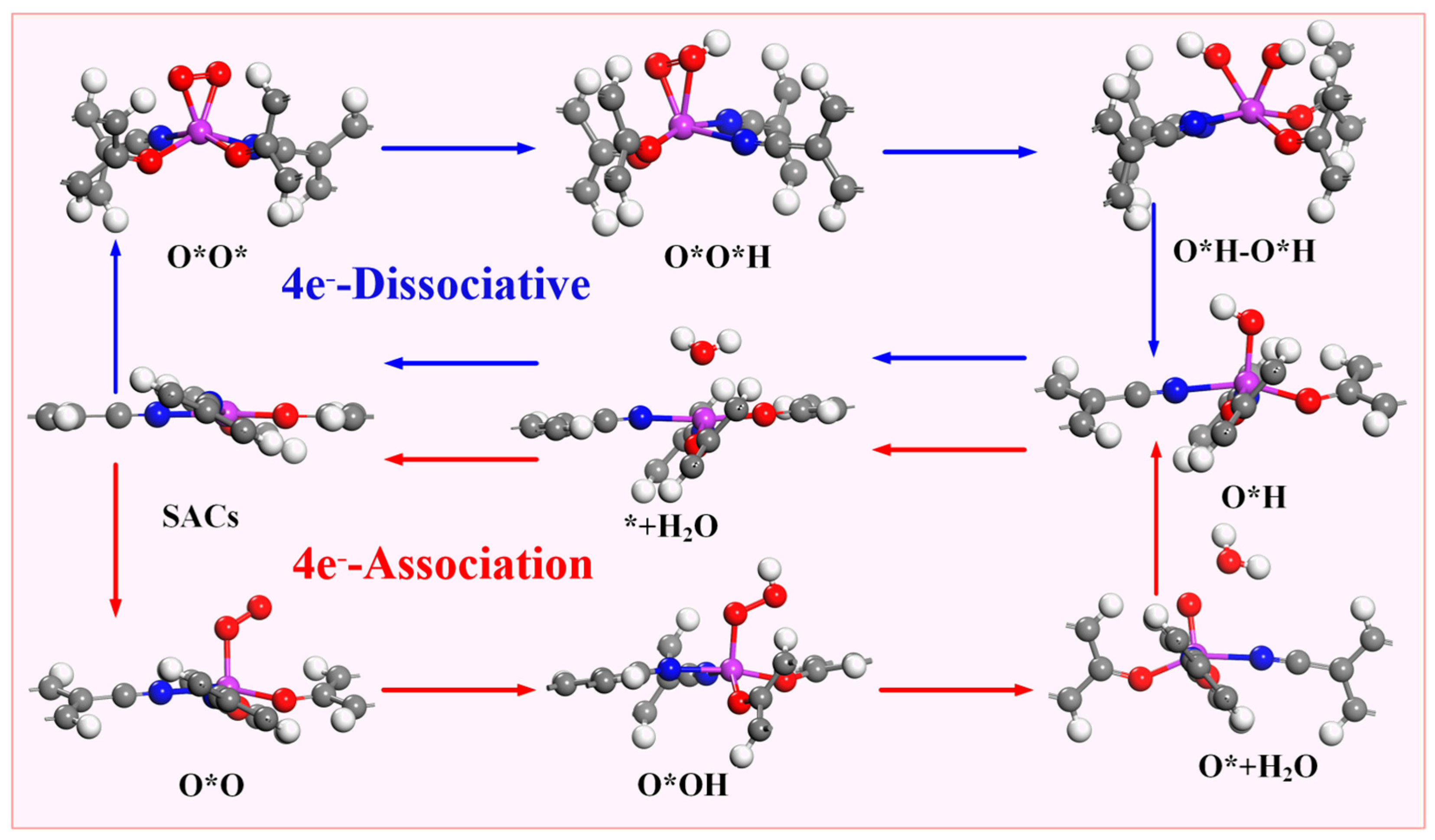
According to the different reduction products, the ORR can be divided into a 2e− pathway to produce H2O2 and a 4e− pathway in which the product is H2O. The 4e− pathway can be divided into a dissociative mechanism and association mechanism (Figure 4). Compared with the 2e− pathway, the 4e−- pathway has better energy conversion efficiency, so suitable ORR catalysts must have good 4e− selectivity. The intermediate O*OH (* + O2 + H+ + e− → O*OH) is obtained by one-step protonation of O2. O*OH can be reduced to H2O2 through the path O*OH + H+ + e− → * + H2O2 or O*OH + H+ + e− →O* + H2O to obtain O*; so, it is necessary to determine whether the catalyst is thermodynamically inclined to the 4e− pathway or the 2e− pathway. Therefore, this paper is determined by comparing the formation barrier of H2O2 and the barrier to obtain O* and H2O.
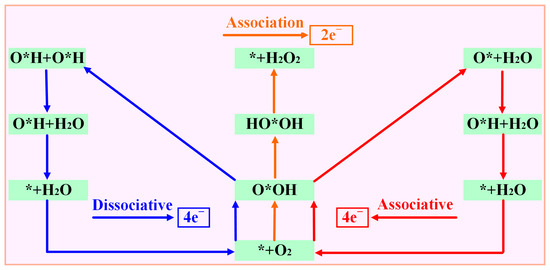
Figure 4.
