
2-Acetyl-5,8-dihydro-6-(4-methyl-3-pentenyl)-1,4-naphthohydroquinone-Derived Chalcones as Potential Anticancer Agents



Abstract
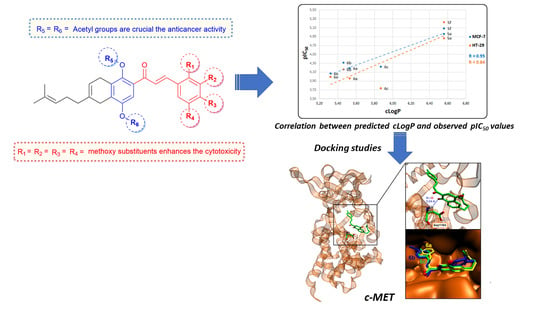
:
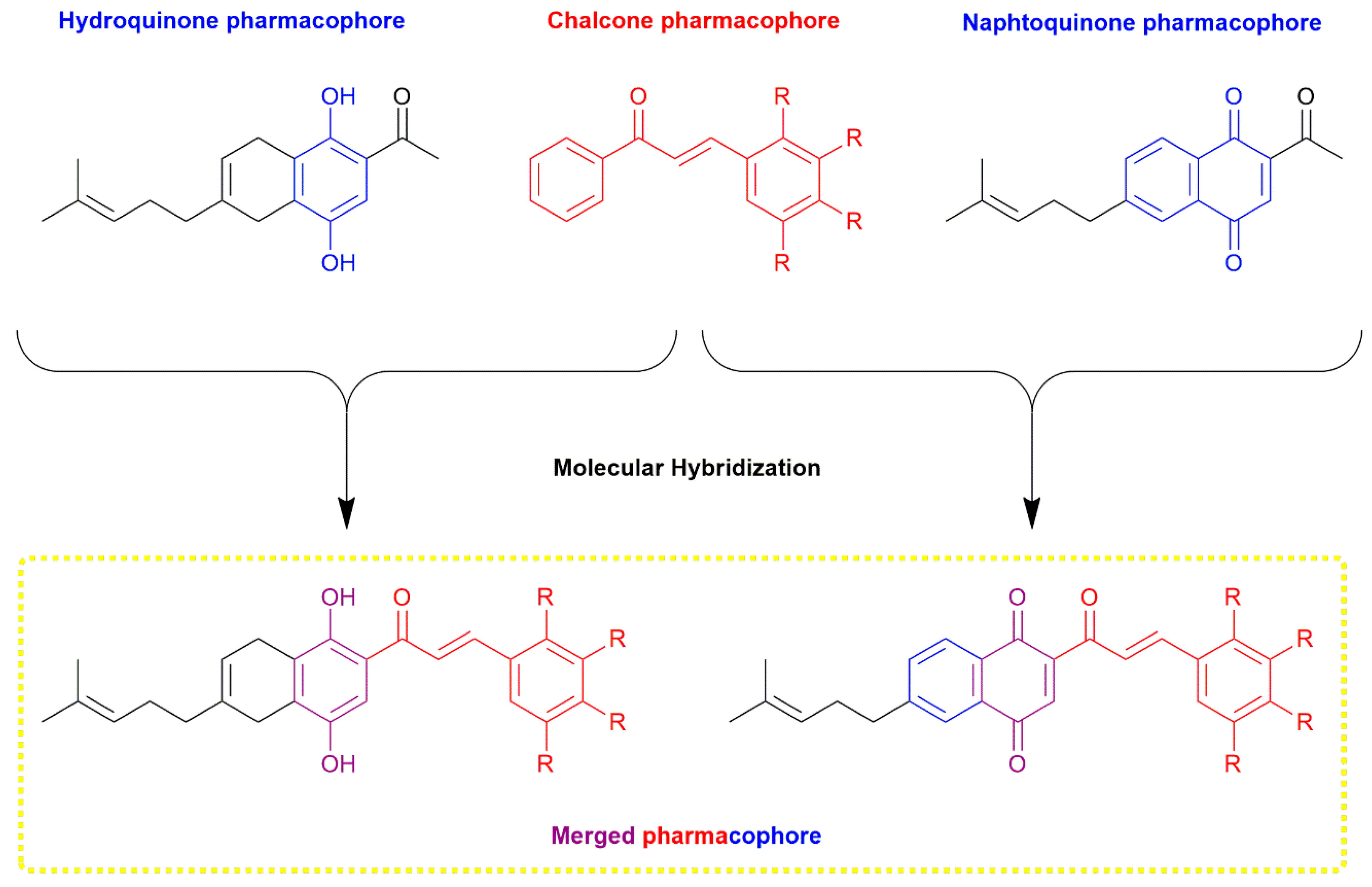
1. Introduction
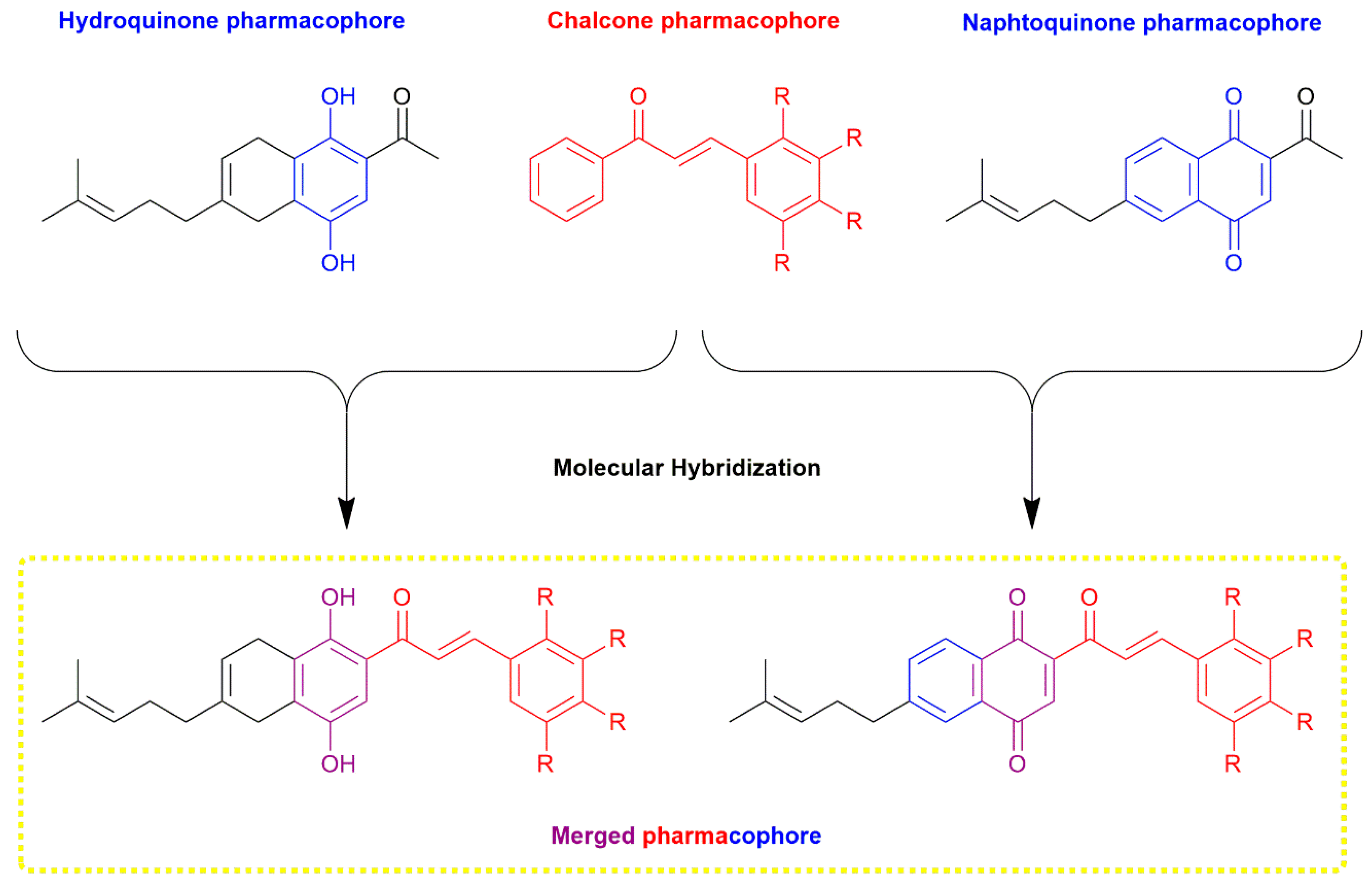
2. Results and Discussion
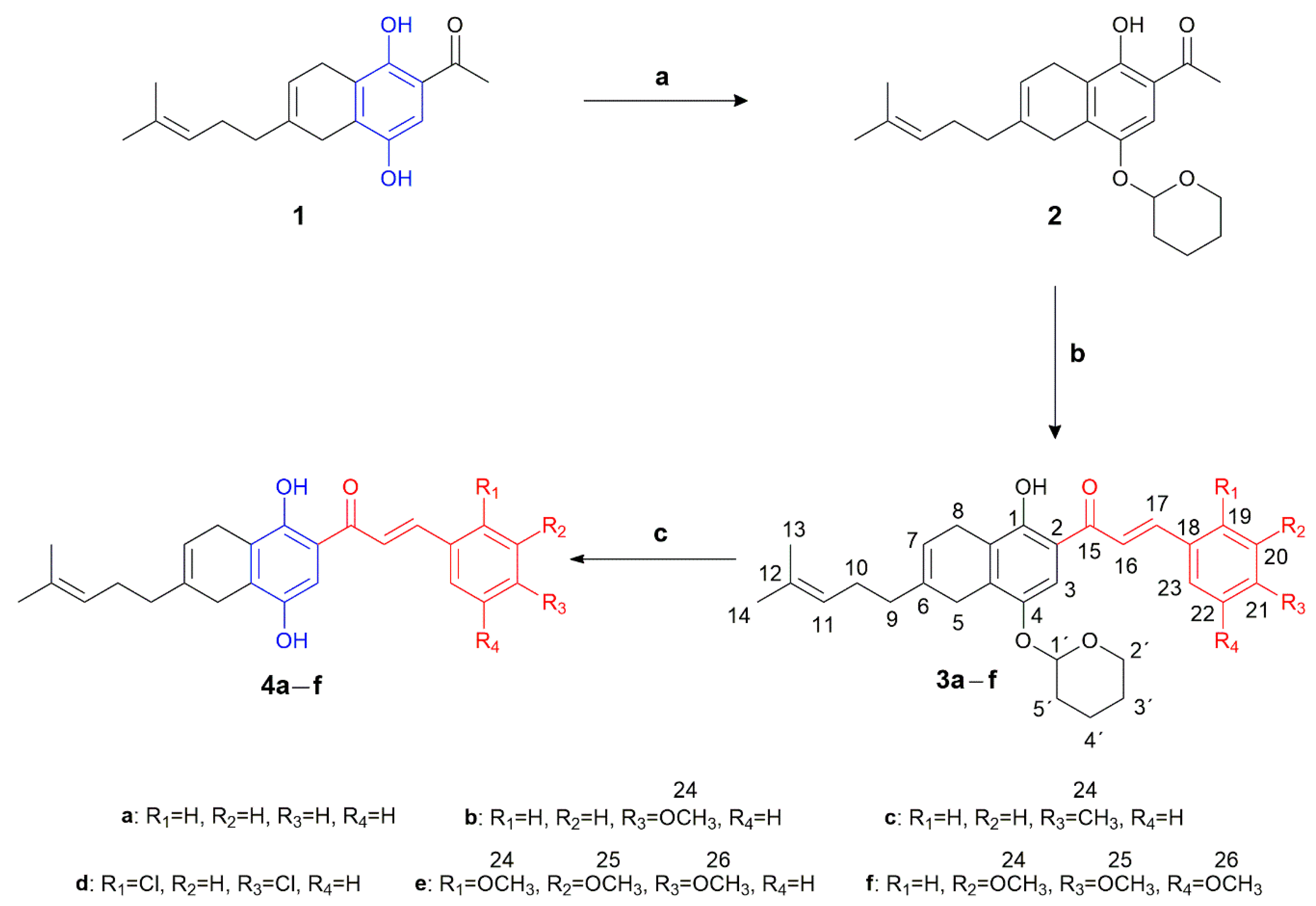
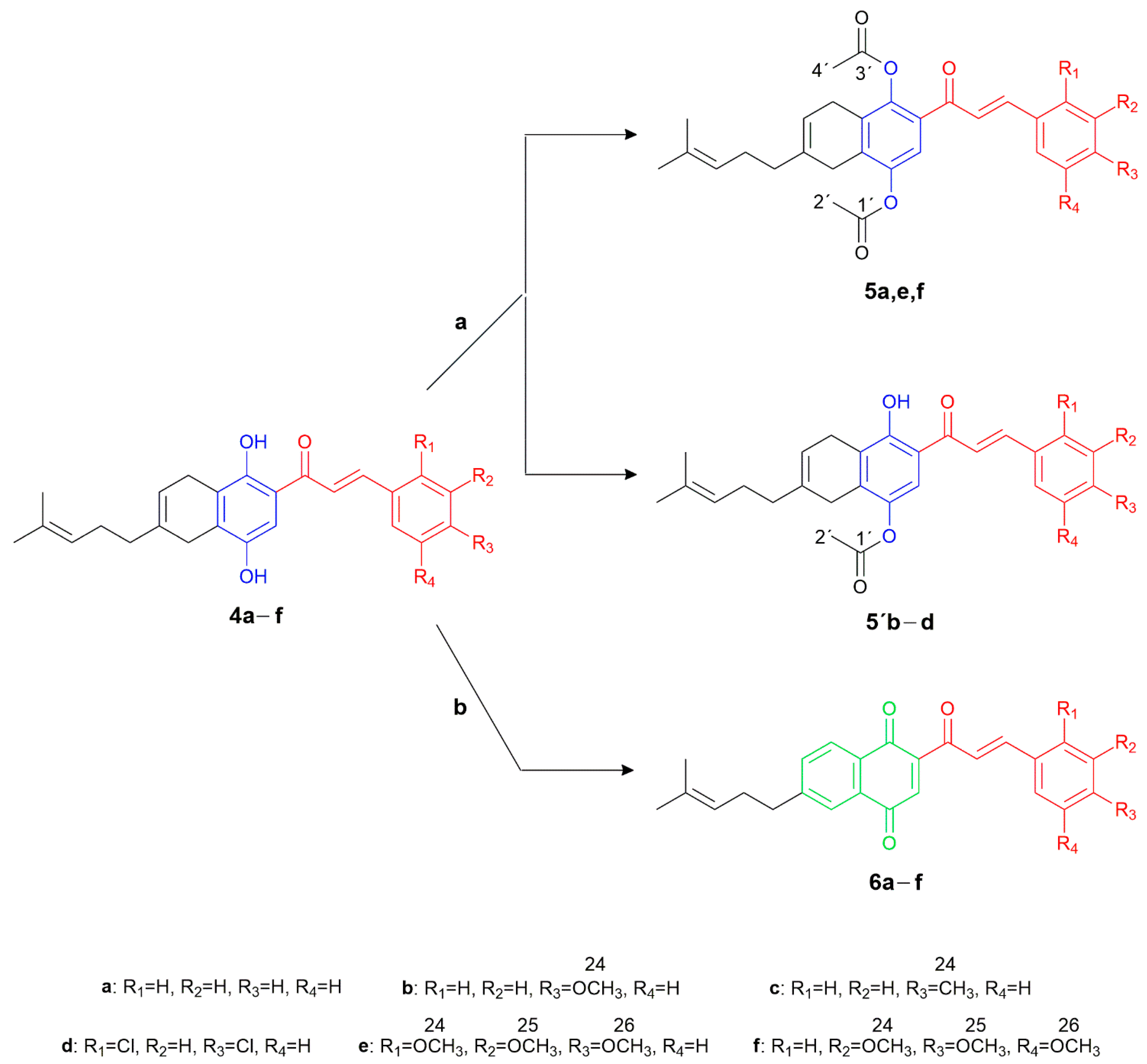
2.1. Chemistry
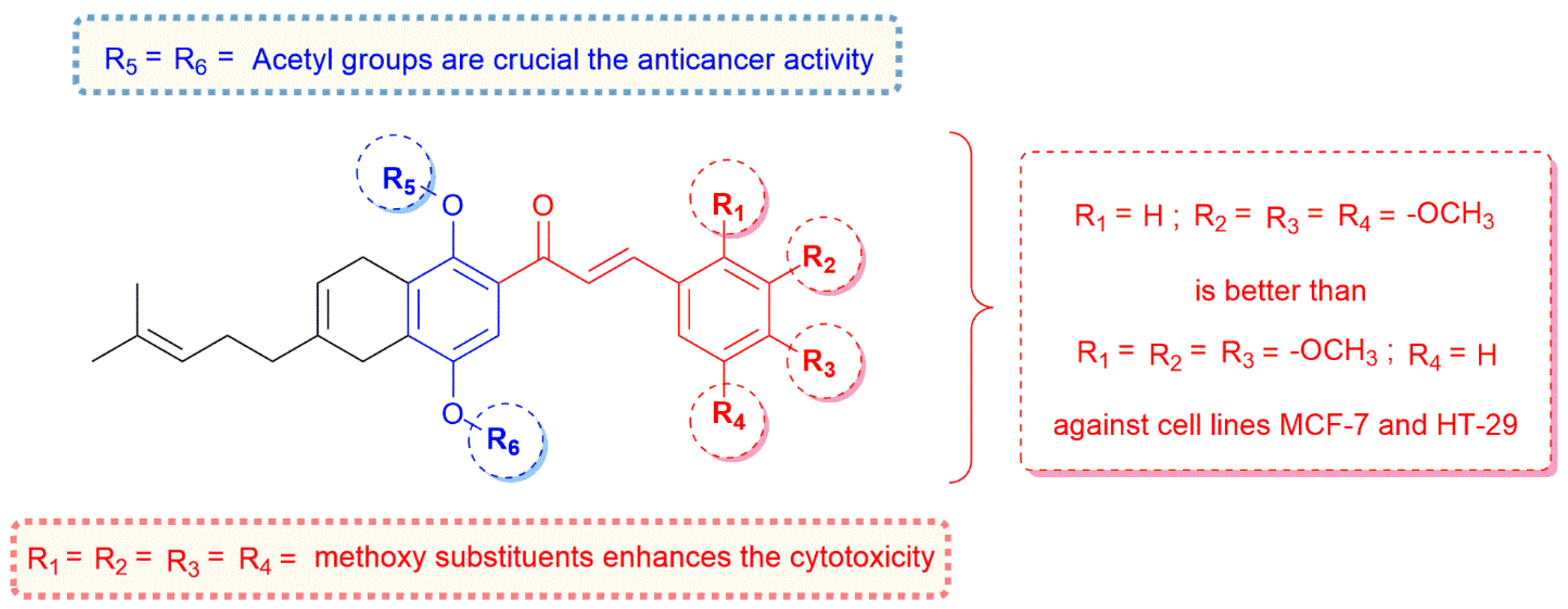
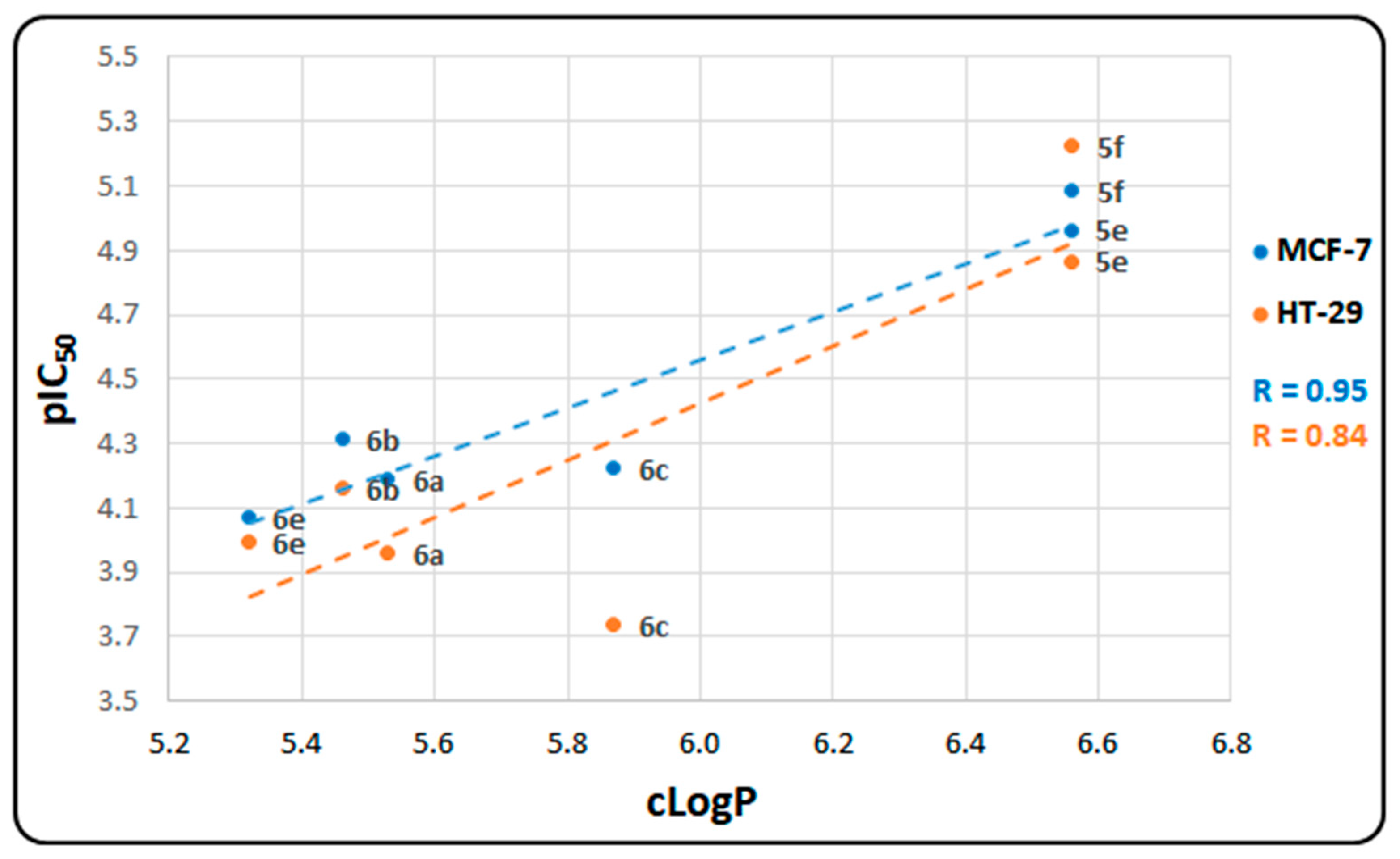
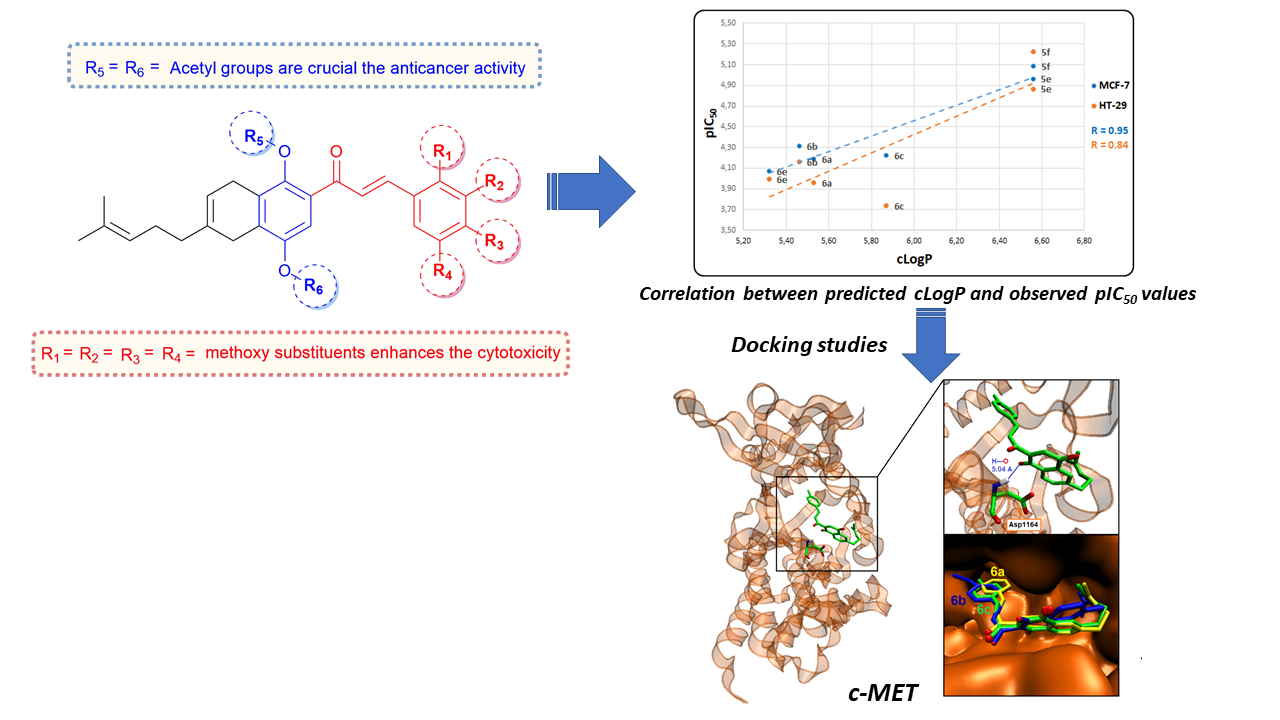
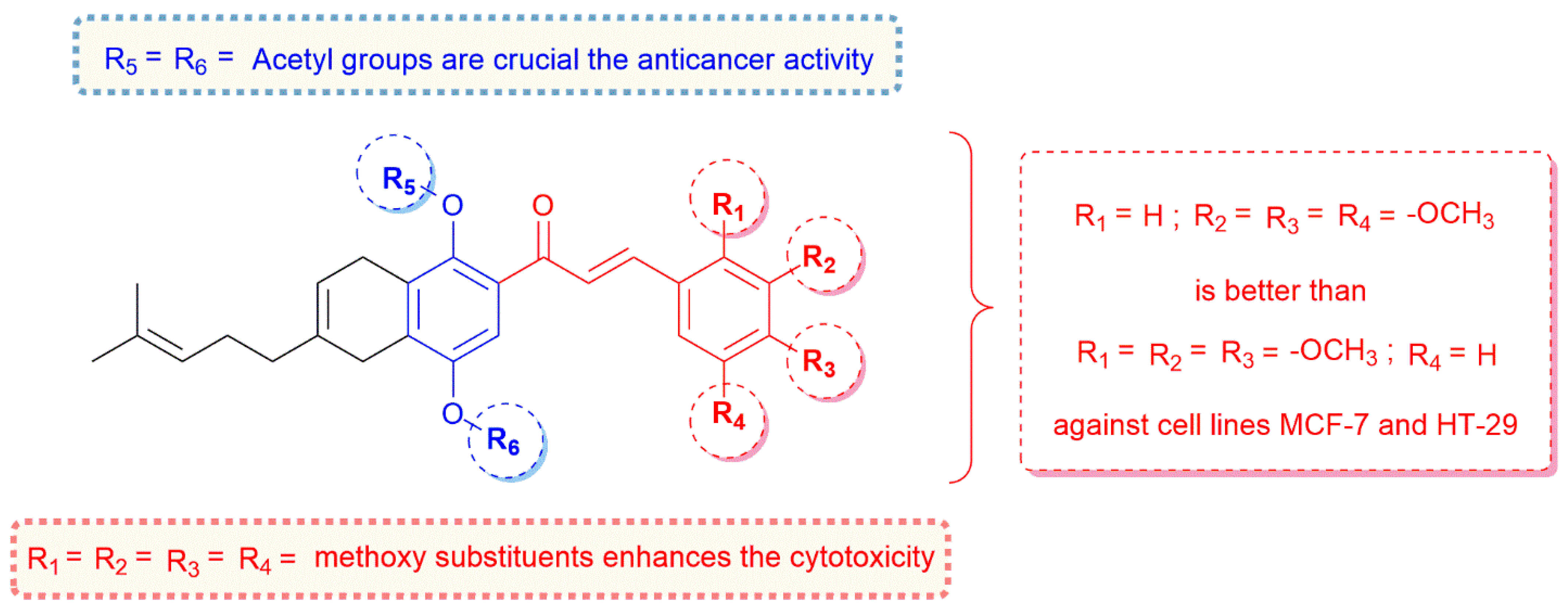
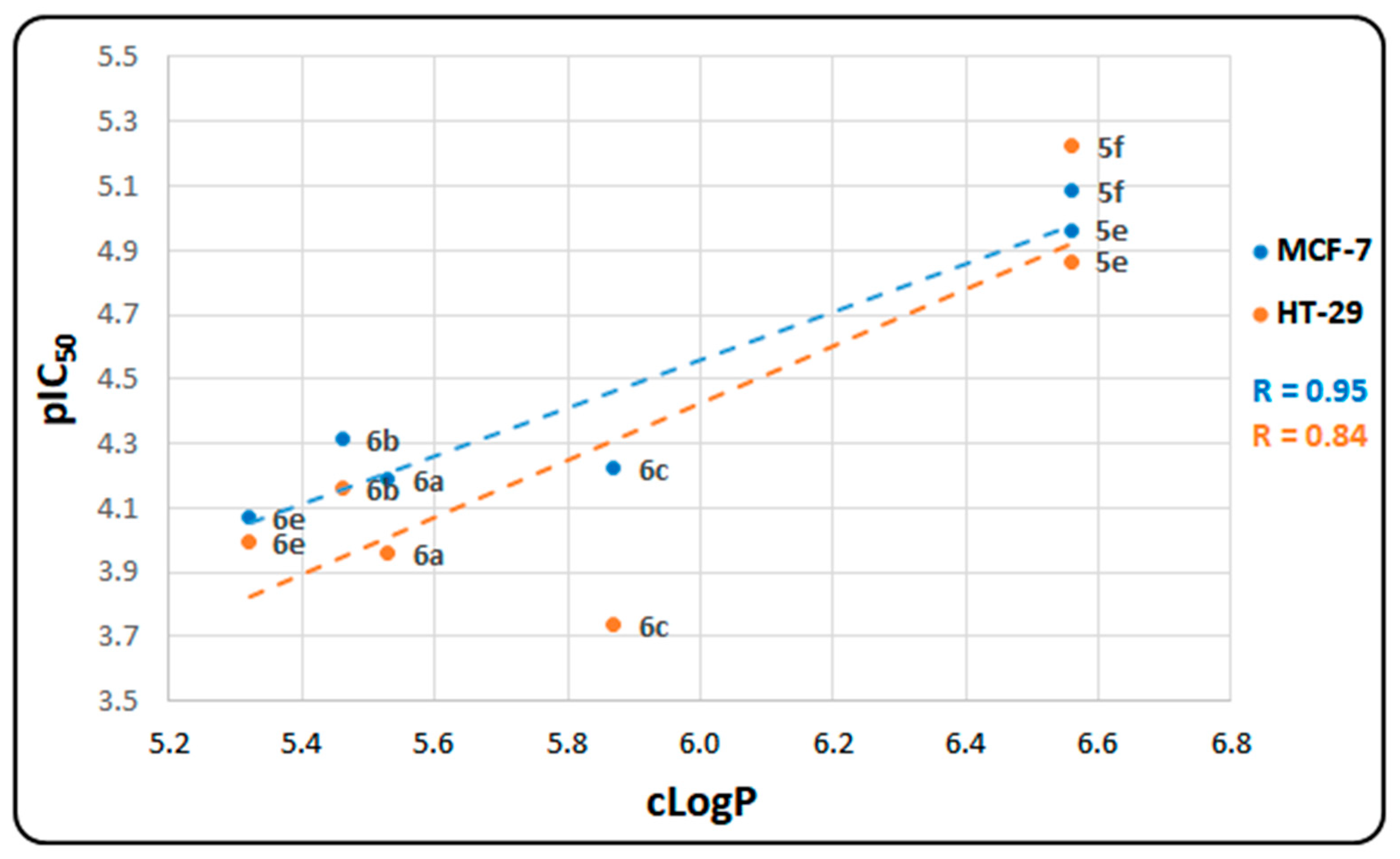
2.2. In Vitro Cytotoxicity Assays
2.3. In Silico Virtual Screening for Potential Antineoplastic Targets of Synthesized Cytotoxic Hybrids
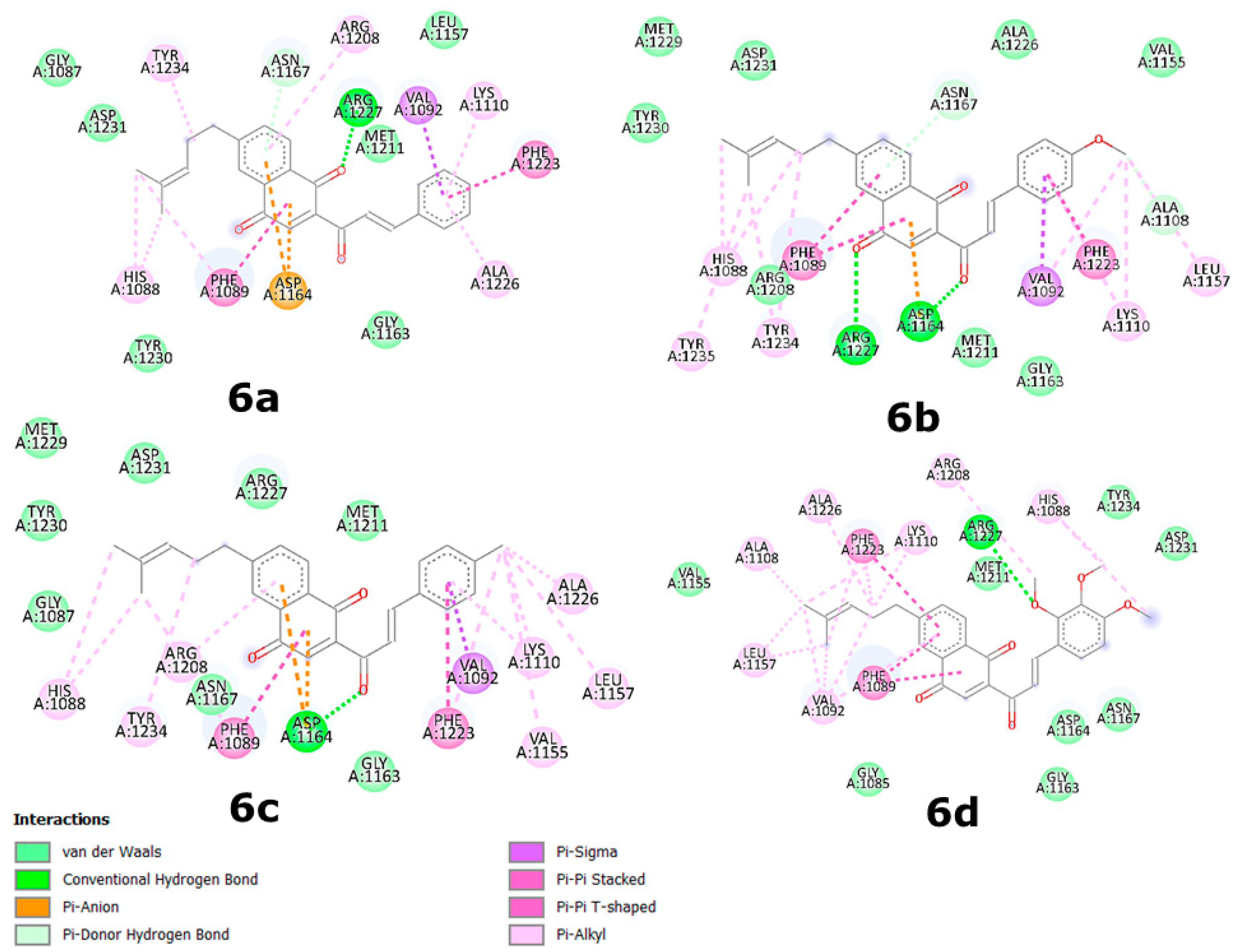
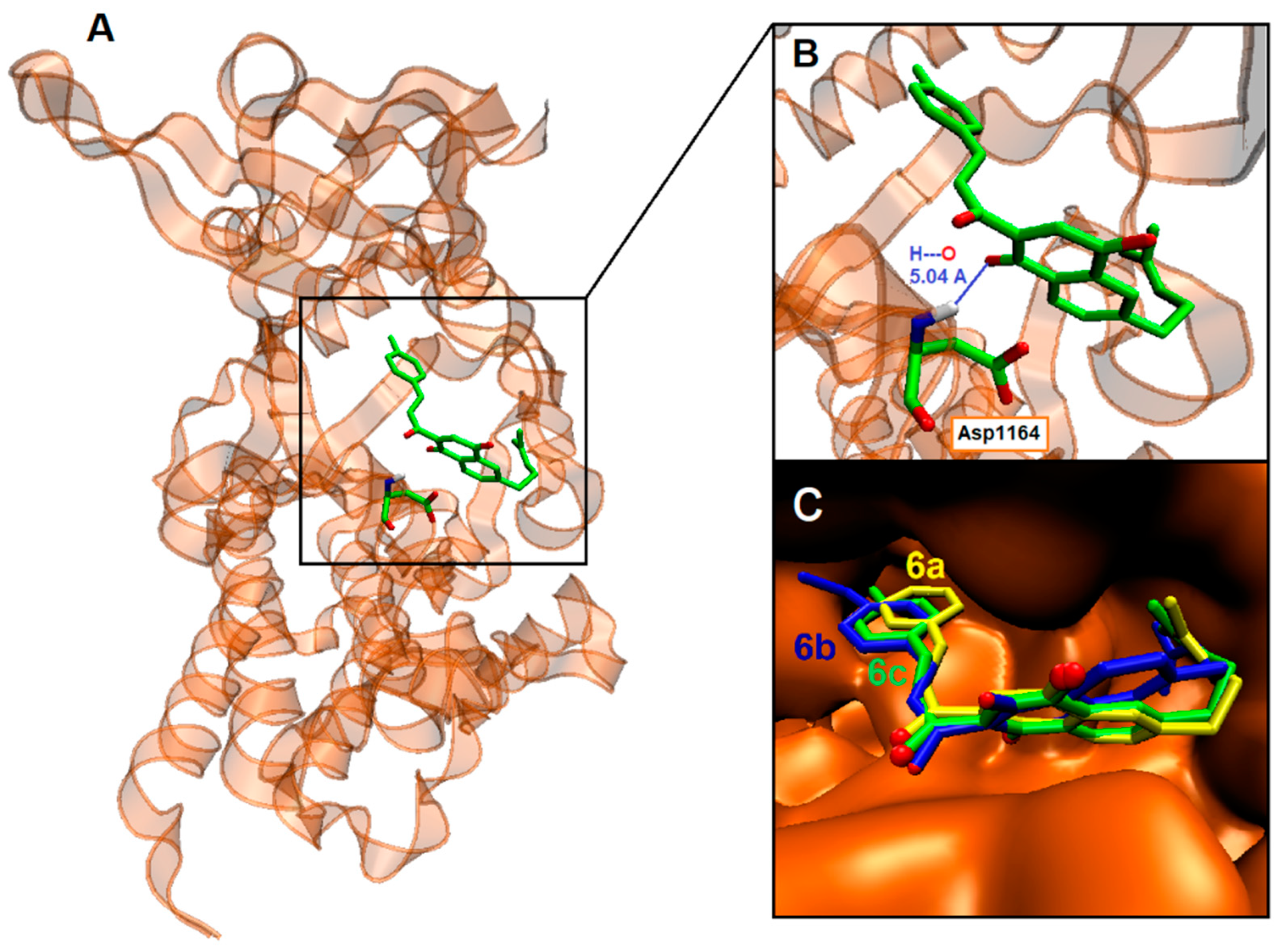
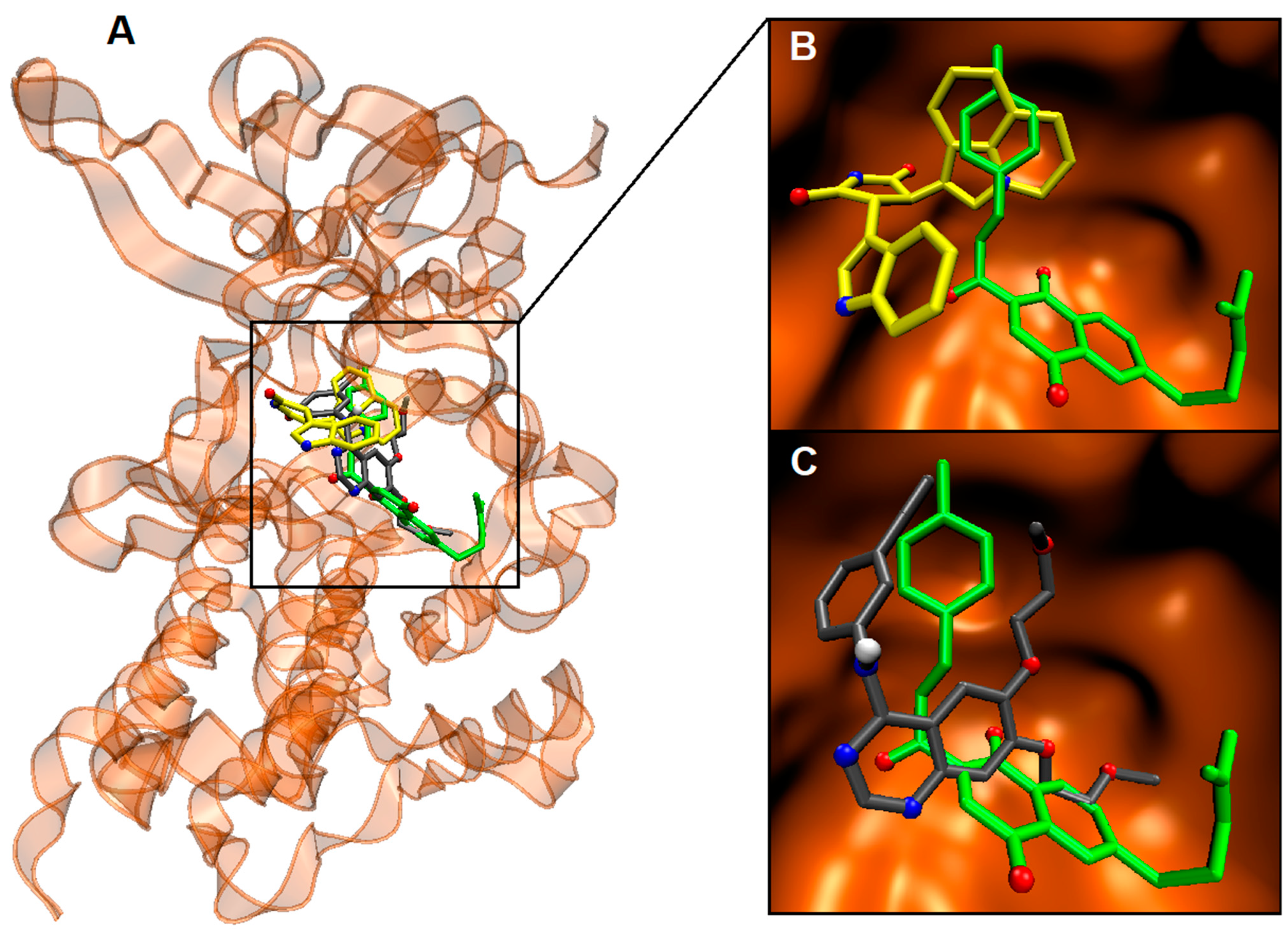
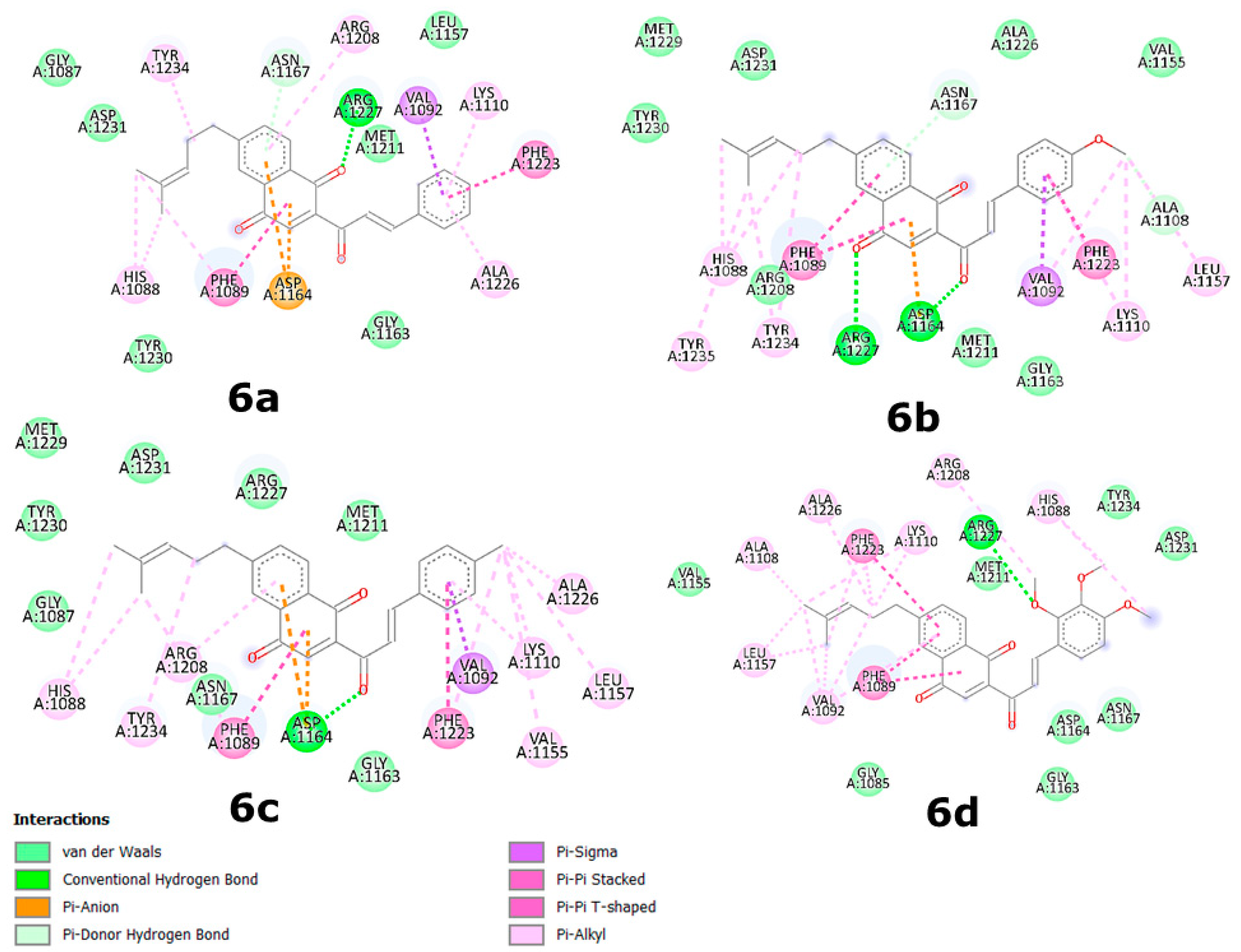
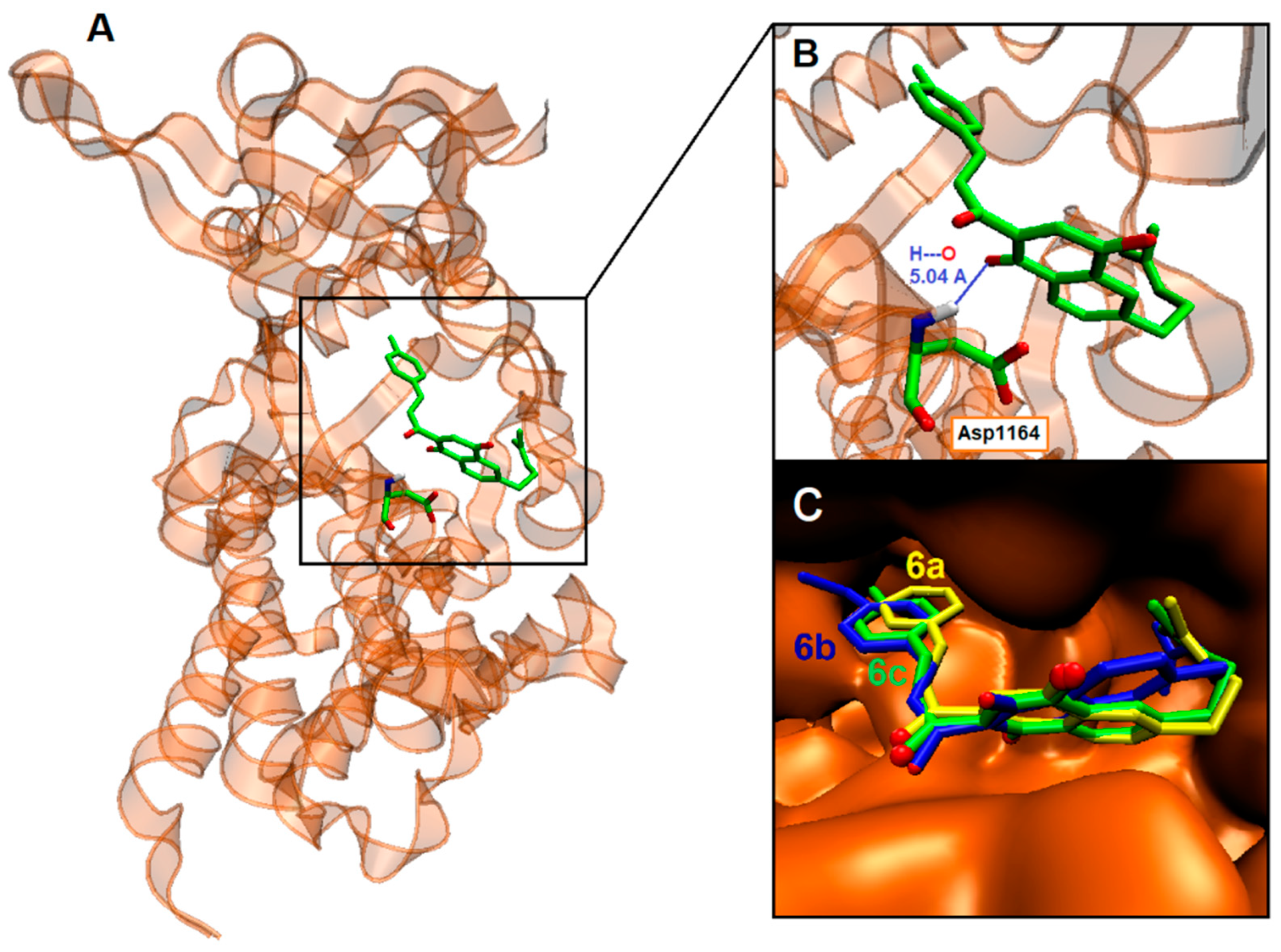
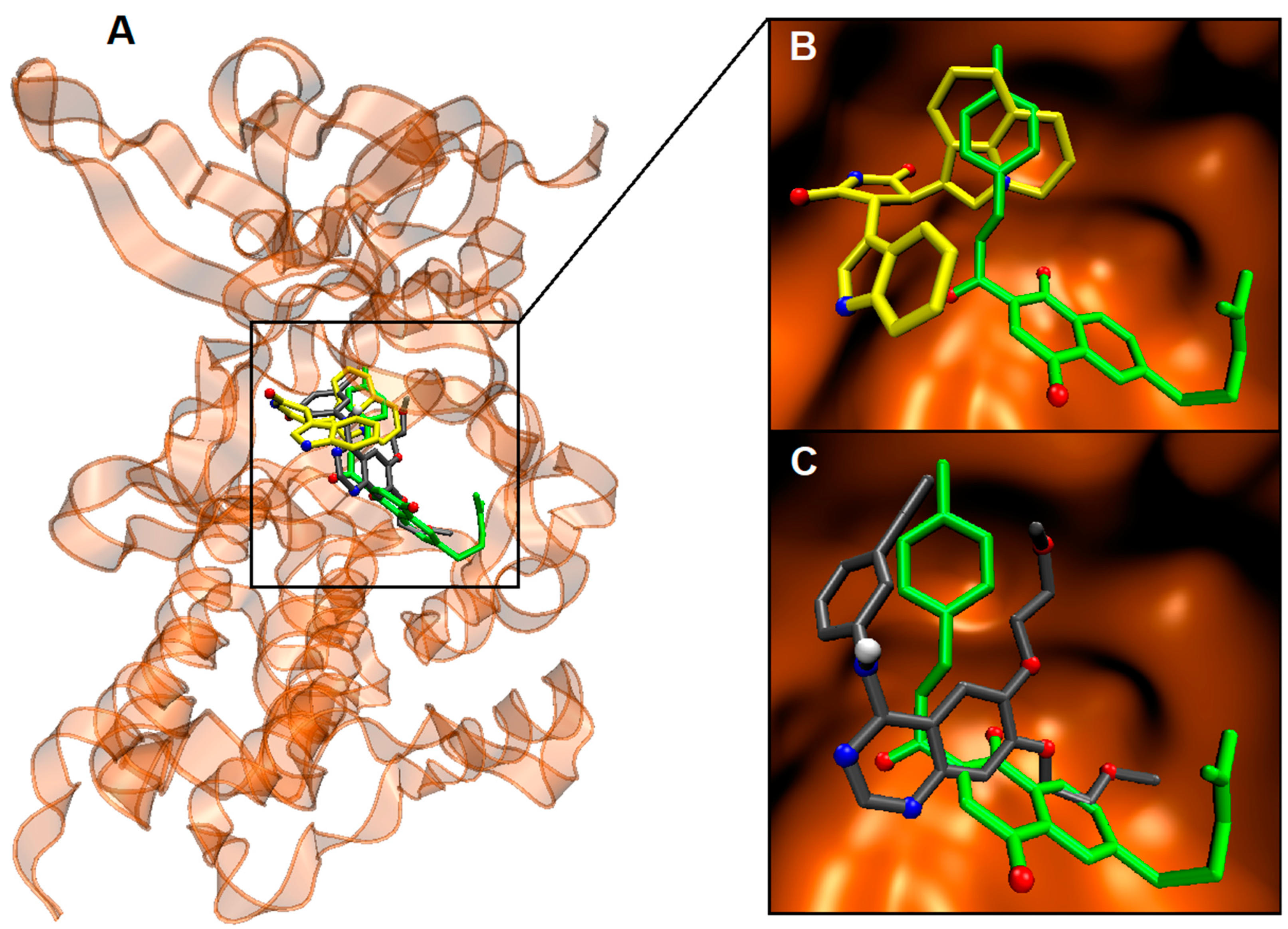
2.4. Binding Site and Docking of Synthesized Cytotoxic Hybrids in c-MET, TRKA, and HER2 Targets
2.5. In Silico Drug-Likeness, Toxicity Risks, and ADME Predictions
3. Materials and Methods
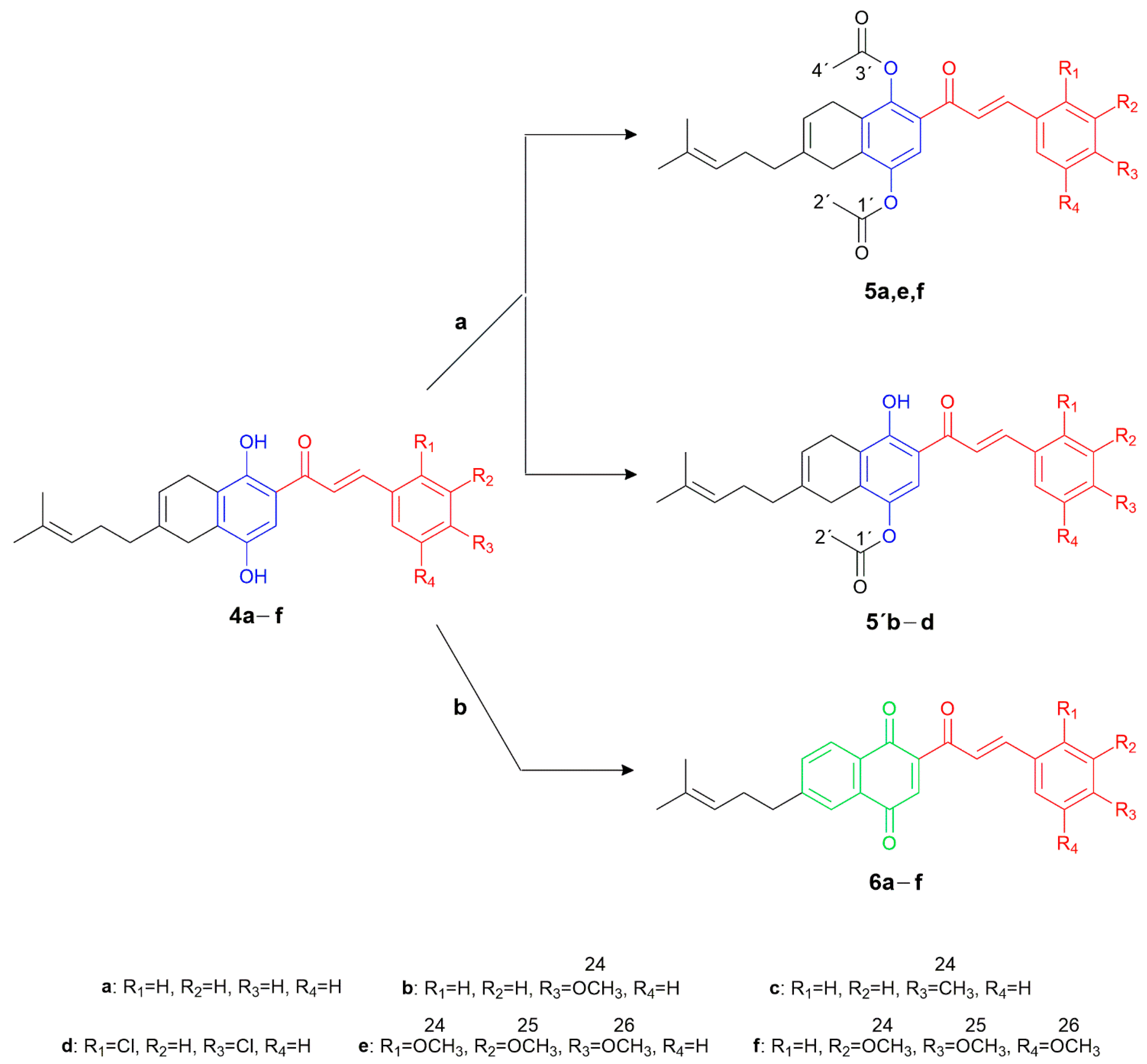
3.1. Chemistry
3.1.1. Procedure for the Synthesis and Molecular Characterization of Precursor 2
Synthesis of 1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}ethanone (2)
3.1.2. General Procedure for the Synthesis and Characterization of Compounds 3a–f
Synthesis of (E)-1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}-3-phenylprop-2-en-1-one (3a)
Synthesis of (E)-1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}-3-(4-methoxyphenyl)prop-2-en-1-one (3b)
Synthesis of (E)-1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}-3-(4-methylphenyl)prop-2-en-1-one (3c)
Synthesis of (E)-3-(2,4-Dichlorophenyl)1-{1-hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}prop-2-en-1-one (3d)
Synthesis of (E)-1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}-3-(2,3,4-trimethoxyphenyl)prop-2-en-1-one (3e)
Synthesis of (E)-1-{1-Hydroxy-6-(4-methylpent-3-en-1-yl)-4-[(tetrahydro-2H-pyran-2-yl)oxy]-5,8-dihydronaphthalen-2-yl}-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (3f)
General Procedure for the Synthesis and Characterization of Compounds 4a–f
Synthesis of (E)-1-[1,4-Dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]-3-phenylprop-2-en-1-one (4a)
Synthesis of (E)-1-[1,4-Dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]-3-(4-methoxyphenyl)prop-2-en-1-one (4b)
Synthesis of (E)-1-[1,4-Dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]-3-(4-methylphenyl)prop-2-en-1-one (4c)
Synthesis of (E)-3-(2,4-Dichlorophenyl)-1-[1,4-dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]prop-2-en-1-one (4d)
Synthesis of (E)-1-[1,4-Dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]-3-(2,3,4-trimethoxyphenyl)prop-2-en-1-one (4e)
Synthesis of (E)-1-[1,4-Dihydroxy-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-2-yl]-3-(3,4,5-trimethoxyphenyl)prop-2-en-1-one (4f)
General Procedure for the Synthesis and Characterization of Compounds 5′b–d and 5e,f
Synthesis of (E)-4-Hydroxy-3-[3-(4-methoxyphenyl)prop-2-enoyl]-7-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-1-yl ethanoate (5′b)
Synthesis of (E)-4-Hydroxy-7-(4-methylpent-3-en-1-yl)-3-[3-(4-methylphenyl)prop-2-enoyl]-5,8-dihydronaphthalen-1-yl ethanoate (5′c)
Synthesis of (E)-3-[3-(2,4-Dichlorophenyl)prop-2-enoyl]-4-hydroxy-7-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-1-yl ethanoate (5′d)
Synthesis of (E)-2-[3-(2,3,4-Trimethoxyphenyl)prop-2-enoyl]-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-1,4-diyl diethanoate (5e)
