
Mechanistic Study on Steric Activity Interplay of Olefin/Polar Monomers for Industrially Selective Late Transition Metal Catalytic Reactions
 , , , ,
, , , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
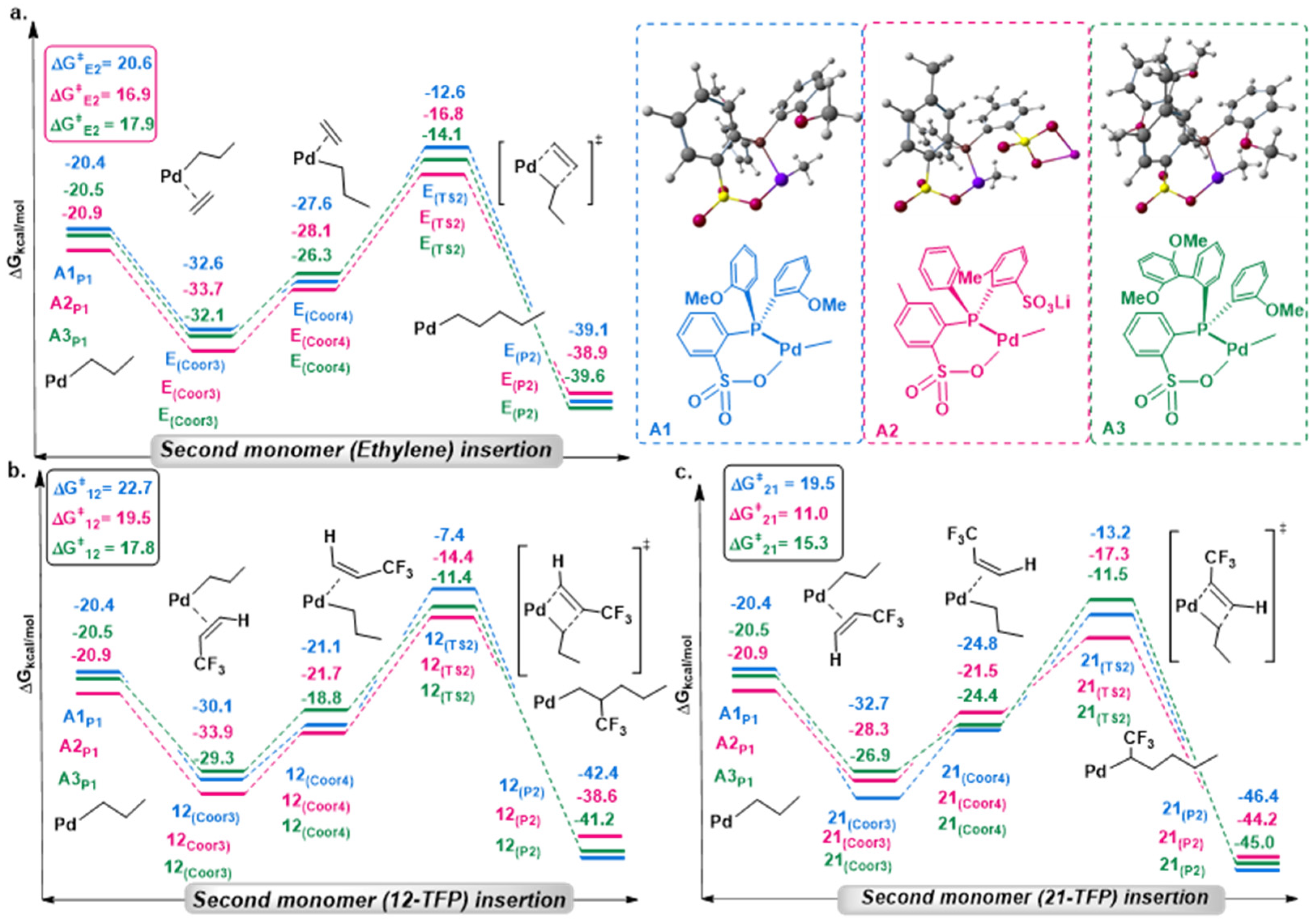
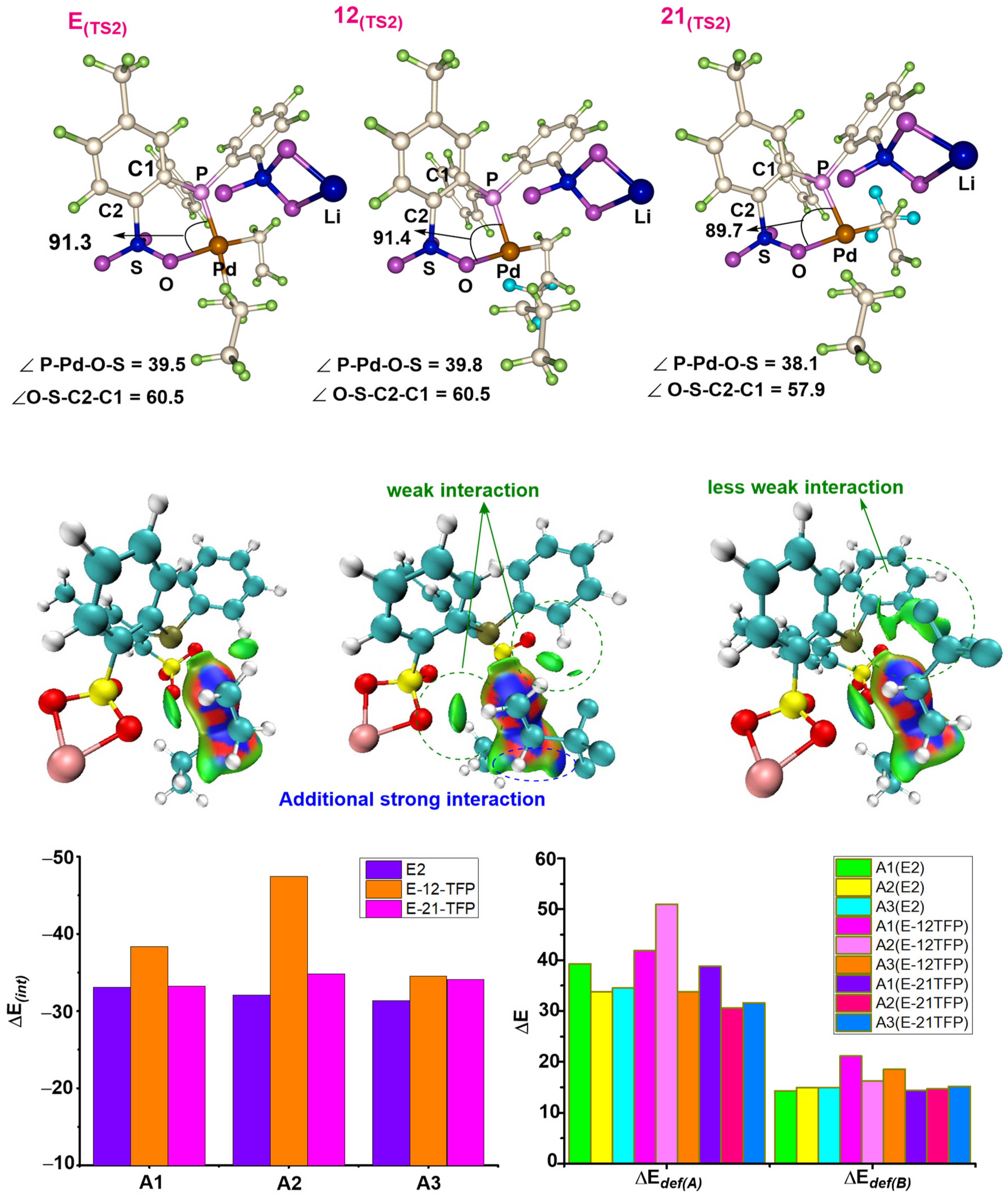
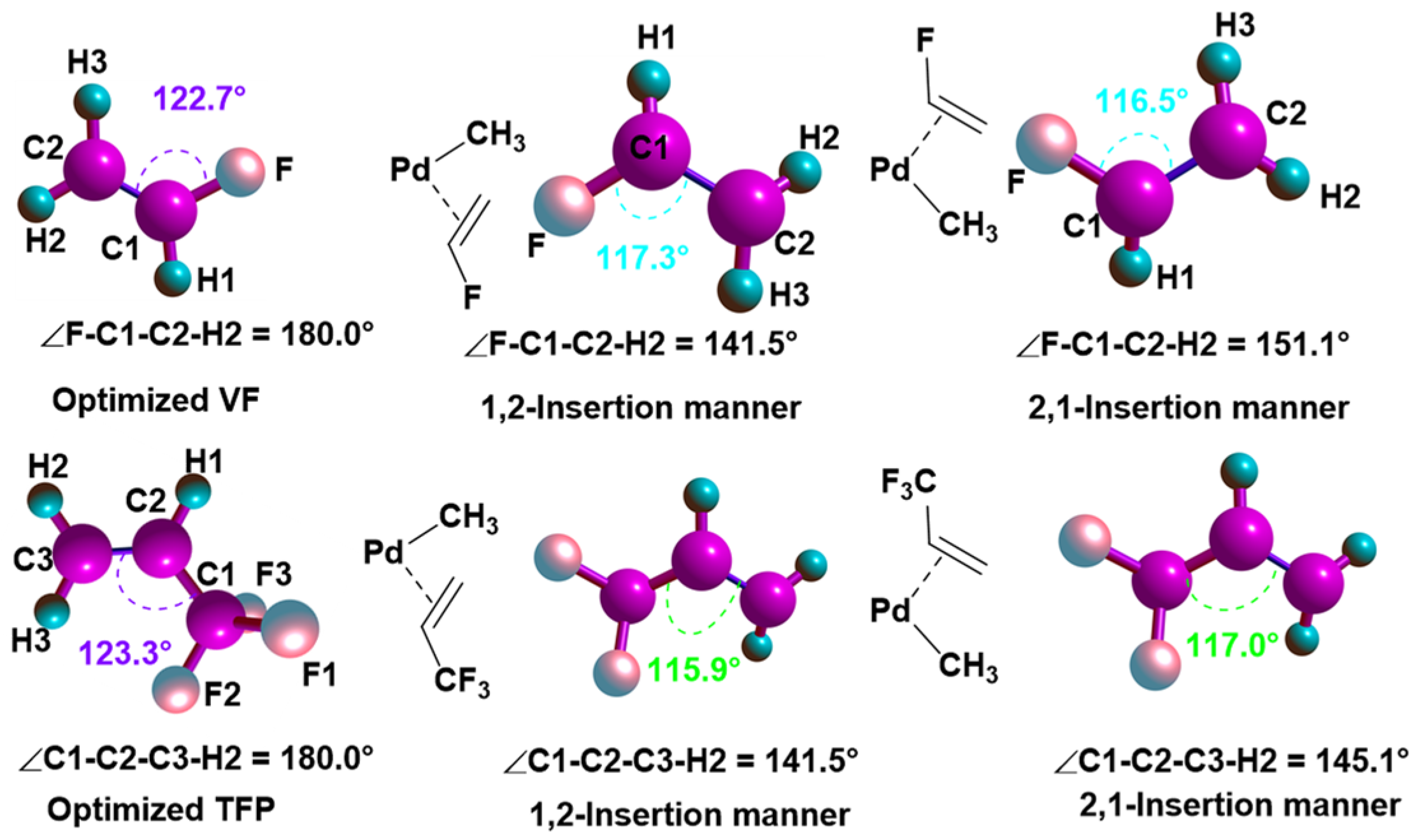
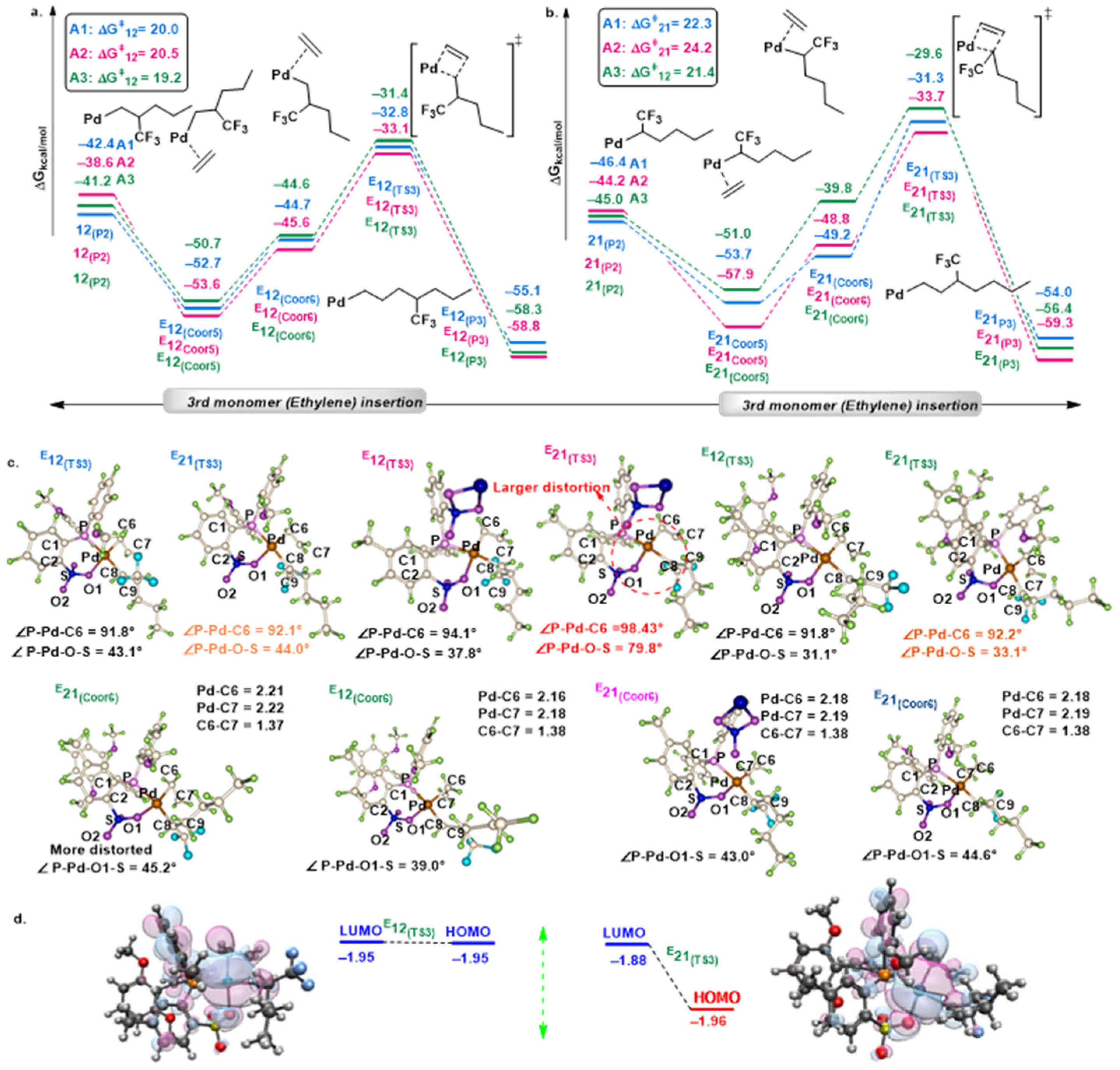
2.1. Chain Initiation and Propagation Using A1, A2, and A3 Complexes
2.2. Effects of Electronic and Steric Parameters on the Polymerization Activity
3. Computational Methods and Software Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kharitonov, A.; Taege, R.; Ferrier, G.; Teplyakov, V.; Syrtsova, D.; Koops, G.