Possible reaction path of oxygen reduction reaction.
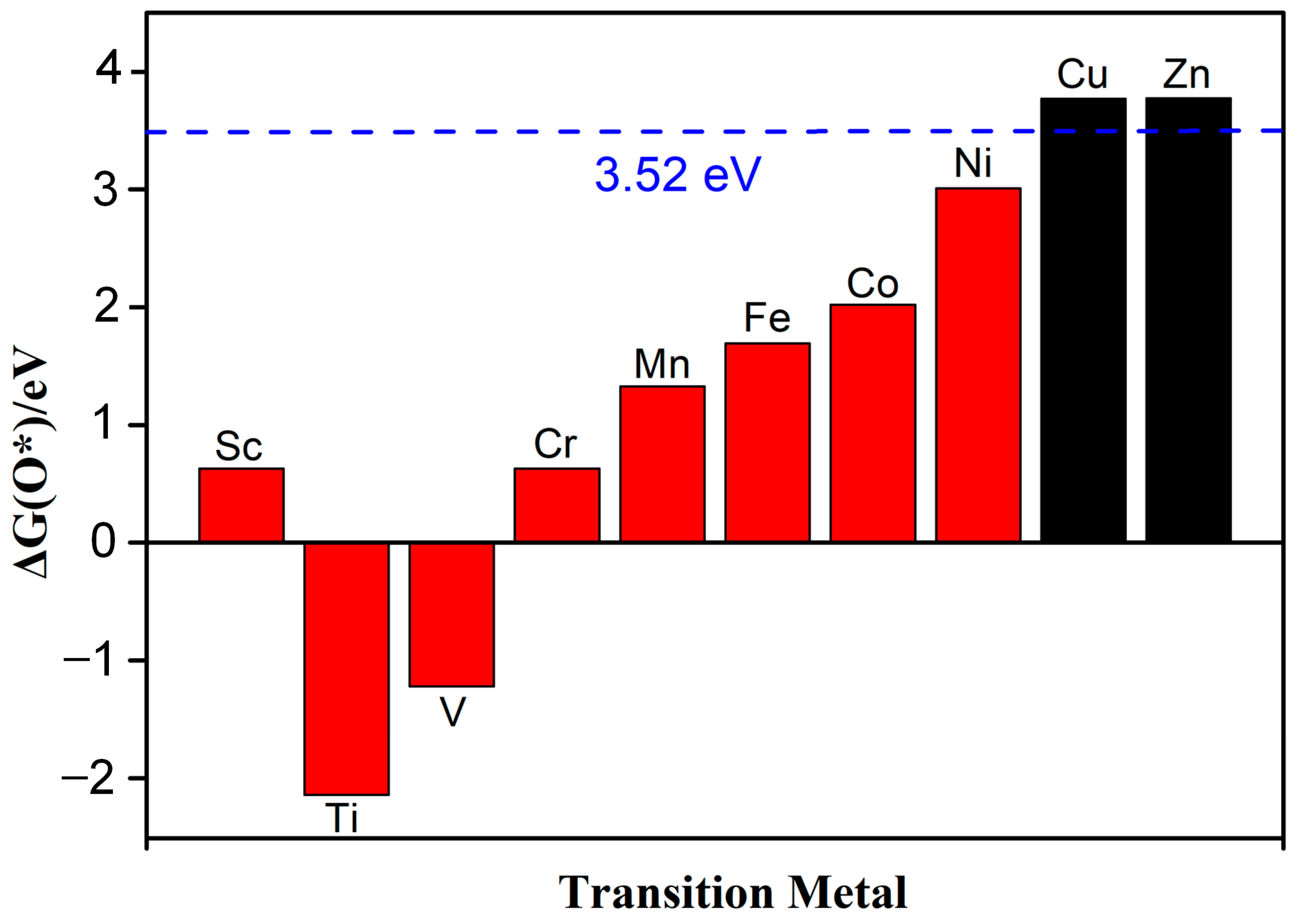
Guo et al. systematically studied the selectivity of ORRs and found that the Gibbs free energy (∆GO*) of O* for the 4e− pathway should be less than 3.52 eV (∆GH2O2 − ∆GH2O) [25]. On the contrary, the Gibbs free energy for the 2e− pathway should be greater than 3.52 eV. The Gibbs free energy of the reaction was determined using the computational hydrogen electrode model (CHE) proposed by Nørskov and co-workers [26], and the details are described in our previous reports [27]. This paper focuses on reactions under strongly acidic conditions (pH = 0). Figure 5 shows that except for Cu-N2O2Cx and Zn-N2O2Cx, the Gibbs free energy of ∆G*O for the other eight catalysts is all less than 3.52 eV, indicating that the ORR mechanism of Cu-N2O2Cx and Zn-N2O2Cx is the 2e− pathway (Figure S2), and the rest of the SACs are more inclined to the 4e− pathway. Next, we focus on exploring the 4e− pathway mechanism of the above eight catalysts.