Synthesis of (E)-2-[3-(3,4,5-Trimethoxyphenyl)prop-2-enoyl]-6-(4-methylpent-3-en-1-yl)-5,8-dihydronaphthalen-1,4-diyl diethanoate (5f)
General Procedure for the Synthesis and Characterization of Compounds 6a–e
Synthesis of (E)-6-(4-Methylpent-3-en-1-yl)-2-(3-phenylprop-2-enoyl)naphthalene-1,4-dione (6a)
Synthesis of (E)-2-[3-(4-Methoxyphenyl)prop-2-enoyl]-6-(4-methylpent-3-en-1-yl) naphthalene-1,4-dione (6b)
Synthesis of (E)-6-(4-Methylpent-3-en-1-yl)-2-[3-(4-methylphenyl)prop-2-enoyl] naphthalene-1,4-dione (6c)
Synthesis of (E)-2-[3-(2,4-Dichlorophenyl)prop-2-enoyl]-6-(4-methylpent-3-en-1-yl) naphthalene-1,4-dione (6d)
Synthesis of (E)-2-[3-(2,3,4-Trimethoxyphenyl)prop-2-enoyl]-6-(4-methylpent-3-en-1-yl) naphthalene-1,4-dione (6e)
3.2. Antiproliferative Assay
3.3. Computational Details
3.3.1. Ligand Preparation
3.3.2. In Silico ADME Prediction
3.3.3. Macromolecule Selection and Retrieval
3.3.4. Molecular Docking of Ligand–Protein Interaction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- World Health Organization. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 7 July 2023).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer Statistics for the Year 2020: An Overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bisio, A.; Pedrelli, F.; D’Ambola, M.; Labanca, F.; Schito, A.M.; Govaerts, R.; De Tommasi, N.; Milella, L. Quinone Diterpenes from Salvia Species: Chemistry, Botany, and Biological Activity. Phytochem. Rev. 2019, 18, 665–842. [Google Scholar] [CrossRef]
- Manickam, M.; Boggu, P.R.; Cho, J.; Nam, Y.J.; Lee, S.J.; Jung, S.-H. Investigation of Chemical Reactivity of 2-Alkoxy-1,4-Naphthoquinones and Their Anticancer Activity. Bioorg. Med. Chem. Lett. 2018, 28, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Zorzanelli, B.C.; Ouverney, G.; Pauli, F.P.; da Fonseca, A.C.C.; de Almeida, E.C.P.; de Carvalho, D.G.; Possik, P.A.; Rabelo, V.W.-H.; Abreu, P.A.; Pontes, B.; et al. Pro-Apoptotic Antitumoral Effect of Novel Acridine-Core Naphthoquinone Compounds against Oral Squamous Cell Carcinoma. Molecules 2022, 27, 5148. [Google Scholar] [CrossRef]
- Sallustio, B.C.; Boddy, A.V. Is There Scope for Better Individualisation of Anthracycline Cancer Chemotherapy? Br. J. Clin. Pharmacol. 2021, 87, 295–305. [Google Scholar] [CrossRef]
- Liang, X.; Wu, Q.; Luan, S.; Yin, Z.; He, C.; Yin, L.; Zou, Y.; Yuan, Z.; Li, L.; Song, X.; et al. A Comprehensive Review of Topoisomerase Inhibitors as Anticancer Agents in the Past Decade. Eur. J. Med. Chem. 2019, 171, 129–168. [Google Scholar] [CrossRef]
- Kuete, V.; Donfack, A.R.N.; Mbaveng, A.T.; Zeino, M.; Tane, P.; Efferth, T. Cytotoxicity of Anthraquinones from the Roots of Pentas Schimperi towards Multi-Factorial Drug-Resistant Cancer Cells. Investig. New Drugs 2015, 33, 861–869. [Google Scholar] [CrossRef]
- Mancini, I.; Vigna, J.; Sighel, D.; Defant, A. Hybrid Molecules Containing Naphthoquinone and Quinolinedione Scaffolds as Antineoplastic Agents. Molecules 2022, 27, 4948. [Google Scholar] [CrossRef]
- Kabakci, Z.; Käppeli, S.; Cantù, C.; Jensen, L.D.; König, C.; Toggweiler, J.; Gentili, C.; Ribaudo, G.; Zagotto, G.; Basler, K.; et al. Pharmacophore-Guided Discovery of CDC25 Inhibitors Causing Cell Cycle Arrest and Tumor Regression. Sci. Rep. 2019, 9, 1335. [Google Scholar] [CrossRef]
- Hsu, M.-J.; Chen, H.-K.; Lien, J.-C.; Huang, Y.-H.; Huang, S.-W. Suppressing VEGF-A/VEGFR-2 Signaling Contributes to the Anti-Angiogenic Effects of PPE8, a Novel Naphthoquinone-Based Compound. Cells 2022, 11, 2114. [Google Scholar] [CrossRef] [PubMed]