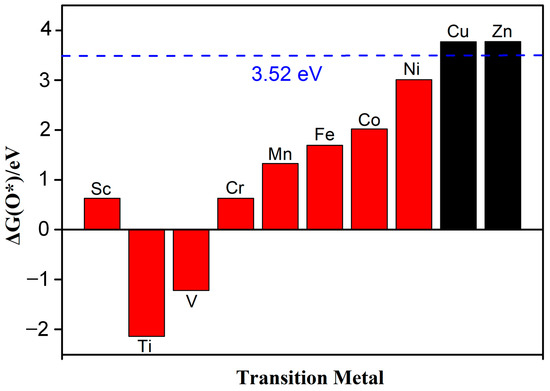
Figure 5.
The Gibbs free energy of ∆G(O*), and the dotted line represents the free energy value of 3.52 eV, which is obtained by G (O *) = G (H2O2) − G (H2O).
2.4.2. 4e− Pathway
As previously mentioned, the 4e− pathway encompasses both associative and dissociative mechanisms, with the specific reaction mechanism employed in the 4e− process being dependent on the stable adsorption configuration of O2 on the catalyst. Figure 6 demonstrates that when the stable adsorption configuration of O2 is side-on, the 4e− reaction tends to proceed via the dissociative mechanism, whereas when the stable adsorption configuration of O2 is end-on, the 4e− reaction follows the associative mechanism. Due to the fact that the stabilization of the adsorption configuration on Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx is side-on, the electrocatalytic O2 reduction on these catalysts adopts the 4e− dissociative mechanism. In contrast, the five catalysts from Cr- N2O2Cx to Ni- N2O2Cx exhibit associative mechanisms for the 4e− pathway.
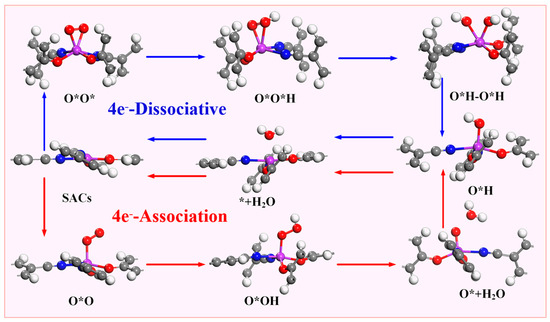
Figure 6.
Two 4e− pathways and corresponding intermediates of oxygen reduction reaction.
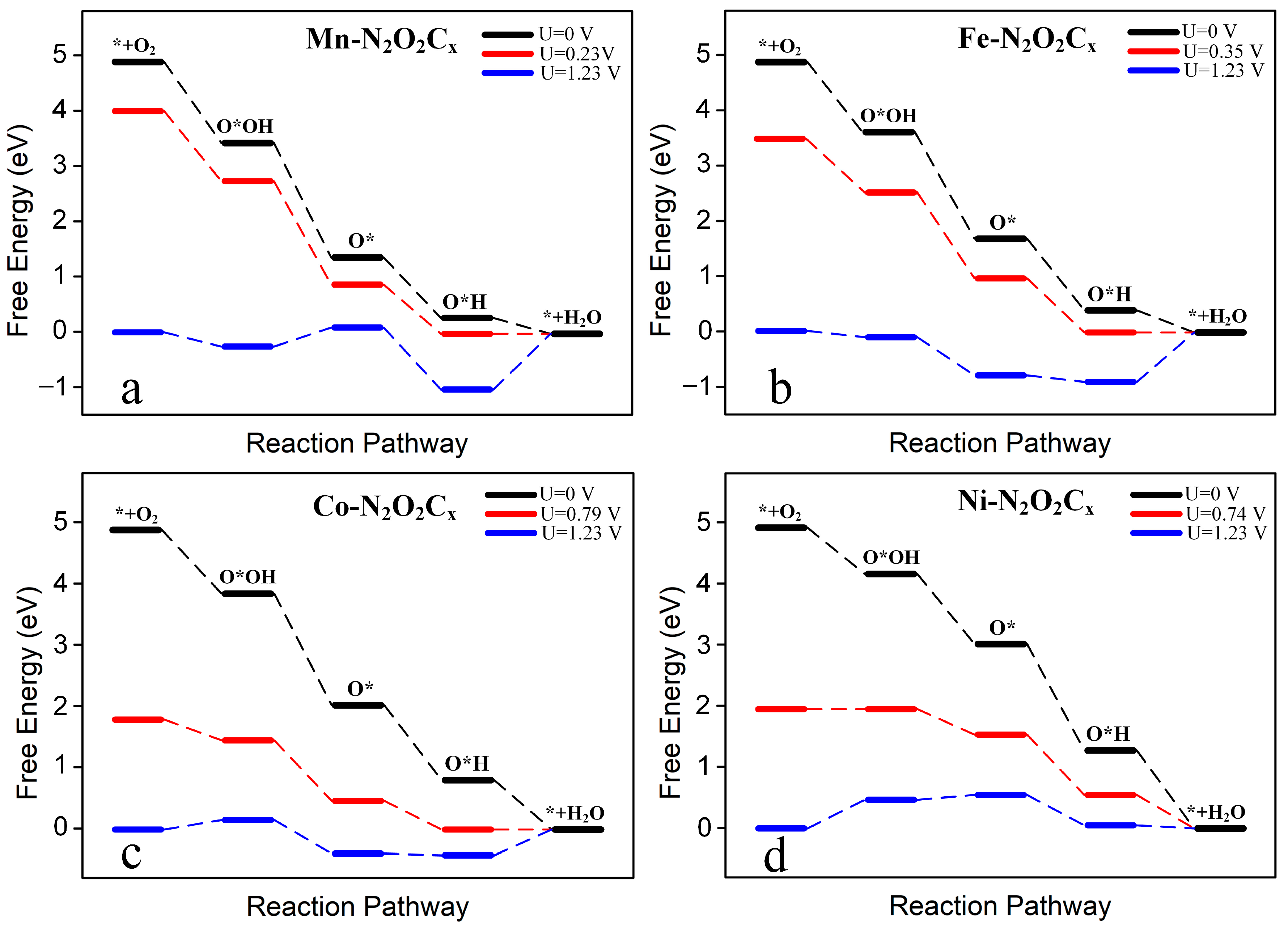
Figure 7 and Figure S3 show the free energy diagrams of the 4e− pathway of electrocatalytic O2 reduction for the eight TM-N2O2Cx single-atom catalysts at three different applied potentials. The black and blue lines represent the applied potentials of 0 V and 1.23 V, respectively, while the red line represents the limiting potential. Figure 7 shows that each step of the protonation process of O2 reduction by Mn-N2O2Cx, Fe-N2O2Cx, Co-N2O2Cx, and Ni-N2O2Cx without applied potential is exothermic; however, the final step of protonation (O*H + H+ + e− → * + H2O) in the 4e− reaction catalyzed by Sc-N2O2Cx, Ti-N2O2Cx, V-N2O2Cx, and Cr-N2O2Cx is an endothermic reaction (Figure S3).
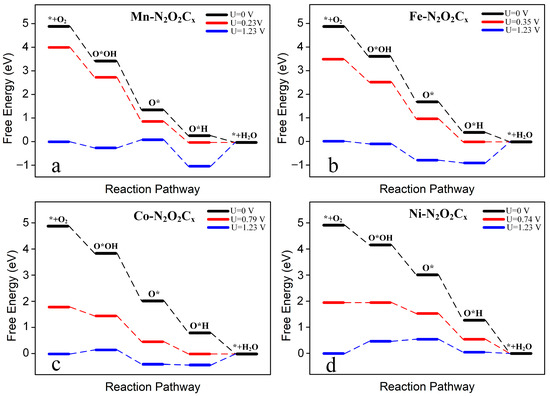
Figure 7.
Free energy diagram of 4e− ORR on Mn-N2O2Cx (a), Fe-N2O2Cx (b), Co-N2O2Cx (c), and Ni-N2O2Cx (d) under different potentials. The black line, blue line, and red line represent U = 0 V, U = limiting potential, and U = 1.23 V, respectively.
The rate-determining step was determined by comparing the maximum increase in the protonation-free energy in the 4e− reaction. It was found that except for Ni-N2O2Cx, of which the rate-determining step was * + O2 + H+ + e− → O*OH (Figure 7d), the remaining seven catalysts were all O*H + H+ + e− → * + H2O (Table 1). The limiting potential (UL) of the ORR can be determined according to the formula UL = −ΔGmax/ne (4), where ΔGmax and n are the increase in the free energy in the rate-determining step and the number of electrons transferred in the reaction, respectively. Finally, the overpotential (ƞ) of the ORR was determined by the difference between UL and 1.23 V (ƞ = UL − 1.23 V), and the detailed data statistics are listed in Table 1.

Table 1.
The calculated rate-determining steps, limiting potentials (UL/V), and over-potential (ƞ/V) for the ORR of the TM-N2O2Cx SACs are listed.
Among the eight SACs investigated, the limiting potentials of Mn-N2O2Cx, Fe-N2O2Cx, Co-N2O2Cx, and Ni-N2O2Cx are all greater than 0 V. At the same time, Sc-N2O2Cx, Ti-N2O2Cx, V-N2O2Cx, and Cr-N2O2Cx display limiting potentials lower than 0 V; especially, Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx are all lower than −0.80 V, which results in their overpotentials all being greater than 2 V, meaning that more external potentials need to be applied for the occurrence of the ORR. In comparison, the limiting potential values of the other five catalysts are relatively positive, lowering the corresponding overpotential. With the exception of Cr-N2O2Cx, whose overpotential exceeds 1 V, the overpotentials of the other catalysts are all lower than 1 V. Notably, Co-N2O2Cx and Ni-N2O2Cx possess limiting potentials of 0.79 V and 0.74 V, respectively, leading to overpotentials below 0.50 V, which means that the ORR catalytic performance of Co-N2O2Cx and Ni-N2O2Cx is comparable to that of Pt catalysts with a working potential of 0.78 V [26] and is better than that of FeN4-doped graphene (0.35 V) [28], The above results show that Co-N2O2Cx and Ni-N2O2Cx are expected to be promising ORR catalysts.