- Nursamsiar; Asnawi, A.; Kartasasmita, R.E.; Ibrahim, S.; Tjahjono, D.H. Synthesis, Biological Evaluation, and Docking Analysis of Methyl Hydroquinone and Bromo Methyl Hydroquinone as Potent Cyclooxygenase (COX-1 and COX-2) Inhibitors. J. Appl. Pharm. Sci. 2018, 8, 16–20. [Google Scholar] [CrossRef]
- Byeon, S.; Yi, Y.-S.; Lee, J.; Yang, W.; Kim, J.; Kim, J.; Hong, S.; Kim, J.-H.; Cho, J. Hydroquinone Exhibits In Vitro and In Vivo Anti-Cancer Activity in Cancer Cells and Mice. Int. J. Mol. Sci. 2018, 19, 903. [Google Scholar] [CrossRef]
- Rudrapal, M.; Khan, J.; Dukhyil, A.A.B.; Alarousy, R.M.I.I.; Attah, E.I.; Sharma, T.; Khairnar, S.J.; Bendale, A.R. Chalcone Scaffolds, Bioprecursors of Flavonoids: Chemistry, Bioactivities, and Pharmacokinetics. Molecules 2021, 26, 7177. [Google Scholar] [CrossRef]
- Lou, H.; Hu, L.; Lu, H.; Wei, T.; Chen, Q. Metabolic Engineering of Microbial Cell Factories for Biosynthesis of Flavonoids: A Review. Molecules 2021, 26, 4522. [Google Scholar] [CrossRef] [PubMed]
- Kostopoulou, I.; Tzani, A.; Polyzos, N.-I.; Karadendrou, M.-A.; Kritsi, E.; Pontiki, E.; Liargkova, T.; Hadjipavlou-Litina, D.; Zoumpoulakis, P.; Detsi, A. Exploring the 2′-Hydroxy-Chalcone Framework for the Development of Dual Antioxidant and Soybean Lipoxygenase Inhibitory Agents. Molecules 2021, 26, 2777. [Google Scholar] [CrossRef] [PubMed]
- Elkanzi, N.A.A.; Hrichi, H.; Alolayan, R.A.; Derafa, W.; Zahou, F.M.; Bakr, R.B. Synthesis of Chalcones Derivatives and Their Biological Activities: A Review. ACS Omega 2022, 7, 27769–27786. [Google Scholar] [CrossRef]
- Sahu, N.K.; S. Balbhadra, S.; Choudhary, J.; V. Kohli, D. Exploring Pharmacological Significance of Chalcone Scaffold: A Review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Ducki, S. The Development of Chalcones as Promising Anticancer Agents. IDrugs 2007, 10, 42–46. [Google Scholar]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A Privileged Structure in Medicinal Chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; He, M.; Li, Y.; Peng, Z.; Wang, G. A Review on Synthetic Chalcone Derivatives as Tubulin Polymerisation Inhibitors. J. Enzyme Inhib. Med. Chem. 2022, 37, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S. Antimitotic Chalcones and Related Compounds as Inhibitors of Tubulin Assembly. Anticancer. Agents Med. Chem. 2009, 9, 336–347. [Google Scholar] [CrossRef]
- Michalkova, R.; Mirossay, L.; Gazdova, M.; Kello, M.; Mojzis, J. Molecular Mechanisms of Antiproliferative Effects of Natural Chalcones. Cancers 2021, 13, 2730. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-Cancer Chalcones: Structural and Molecular Target Perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef]
- Noser, A.A.; Shehadi, I.A.; Abdelmonsef, A.H.; Salem, M.M. Newly Synthesized Pyrazolinone Chalcones as Anticancer Agents via Inhibiting the PI3K/Akt/ERK1/2 Signaling Pathway. ACS Omega 2022, 7, 25265–25277. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational Approaches, Design Strategies, Structure Activity Relationship and Mechanistic Insights for Anticancer Hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Viegas-Junior, C.; Barreiro, E.J. Carlos Alberto Manssour Fraga Molecular Hybridization: A Useful Tool in the Design of New Drug Prototypes. Curr. Med. Chem. 2007, 14, 1829–1852. [Google Scholar] [CrossRef]
- Karthikeyan, C.; Solomon, V.R.; Lee, H.; Trivedi, P. Design, Synthesis and Biological Evaluation of Some Isatin-Linked Chalcones as Novel Anti-Breast Cancer Agents: A Molecular Hybridization Approach. Biomed. Prev. Nutr. 2013, 3, 325–330. [Google Scholar] [CrossRef]
- Mohamed, M.F.A.; Abuo-Rahma, G.E.-D.A. Molecular Targets and Anticancer Activity of Quinoline–Chalcone Hybrids: Literature Review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef] [PubMed]