2.5. Scaling Relationship between Oxygen-Containing Intermediates
The catalytic performance of the catalyst is determined by its electronic structure. According to Sabatier’s principle, an excellent catalyst has appropriate adsorption capacity for reaction intermediates, which is neither strong nor weak. If the catalyst’s adsorption capacity is too high, it prevents intermediate detachment, causing the catalytic active site to become passivated and, finally, leads to a considerable loss in catalytic performance [29]. Conversely, if the adsorption capacity is too weak, the adsorption of intermediates on the catalyst surface is not favorable, making the reaction unable to proceed. Therefore, if there is a relationship between the adsorption energies of the ORR intermediates, we can effectively discover and design the most suitable ORR catalysts by the descriptor-based method.
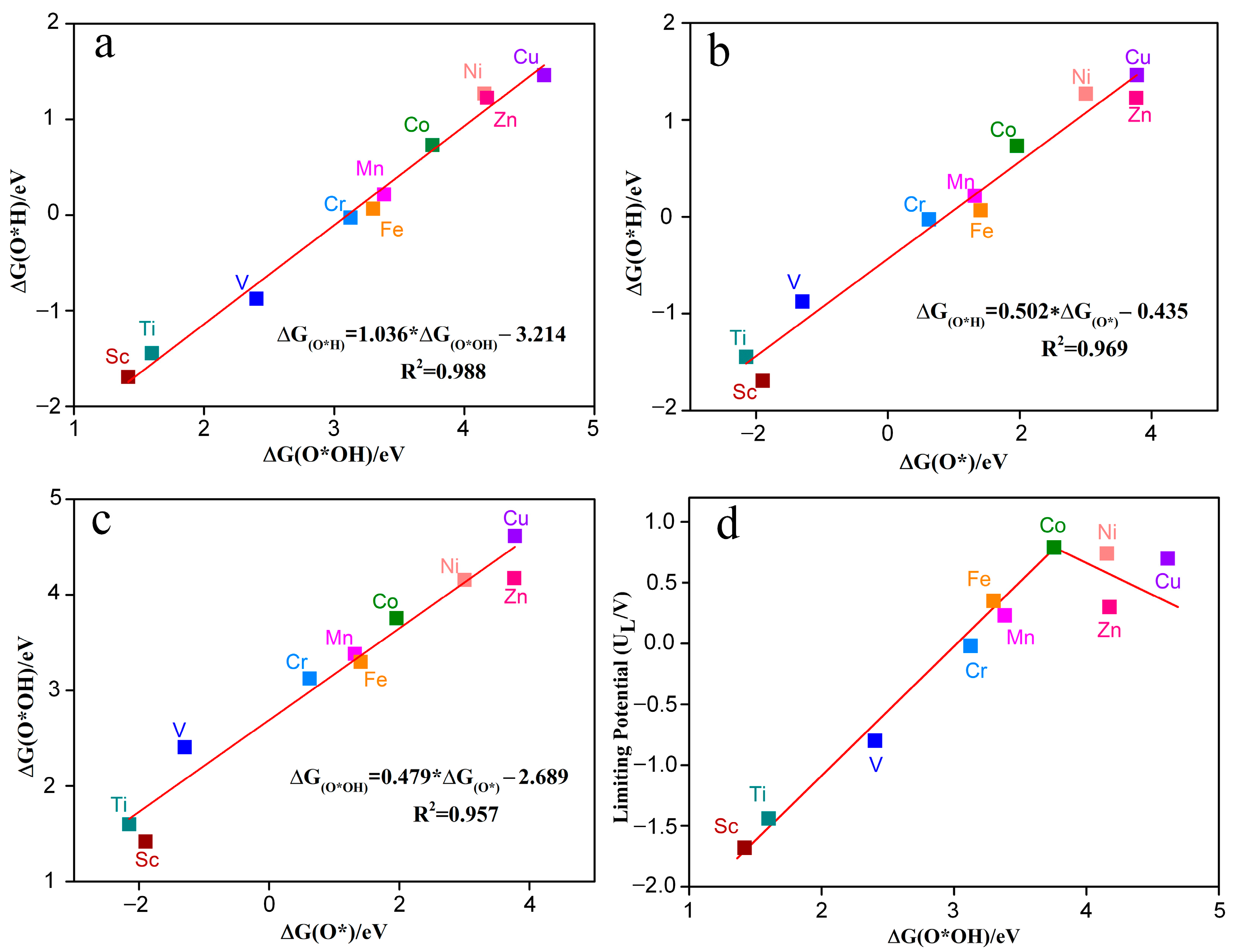
Figure 8a–c shows the Gibbs adsorption free energy relationships between different reaction intermediates (such as O*OH, O*H, O*) for the electrocatalytic O2 reduction by TM-N2O2Cx single-atom catalysts. The Gibbs adsorption free energy relative to H2O and H2 were calculated based on the following equations: ΔGO*H = GO*H + 0.5GH2 − G* − GH2O (6); ΔGO* = GO* + GH2 − G* − GH2O (7); ΔGO*OH = GO*OH + 1.5GH2 − G* − 2GH2O (8). G* is the free energy of TM-N2O2Cx, while GH2 and GH2O represent the gas phase free energy of H2 and H2O molecules, respectively. The results indicate that no matter the active metal atom, there is a universal scaling relation between O*OH, O*H, and O*. ΔGO*H can be represented by ΔGO*OH through the function ΔGO*H = 1.036ΔGO*OH − 3.214 eV (Figure 8a) or by ΔGO* through the function ΔGO*H = 0.502ΔGO* − 0.435 eV (Figure 8b). The coefficients of determination (R2) are 0.988 and 0.969, respectively, indicating a very strong linear relationship between ΔGO*H and ΔGO*OH or ΔGO*. In addition, Δ GO*OH can be created by Δ GO* through the function Δ GO*OH = 0.479 Δ GO* −2.689 eV (Figure 8c), and the same strong linear relationship exists between ΔGO*OH and ΔGO* (R2 = 0.957). These results are in agreement with previous studies [30,31]. Thus, the catalytic activity of TM-N2O2Cx single-atom catalysts for electrocatalytic O2 reduction can be described by the descriptors ΔGO*H, ΔGO*OH, or ΔGO*.
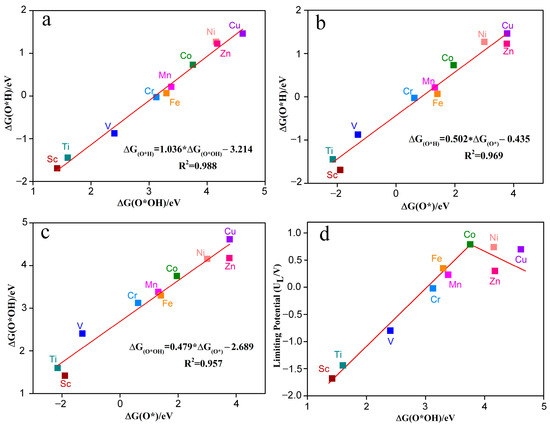
Figure 8.
The Gibbs free energy of intermediates O*OH and O*H (a), O* and O*H (b), O* and O*OH (c) on the catalyst of TM-N2O2Cx. (d) Volcano plots between limiting potential and ∆G(O*OH).
Figure 8d shows that the adsorption strength of TM-N2O2Cx to O*OH (ΔGO*OH) significantly affects the limiting potential of ORR. With the increasing value of ΔGO*OH, the limiting potential (UL) shows a trend of increasing and then decreasing, and there is a significant volcanic relationship between ΔGO*OH and UL. Among the ten TM-N2O2Cx single-atom catalysts studied in this paper, the limiting potentials of Co-N2O2Cx and Ni-N2O2Cx are very close to the top of the volcano diagram, making their limiting potential values closest to the potential equilibrium value of the ORR (1.23 V) and allowing a small overpotential of 0.44 V and 0.49 V, respectively (Table 1), which is consistent with the overpotential of commercial Pt (0.45 V). In contrast, the limiting potentials of Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx are far from the top of the volcano plot, leading to an ORR with higher overpotentials (>2 V). Therefore, the volcano curve can effectively help us to screen for superior ORR catalysts.
3. Computational Methods
This study is carried out based on the density functional theory of spin polarization with the help of the Dmol3 module [32]. The structure optimization, total energy, and electronic properties of the TM-N2O2Cx models are treated using the Perdew–Burke–Ernzerhof (PBE) exchange-related energy generalization in the generalized gradient approximation (GGA) [33], the basis group is used as a double numerical plus polarization basis group, and the core electrons are treated in an all-electron way. To obtain a high accuracy, the energy convergence criterion is 10−6 Ha, the Monkhorst–Pack grid uses 5 × 5 × 1 K points for structure optimization and 10 × 10 × 1 for performance calculation, and the vacuum layer is set to 15 Å to eliminate the interaction between TM-N2O2Cx layers. In addition, Van der Waals dispersion was introduced to better describe the adsorption of O2 and reaction intermediates on the TM-N2O2Cx surface. Since the ORR occurs in solution, a conductor approximation shielding model (COSMO) is usually used for electrocatalytic reactions to better simulate the real reaction environment, and the dielectric constant of the solvent is set to 78.54 in this paper [34].