- Kurt, B.Z.; Ozten Kandas, N.; Dag, A.; Sonmez, F.; Kucukislamoglu, M. Synthesis and Biological Evaluation of Novel Coumarin-Chalcone Derivatives Containing Urea Moiety as Potential Anticancer Agents. Arab. J. Chem. 2020, 13, 1120–1129. [Google Scholar] [CrossRef]
- Gao, F.; Huang, G.; Xiao, J. Chalcone Hybrids as Potential Anticancer Agents: Current Development, Mechanism of Action, and Structure-activity Relationship. Med. Res. Rev. 2020, 40, 2049–2084. [Google Scholar] [CrossRef]
- Bahia, S.B.B.B.; Reis, W.J.; Jardim, G.A.M.; Souto, F.T.; de Simone, C.A.; Gatto, C.C.; Menna-Barreto, R.F.S.; de Castro, S.L.; Cavalcanti, B.C.; Pessoa, C.; et al. Molecular Hybridization as a Powerful Tool towards Multitarget Quinoidal Systems: Synthesis, Trypanocidal and Antitumor Activities of Naphthoquinone-Based 5-Iodo-1,4-Disubstituted-, 1,4- and 1,5-Disubstituted-1,2,3-Triazoles. Medchemcomm 2016, 7, 1555–1563. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Chrobak, E.; Bębenek, E.; Latocha, M. Hybrids of 1,4-Quinone with Quinoline Derivatives: Synthesis, Biological Activity, and Molecular Docking with DT-Diaphorase (NQO1). Molecules 2022, 27, 6206. [Google Scholar] [CrossRef]
- Maldonado, J.; Acevedo, W.; Molinari, A.; Oliva, A.; Knox, M.; San Feliciano, A. Synthesis, In Vitro Evaluation and Molecular Docking Studies of Novel Naphthoisoxazolequinone Carboxamide Hybrids as Potential Antitumor Agents. Polycycl. Aromat. Compd. 2022, 43, 4960–4983. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Arismendi, M.; Imbarack, E.; Gálvez, C.; Maldonado, J.; Feliciano, A.S. The Synthesis of Some Fused Pyrazolo-1,4-Naphthoquinones. J. Heterocycl. Chem. 2015, 52, 620–622. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Arismendi-Macuer, M.; Guzmán, L.; Fuentealba, M.; Knox, M.; Vinet, R.; San Feliciano, A. New 1H-Benzo[f]Indazole-4,9-Diones Conjugated with C-Protected Amino Acids and Other Derivatives: Synthesis and in Vitro Antiproliferative Evaluation. Molecules 2015, 20, 21924–21938. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; del Corral, J.M.M.; Castro, M.A.; Araya, C.; García-Grávalos, M.D.; San Feliciano, A. Cytotoxic–Antineoplastic Activity of Acetyl Derivatives of Prenylnaphthohydroquinone. Farm. 2004, 59, 651–656. [Google Scholar] [CrossRef]
- Cooper, S.C.; Sammes, P.G. Acyl Rearrangements in Acylbenzoquinone Cycloadducts. J. Chem. Soc. Perkin Trans. 1984, 1, 2407–2414. [Google Scholar] [CrossRef]
- Andrade, J.T.; Santos, F.R.S.; Lima, W.G.; Sousa, C.D.F.; Oliveira, L.S.F.M.; Ribeiro, R.I.M.A.; Gomes, A.J.P.S.; Araújo, M.G.F.; Villar, J.A.F.P.; Ferreira, J.M.S. Design, Synthesis, Biological Activity and Structure-Activity Relationship Studies of Chalcone Derivatives as Potential Anti-Candida Agents. J. Antibiot. 2018, 71, 702–712. [Google Scholar] [CrossRef]
- Hsieh, H.K.; Lee, T.H.; Wang, J.P.; Wang, J.J.; Lin, C.N. Synthesis and Anti-Inflammatory Effect of Chalcones and Related Compounds. Pharm. Res. 1998, 15, 39–46. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Ojeda, C.; Miguel del Corral, J.M.; Castro, M.A.; Cuevas, C.; San Feliciano, A. New Cytotoxic-Antineoplastic Prenyl-1,2-Naphthohydroquinone Derivatives. Bioorg. Med. Chem. 2005, 13, 6645–6650. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, T.; Mihis, A.G. Two Important Anticancer Mechanisms of Natural and Synthetic Chalcones. Int. J. Mol. Sci. 2022, 23, 11595. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.I.; Yamali, C.; Gunesacar, G.; Sakagami, H.; Okudaira, N.; Uesawa, Y.; Kagaya, H. Cytotoxicity, Apoptosis, and QSAR Studies of Phenothiazine Derived Methoxylated Chalcones as Anticancer Drug Candidates. Med. Chem. Res. 2018, 27, 2366–2378. [Google Scholar] [CrossRef]
- Sharma, R.; Kumar, R.; Kodwani, R.; Kapoor, S.; Khare, A.; Bansal, R.; Khurana, S.; Singh, S.; Thomas, J.; Roy, B.; et al. A Review on Mechanisms of Anti Tumor Activity of Chalcones. Anticancer. Agents Med. Chem. 2015, 16, 200–211. [Google Scholar] [CrossRef]
- Albright, C.F.; Graciani, N.; Han, W.; Yue, E.; Stein, R.; Lai, Z.; Diamond, M.; Dowling, R.; Grimminger, L.; Zhang, S.-Y.; et al. Matrix Metalloproteinase–Activated Doxorubicin Prodrugs Inhibit HT1080 Xenograft Growth Better than Doxorubicin with Less Toxicity. Mol. Cancer Ther. 2005, 4, 751–760. [Google Scholar] [CrossRef]
- Negi, R.R.; Rana, S.V.; Gupta, V.; Gupta, R.; Chadha, V.D.; Prasad, K.K.; Dhawan, D.K. Over-Expression of Cyclooxygenase-2 in Colorectal Cancer Patients. Asian Pacific J. Cancer Prev. 2019, 20, 1675–1681. [Google Scholar] [CrossRef]
- Singh, B.; Berry, J.A.; Shoher, A.; Ramakrishnan, V.; Lucci, A. COX-2 Overexpression Increases Motility and Invasion of Breast Cancer Cells. Int. J. Oncol. 2005, 26, 1393–1399. [Google Scholar]
- Guo, S.; Colbert, L.S.; Fuller, M.; Zhang, Y.; Gonzalez-Perez, R.R. Vascular Endothelial Growth Factor Receptor-2 in Breast Cancer. Biochim. Biophys. Acta—Rev. Cancer 2010, 1806, 108–121. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen Receptor Alpha in Human Breast Cancer: Occurrence and Significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB Network: At Last, Cancer Therapy Meets Systems Biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, S.; Ko, S.; In, Y.; Moon, H.-G.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.-H.; Ju, Y.S.; et al. Recurrent Fusion Transcripts Detected by Whole-Transcriptome Sequencing of 120 Primary Breast Cancer Samples. Genes, Chromosom. Cancer 2015, 54, 681–691. [Google Scholar] [CrossRef]
- Ahmad, D.A.J.; Negm, O.H.; Alabdullah, M.L.; Mirza, S.; Hamed, M.R.; Band, V.; Green, A.R.; Ellis, I.O.; Rakha, E.A. Clinicopathological and Prognostic Significance of Mitogen-Activated Protein Kinases (MAPK) in Breast Cancers. Breast Cancer Res. Treat. 2016, 159, 457–467. [Google Scholar] [CrossRef]
- Pashirzad, M.; Khorasanian, R.; Fard, M.M.; Arjmand, M.-H.; Langari, H.; Khazaei, M.; Soleimanpour, S.; Rezayi, M.; Ferns, G.A.; Hassanian, S.M.; et al. The Therapeutic Potential of MAPK/ERK Inhibitors in the Treatment of Colorectal Cancer. Curr. Cancer Drug Targets 2021, 21, 932–943. [Google Scholar] [CrossRef]
- Abourehab, M.A.S.; Alqahtani, A.M.; Youssif, B.G.M.; Gouda, A.M. Globally Approved EGFR Inhibitors: Insights into Their Syntheses, Target Kinases, Biological Activities, Receptor Interactions, and Metabolism. Molecules 2021, 26, 6677. [Google Scholar] [CrossRef]
- Liu, F.; Wei, Y.; Zhang, H.; Jiang, J.; Zhang, P.; Chu, Q. NTRK Fusion in Non-Small Cell Lung Cancer: Diagnosis, Therapy, and TRK Inhibitor Resistance. Front. Oncol. 2022, 12, 864666. [Google Scholar] [CrossRef]
- Li, S. Anlotinib: A Novel Targeted Drug for Bone and Soft Tissue Sarcoma. Front. Oncol. 2021, 11, 664853. [Google Scholar] [CrossRef]
- Sylvester, P.W. Targeting Met Mediated Epithelial-mesenchymal Transition in the Treatment of Breast Cancer. Clin. Transl. Med. 2014, 3, 30. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, B.P. Epithelial-Mesenchymal Transition in Breast Cancer Progression and Metastasis. Chin. J. Cancer 2011, 30, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, G.; Smith, C.; Goddard, L.; Jordan, N.; McClelland, R.; Barrett-Lee, P.; Nicholson, R.I.; Hiscox, S. Targeting Focal Adhesion Kinase in ER+/HER2+ Breast Cancer Improves Trastuzumab Response. Endocr. Relat. Cancer 2013, 20, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Brylinski, M. Aromatic Interactions at the Ligand-Protein Interface: Implications for the Development of Docking Scoring Functions. Chem. Biol. Drug Des. 2018, 91, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liu, D. Suppression of C-MET Overcomes Erlotinib Resistance in Tongue Cancer Cells. Onco. Targets. Ther. 2018, 11, 5499–5508. [Google Scholar] [CrossRef]