4. Conclusions
We designed a single-atom catalyst (TM-N2O2Cx) with a TM-N2O2 coordination unit and investigated its catalytic performance for electrocatalytic O2 reduction based on density functional theory. The obtained results demonstrate the outstanding thermodynamic and electrochemical stability of TM-N2O2Cx. Notably, most catalysts examined in this study follow the 4e− pathway for O2 reduction, with the exception of Cu-N2O2Cx and Zn-N2O2Cx which exhibit a 2e− ORR pathway. Sc-N2O2Cx, Ti-N2O2Cx, and V-N2O2Cx conform to the dissociative mechanism, while the rest follow the associative mechanism. Among the ten catalysts studied in this paper, Co-N2O2Cx and Ni-N2O2Cx display the lowest overpotential (less than 0.5 V), which is equivalent to the overpotential of the benchmark catalyst Pt, making Co-N2O2Cx and Ni-N2O2Cx possible as alternative candidates to Pt. It is found that there is a scaling relationship between the adsorption-free energy of oxygen-containing intermediates (O*OH, O*H, O*) and a volcano curve between the adsorption-free energy of O*OH and the limiting potential, providing a foundation for the selection of exceptional ORR catalysts.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28217264/s1. Figure S1. Projected partial density of states (PDOS) of TM-N2O2Cx, (a) Sc-N2O2Cx, (b) Ti-N2O2Cx, (c) V-N2O2Cx, (d) Cr-N2O2Cx, (e) Mn-N2O2Cx, (f) Fe-N2O2Cx, (bg) Co-N2O2Cx, (bh) Ni-N2O2Cx,(i)Cu-N2O2Cx, (j)Zn-N2O2Cx. Figure S2. Free energy diagram of 2e− ORR on Cu-N2O2Cx (a) and Zn-N2O2Cx (b) under 0 V. Figure S3. Free energy diagram of 4e− ORR on Sc-N2O2Cx (a), Ti-N2O2Cx (b), V-N2O2Cx (c), and Cr-N2O2Cx (d) under different potentials. Table S1 The lattice constants, top, and side views of optimized crystal structures of TM-N2O2Cx. Table S2 Computed formation energy (Ef) and dissolution potential (Udiss) of TM-N2O2Cx. Table S3. The top and side views of the most stable adsorption state of O2 on TM-N2O2Cx. Table S4. The calculated free energy corrections (Gc) for adsorbates.
Author Contributions
J.-H.L. and H.J. designed and built the models; L.Y. and H.J. performed the DFT calculation; B.L. and X.C. (Xiaohua Cao) performed the data and theoretical analysis; J.-H.L. wrote the manuscript; X.C. (Xiudong Chen) proposed the topic and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded by the National Natural Science Foundation of China (22163003), the Jiangxi Provincial Natural Science Foundation (20224BAB214019, 20232BAB204024, 20232BAB203024), College Student Innovation and Entrepreneurship Program (202111843004), Science and Technology Project of Jiangxi Provincial Department of Education (GJJ2201937).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
We acknowledge the National Supercomputing Center in Shenzhen for providing the computational resources and materials studio (version, module).
Conflicts of Interest
There are no conflicts to declare.
Sample Availability
Not applicable.
References
- Ong, S.; Al-Othman, A.; Tawalbeh, M. Emerging technologies in prognostics for fuel cells including direct hydrocarbon fuel cells. Energy 2023, 277, 127721. [Google Scholar] [CrossRef]
- Stephens, I.E.L.; Rossmeisl, J.; Chorkendorff, I. Toward sustainable fuel cells. Science 2016, 354, 1378–1379. [Google Scholar] [CrossRef]
- Allendorf, M.D. Oxygen reduction reaction: A framework for success. Nat. Energy 2016, 1, 16058. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Wei, Z. Transition-metal-oxide-based catalysts for the oxygen reduction reaction. J. Mater. Chem. A 2018, 6, 8194–8209. [Google Scholar] [CrossRef]
- Chen, S.; Zhao, J.; Su, H.; Li, H.; Wang, H.; Hu, Z.; Bao, J.; Zeng, J. Pd-Pt Tesseracts for the Oxygen Reduction Reaction. J. Am. Chem. Soc. 2021, 143, 496–503. [Google Scholar] [CrossRef]
- Cui, P.; Zhao, L.; Long, Y.; Dai, L.; Hu, C. Carbon-Based Electrocatalysts for Acidic Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2023, 62, e202218269. [Google Scholar] [CrossRef]
- Tian, X.L.; Xu, Y.Y.; Zhang, W.; Wu, T.; Xia, B.Y.; Wang, X. Unsupported Platinum-Based Electrocatalysts for Oxygen Reduction Reaction. ACS Energy Lett. 2017, 2, 2035–2043. [Google Scholar] [CrossRef]
- Shao, M.; Chang, Q.; Dodelet, J.-P.; Chenitz, R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev. 2016, 116, 3594–3657. [Google Scholar] [CrossRef]
- Gawande, M.B.; Ariga, K.; Yamauchi, Y. Single-Atom Catalysts. Small 2021, 17, 2101584. [Google Scholar] [CrossRef]