- Han, S.-Y. TRK Inhibitors: Tissue-Agnostic Anti-Cancer Drugs. Pharmaceuticals 2021, 14, 632. [Google Scholar] [CrossRef]
- Schaefer, G.; Shao, L.; Totpal, K.; Akita, R.W. Erlotinib Directly Inhibits HER2 Kinase Activation and Downstream Signaling Events in Intact Cells Lacking Epidermal Growth Factor Receptor Expression. Cancer Res. 2007, 67, 1228–1238. [Google Scholar] [CrossRef]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An Open-Source Program For Chemistry Aware Data Visualization And Analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of Drug Solubility from Structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Nakano, M.; Fukami, T.; Gotoh, S.; Nakajima, M. A-to-I RNA Editing Up-Regulates Human Dihydrofolate Reductase in Breast Cancer. J. Biol. Chem. 2017, 292, 4873–4884. [Google Scholar] [CrossRef]
- Lei, H.; Deng, C.-X. Fibroblast Growth Factor Receptor 2 Signaling in Breast Cancer. Int. J. Biol. Sci. 2017, 13, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Haldosén, L.-A.; Zhao, C.; Dahlman-Wright, K. Estrogen Receptor Beta in Breast Cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, G.; Li, Y.; Padia, R.N.; Dong, Z.; Pan, Z.K.; Liu, K.; Huang, S. Extracellular Signal–Regulated Kinase Signaling Pathway Regulates Breast Cancer Cell Migration by Maintaining Slug Expression. Cancer Res. 2009, 69, 9228–9235. [Google Scholar] [CrossRef]
- Zhang, X.; Li, B.; Song, M.; Song, J. Expression and Significance of ERK Protein in Human Breast Carcinoma. Chinese J. Cancer Res. 2004, 16, 269–273. [Google Scholar] [CrossRef]
- Sultan, A.S.; Brim, H.; Sherif, Z.A. Co-Overexpression of Janus Kinase 2 and Signal Transducer and Activator of Transcription 5a Promotes Differentiation of Mammary Cancer Cells through Reversal of Epithelial–Mesenchymal Transition. Cancer Sci. 2008, 99, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Molinari, A.; Oliva, A.; Arismendi-Macuer, M.; Guzmán, L.; Acevedo, W.; Aguayo, D.; Vinet, R.; San Feliciano, A. Antiproliferative Benzoindazolequinones as Potential Cyclooxygenase-2 Inhibitors. Molecules 2019, 24, 2261. [Google Scholar] [CrossRef]
- Acevedo, W.; González-Nilo, F.; Agosin, E. Docking and Molecular Dynamics of Steviol Glycoside–Human Bitter Receptor Interactions. J. Agric. Food Chem. 2016, 64, 7585–7596. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Visualizer; V20.1.0, Vol19295; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | MCF-7 | HT-29 | ||
|---|---|---|---|---|
| IC50, µM [a] | pIC50 [b] | IC50, µM | pIC50 | |
| 4a | >300 | - | >300 | - |
| 4b | >300 | - | >300 | - |
| 4c | >300 | - | >300 | - |
| 4d | >300 | - | >300 | - |
| 4e | >300 | - | >300 | - |
| 4f | >300 | - | >300 | - |
| 5a | nt | - | nt | - |
| 5′b | >300 | - | >300 | - |
| 5′c | >300 | - | >300 | - |
| 5′d | >300 | - | >300 | - |
| 5e | 10.9 ± 0.21 | 4.96 | 13.6 ± 0.14 | 4.87 |
| 5f | 8.2 ± 0.42 | 5.09 | 6.0 ± 0.53 | 5.22 |
| 6a | 64.8 ± 1.09 | 4.19 | 110.5 ± 1.93 | 3.96 |
| 6b | 48.7 ± 0.76 | 4.31 | 69.0 ± 0.21 | 4.16 |
| 6c | 59.8 ± 1.83 | 4.22 | 183.3 ± 1.21 | 3.74 |
| 6d | >300 | - | >300 | - |
| 6e | 85.0 ± 0.93 | 4.07 | 101.7 ± 1.10 | 3.99 |
| 6f | nt | - | nt | - |
| Doxorubicin | 0.27 ± 0.08 | 6.57 | 4.07 ± 0.92 | 5.39 |
| Compounds | Target Proteins | |||||
|---|---|---|---|---|---|---|
| EGFR | HER2 | c-MET | TRKA | MEK1 | TPK | |
| 5e | −7.2 | −8.3 | −9.8 | −8.9 | −9.2 | −8.6 |
| 5f | −7.4 | −8.6 | −9.7 | −9.1 | −8.7 | −8.2 |
| 6a | −10.6 | −10.8 | −10.3 | −10.5 | −10.5 | −10.3 |
| 6b | −10.6 | −10.2 | −10.3 | −10.7 | −9.8 | −10.3 |
| 6c | −11.1 | −10.4 | −10.6 | −11.0 | −10.2 | −10.4 |
| 6e | −10.0 | −10.2 | −9.8 | −9.6 | −9.9 | −9.9 |
| P avge. | −9.53 | −9.75 | −10.08 | −9.97 | −9.72 | −9.62 |
| Erlotinib | −8.6 | −8.0 | −9.1 | −8.8 | −8.0 | −8.5 |
| Larotrectinib | −10.3 | −8.8 | −10.8 | −11.0 | −9.4 | −9.5 |
| Almonertinib | −7.7 | −8.4 | −10.4 | −9.8 | −8.8 | −10.1 |
| Anlotinib | −9.2 | −10.3 | −9.0 | −10.7 | −9.7 | −9.0 |
| Compounds | ΔGbin | H-Bonds and Hydrophobic Contacts in the Binding Site * |
|---|---|---|
| c-MET (mean ΔGbin = −10.08 kcal/mol) | ||
| 5e | −9.8 | Gly1028, Ile1084, Gly1085, His1088, Phe1089 *, Val1092/Ala1108, Lys1110, Val1155, Leu1157, Asp1164, Gly1163/Arg1208, Met1211, Phe1223 *, Ala 1226, Arg1227, Asp1231, Tyr1234 * |
| 5f | −9.7 | Ile1084, Gly1085, Arg1086, Gly1087, His1088, Phe1089 *, Val1092/Ala1108, Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167 /Arg1208, Met1211, Phe1223 *, Ala1226, Arg1227, Asp1231, Tyr1234 |
| 6a | −10.3 | Gly1087, His1088, Phe1089 *, Val1092 */Lys1110 *, Leu1157, Gly1163, Asp1164/Arg1208, Val1092, Met1211, Phe1223 *, Ala1226 *, Arg1227, Tyr1230, Asp1231, Tyr1234 * |
| 6b | −10.3 | His1088, Phe1089 *, Val1092 */Val1155, Leu1157, Gly1163, Asp1164/Arg1208, Met1211, Phe1223 *, Ala1226, Arg1227, Met1229, Tyr1230, Asp1231/Tyr1234 *, Tyr1235 * |
| 6c | −10.6 | Gly1087, His1088, Phe1089 *, Val1092 */Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167/Arg1208, Met1211, Ala1226, Arg1227, Met1229, Tyr1230, Asp1231 |
| 6e | −9.8 | Gly1085, His1088, Phe1089 *, Val1092/Ala1108, Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167/Arg1208, Met1211, Phe1223 *, Ala1226, Arg1227, Asp1231, Tyr1234 |
| TRKA (mean ΔGbin = −9.97 kcal/mol) | ||
| 5e | −8.9 | Leu516 *, Gly517, Glu518, Val524 */Ala542, Lys544, Glu560/Leu564, Val573 *, Phe589 *, Tyr591/Gly595, Asp596, Arg599, Leu657, Gly667/Asp668, Phe669 *, Arg673, Ile675, Tyr676 |
| 5f | −9.1 | Leu516 *, Gly517, Gly519, Phe521 *, Gly522, Val524 */Ala542 *, Lys544, Glu560/Phe589, Glu590, Tyr591/Met592, Gly595, Asp596, Arg599, Leu657/Gly670, Ser672, Arg673, Ile675, Phe669 * |
| 6a | −10.5 | Leu516, Val524 */Lys544, Glu560/Leu564 *, Ile572, Val573, Phe589, Glu590, Tyr591/Gly595, Asp596, Leu657/Ile666, Gly667, Asp668, Phe669 *, Arg673 |
| 6b | −10.7 | Leu516, Val524 */Ala542 *, Lys544,/Leu564 *, Ile572, Val573 *, Glu590, Phe589, Tyr591/Gly595, Asp596, Leu641, His648, Leu657/Gly667, Asp668, Phe669 * |
| 6c | −11.0 | Leu516, Val524 */Ala542 *, Lys544, Glu560/Leu564 *, Il572, Val573, Phe589, Glu590, Tyr591/Gly595, Asp596, Leu657/Ille666, Gly667, Asp668, Phe669 * |
| 6e | −9.5 | Leu516, Gly519, Phe521, Gly522, Val524 */Ala542, Lys544 *, Glu560/Phe589, Tyr591/Asp596, Arg599, Leu657 */Asp668, Phe669 *, Gly670, Ser672, Arg673, Ile675, Tyr676 |
| HER2 (mean ΔGbin = −9.75 kcal/mol) | ||
| 5e | −8.3 | Leu726 *, Val734 *, Ala751 */Lys753 *, Ile767, Glu770, Ala771, Met774/Ser783, Arg784, Leu785, Leu796, Thr798/Leu800, Met801, Gly804, Cys805, Leu852 */Thr862, Asp863, Phe864 * |
| 5f | −8.6 | Leu726, Val734 *, Ala751 */Ile752, Lys753, Ile767, Glu770, Ala771, Met774/Arg784, Leu785 *, Leu796 *, Thr798/Gln799, Leu800, Met801, Gly804, Cys805, Leu852 */Phe864, Gly865 |
| 6a | −10.8 | Leu726, Gly727, Val734, Ala751 */Lys753, Ile767, Glu770, Ala771 */Ser783, Arg784, Leu785 *, Leu796 *, Thr798/Glu799, Leu800, Met801, Leu852 */Thr862, Asp863, Phe864 |