- Su, J.; Zhuang, L.; Zhang, S.; Liu, Q.; Zhang, L.; Hu, G. Single atom catalyst for electrocatalysis. Chin. Chem. Lett. 2021, 32, 2947–2962. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, J. Single-Atom Catalysts for Electrocatalytic Applications. Adv. Funct. Mater. 2020, 30, 2000768. [Google Scholar] [CrossRef]
- Lin, L.; Chen, Z.; Chen, W. Single atom catalysts by atomic diffusion strategy. Nano Res. 2021, 14, 4398–4416. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, T.; Liang, X.; Deng, X.; Guo, W.; Wang, Z.; Lu, X.; Wu, C.-M.L. Single transition metal atoms on nitrogen-doped carbon for CO2 electrocatalytic reduction: CO production or further CO reduction? App. Surf. Sci. 2020, 533, 147466. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, R.; Fan, Z.; Wen, H.; Chen, Y.; Lin, R.; Zhu, Y.; Yang, X.; Chen, Z. Synergistic copper nanoparticles and adjacent single atoms on biomass-derived N-doped carbon toward overall water splitting. Inorg. Chem. Front. 2023, 10, 443–453. [Google Scholar] [CrossRef]
- Yi, J.-D.; Xu, R.; Wu, Q.; Zhang, T.; Zang, K.-T.; Luo, J.; Liang, Y.-L.; Huang, Y.-B.; Cao, R. Atomically Dispersed Iron-Nitrogen Active Sites within Porphyrinic Triazine-Based Frameworks for Oxygen Reduction Reaction in Both Alkaline and Acidic Media. ACS Energy Lett. 2018, 3, 883–889. [Google Scholar] [CrossRef]
- Hunter, M.A.; Fischer, J.M.T.A.; Yuan, Q.; Hankel, M.; Searles, D.J. Evaluating the Catalytic Efficiency of Paired, Single-Atom Catalysts for the Oxygen Reduction Reaction. ACS Catal. 2019, 9, 7660–7667. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, G.; Wang, Q.; Wang, D.; Tao, X.; Zhang, T.; Feng, X.; Müllen, K. Fe-N-C Electrocatalysts with Densely Accessible Fe-N4 Sites for Efficient Oxygen Reduction Reaction. Adv. Funct. Mater. 2021, 31, 2102420. [Google Scholar] [CrossRef]
- Wang, Z.; Jin, X.; Zhu, C.; Liu, Y.; Tan, H.; Ku, R.; Zhang, Y.; Zhou, L.; Liu, Z.; Hwang, S.-J.; et al. Atomically Dispersed Co2-N6 and Fe-N4 Costructures Boost Oxygen Reduction Reaction in Both Alkaline and Acidic Media. Adv. Mater. 2021, 33, 2104718. [Google Scholar] [CrossRef]
- Ge, F.; Qiao, Q.; Chen, X.; Wu, Y. Probing the catalytic activity of M-N4-xOx embedded graphene for the oxygen reduction reaction by density functional theory. Front. Chem. Sci. Eng. 2021, 15, 1206–1216. [Google Scholar] [CrossRef]
- Dong, W.; Zhang, N.; Li, S.; Min, S.; Peng, J.; Liu, W.; Zhan, D.; Bai, H. A Mn single atom catalyst with Mn-N2O2 sites integrated into carbon nanosheets for efficient electrocatalytic CO2 reduction. J. Mater. Chem. A 2022, 10, 10892–10901. [Google Scholar] [CrossRef]
- Wei, X.; Cao, S.; Xu, H.; Jiang, C.; Wang, Z.; Ouyang, Y.; Lu, X.; Dai, F.; Sun, D. Novel Two-Dimensional Metal Organic Frameworks: High-Performance Bifunctional Electrocatalysts for OER/ORR. ACS Mater. Lett. 2022, 4, 1991–1998. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K. Electrochemical dissolution of surface alloys in acids: Thermodynamic trends from first-principles calculations. Electrochim. Acta 2007, 52, 5829–5836. [Google Scholar] [CrossRef]
- Guo, X.; Lin, S.; Gu, J.; Zhang, S.; Chen, Z.; Huang, S. Simultaneously Achieving High Activity and Selectivity toward Two-Electron O2 Electroreduction: The Power of Single-Atom Catalysts. ACS Catal. 2019, 9, 11042–11054. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Liu, J.-H.; Yang, L.-M.; Ganz, E. Electrochemical reduction of CO2 by single atom catalyst TM-TCNQ monolayers. J. Mater. Chem. A 2019, 7, 3805–3814. [Google Scholar] [CrossRef]
- Chai, G.-L.; Hou, Z.; Shu, D.-J.; Ikeda, T.; Terakura, K. Active Sites and Mechanisms for Oxygen Reduction Reaction on Nitrogen-Doped Carbon Alloy Catalysts: Stone-Wales Defect and Curvature Effect. J. Am. Chem. Soc. 2014, 136, 13629–13640. [Google Scholar] [CrossRef]
- Koper, M.T.M. Thermodynamic theory of multi-electron transfer reactions: Implications for electrocatalysis. J. Electroanal. Chem. 2011, 660, 254–260. [Google Scholar] [CrossRef]
- He, T.; Matta, S.K.; Will, G.; Du, A. Transition-Metal Single Atoms Anchored on Graphdiyne as High-Efficiency Electrocatalysts for Water Splitting and Oxygen Reduction. Small Methods 2019, 3, 1800419. [Google Scholar] [CrossRef]
- Meng, Y.; Yin, C.; Li, K.; Tang, H.; Wang, Y.; Wu, Z. Improved Oxygen Reduction Activity in Heteronuclear FeCo-Codoped Graphene: A Theoretical Study. ACS Sustain. Chem. Eng. 2019, 7, 17273–17281. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).