| 6b | −10.2 | Leu726 *, Phe731, Val734 *, Ala751 */Lys753, Leu755, Ile767, Glu770, Met774,/Ser783, Arg784, Leu785 *, Leu796, Thr798/Gln799, Leu800, Met801, Gly804, Cys805, Leu852 */Thr862, Asp863, Phe864 *, Gly865 |
| 6c | −10.4 | Leu726 *, Phe731, Val734 *, Ala751 */Lys753, Leu755, Ile767, Glu770, Met774/Ser783, Arg784, Leu785 *, Leu796, Thr798/Gln799, Leu800, Met801, Gly804, Leu852 */Thr862, Asp863, Phe864 *, Gly865 |
| 6e | −10.2 | Phe731/Glu770, Met774/Ser783, Arg784, Thr798/Gln799, Met801, Gly804/Thr862, Asp863, Gly865, Arg849 |
| Compounds | ΔGbin (kcal/mol) | H-Bonds and Hydrophobic Contacts in the Binding Site |
|---|---|---|
| c-MET | ||
| 6c | −10.6 | Gly1087, His1088, Phe1089, Val1092, Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167, Arg1208, Met1211, Ala1226, Arg1227, Met1229, Tyr1230, Asp1231 |
| Ligand 1 [a] | −14.6 | Ile1084, Gly1085, Phe1089, Val1092, Ala1108, Lys1110, Leu1140, Leu1157, Tyr1159, Met1158, Met1160, Gly1163, Met1211, Phe1223, Ala1226, Arg1227 |
| Erlotinib [b] | −9.1 | Phe1089, Val1092, Ala1108, Lys1110, Val1155, Leu1157, Gly1163, Asp1164, Asn1167, Arg1208, Met1211, Arg1221, Phe1223, Ala1226, Arg1227, Asp1231, Tyr1234 |
| TRKA | ||
| 6c | −11.0 | Leu516, Val524, Ala542, Lys544, Glu560, Leu564, Ile572, Val573, Phe589, Glu590, Tyr591, Gly595, Asp596, Leu657, Ille666, Gly667, Asp668, Phe669 |
| Ligand 2 [a] | −14.2 | Leu516, Val524, Ala542, Lys544, Arg559, Glu560, Leu563, Leu564, Leu567, Ile572, Val573, Phe589, Glu590, Tyr591, Met592, Leu641, Phe646, His648, Leu657, Ile666, Gly667, Asp668, Phe669 |
| Larotrectinib [b] | −11.0 | Gly517, Glu518, Gly519, Phe521, Gly522, Val524, Ala542, Lys544, Glu560, Val573, Met587, Phe589, Leu657, Gly667, Asp668, Phe669, Gly670, Ser672, Arg673 |
| HER2 | ||
| 6c | −10.2 | Leu726, Phe731, Val734, Ala751, Lys753, Leu755, Ile767, Glu770, Met774, Ser783, Arg784, Leu785, Leu796, Thr798, Gln799, Leu800, Met801, Gly804, Leu852, Thr862, Asp863, Phe864, Gly865, |
| Ligand 3 [a] | −14.5 | Leu726, Val734, Ala751, Lys753, Met774, Ser783, Arg784, Leu785, Leu796, Thr798, Gln799, Leu800, Met801, Pro802, Gly804, Cys805, Leu807, Asp808, Arg849, Leu852, Thr862, Asp863, Phe864 |
| Erlotinib [b] | −8.0 | Leu726, Val734, Ala751, Lys753, Leu785, Leu796, Thr798, Gn799, Leu800, Met801, Gly804, Cys805, Asn850, Leu852, Thr862, Asp863, Phe864 |
| Compound | M | T | I | R | Drug-Likeness |
|---|---|---|---|---|---|
| 5e | N | n | h | n | −0.14 |
| 5f | N | n | h | n | 2.15 |
| 6a | N | n | n | n | −5.81 |
| 6b | N | n | h | l | −2.28 |
| 6c | N | n | n | n | −4.04 |
| 6e | N | n | n | n | −1.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, J.; Oliva, A.; Molinari, A.; Acevedo, W. 2-Acetyl-5,8-dihydro-6-(4-methyl-3-pentenyl)-1,4-naphthohydroquinone-Derived Chalcones as Potential Anticancer Agents. Molecules 2023, 28, 7172. https://doi.org/10.3390/molecules28207172
Maldonado J, Oliva A, Molinari A, Acevedo W. 2-Acetyl-5,8-dihydro-6-(4-methyl-3-pentenyl)-1,4-naphthohydroquinone-Derived Chalcones as Potential Anticancer Agents. Molecules. 2023; 28(20):7172. https://doi.org/10.3390/molecules28207172
Chicago/Turabian StyleMaldonado, Javier, Alfonso Oliva, Aurora Molinari, and Waldo Acevedo. 2023. "2-Acetyl-5,8-dihydro-6-(4-methyl-3-pentenyl)-1,4-naphthohydroquinone-Derived Chalcones as Potential Anticancer Agents" Molecules 28, no. 20: 7172. https://doi.org/10.3390/molecules28207172
APA StyleMaldonado, J., Oliva, A., Molinari, A., & Acevedo, W. (2023). 2-Acetyl-5,8-dihydro-6-(4-methyl-3-pentenyl)-1,4-naphthohydroquinone-Derived Chalcones as Potential Anticancer Agents. Molecules, 28(20), 7172. https://doi.org/10.3390/molecules28207172