-H. Direct fluorination—Useful tool to enhance commercial properties of polymer articles. J. Fluor. Chem. 2005, 126, 251–263. [Google Scholar] [CrossRef]
- Kharitonov, A.; Moskvin, Y.L.; Teplyakov, V.; Le Roux, J. Direct fluorination of poly (vinyl trimethylsilane) and poly (phenylene oxide). J. Fluor. Chem. 1999, 93, 129–137. [Google Scholar] [CrossRef]
- Boz, E.; Nemeth, A.J.; Ghiviriga, I.; Jeon, K.; Alamo, R.G.; Wagener, K.B. Precision ethylene/vinyl chloride polymers via condensation polymerization. Macromolecules 2007, 40, 6545–6551. [Google Scholar] [CrossRef]
- Boz, E.; Nemeth, A.J.; Alamo, R.G.; Wagener, K.B. Precision ethylene/vinyl bromide polymers. Adv. Synth. Catal. 2007, 349, 137–141. [Google Scholar] [CrossRef]
- Boz, E.; Wagener, K.B.; Ghosal, A.; Fu, R.; Alamo, R.G. Synthesis and crystallization of precision ADMET polyolefins containing halogens. Macromolecules 2006, 39, 4437–4447. [Google Scholar] [CrossRef]
- Yang, H.; Islam, M.; Budde, C.; Rowan, S.J. Ring-opening metathesis polymerization as a route to controlled copolymers of ethylene and polar monomers: Synthesis of ethylene–vinyl chloride-like copolymers. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 2107–2116. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, C.; Tanaka, R.; Shiono, T.; Cai, Z. Copolymerization of Ethylene and Fluoroalkylnorbornene Using Highly Active ansa-(Fluorenyl)(amido)titanium-Based Catalysts. Macromol. Chem. Phys. 2019, 220, 1900306. [Google Scholar] [CrossRef]
- Nakamura, A.; Anselment, T.M.; Claverie, J.; Goodall, B.; Jordan, R.F.; Mecking, S.; Rieger, B.; Sen, A.; Van Leeuwen, P.W.; Nozaki, K. Ortho-phosphinobenzenesulfonate: A superb ligand for palladium-catalyzed coordination–insertion copolymerization of polar vinyl monomers. Acc. Chem. Res. 2013, 46, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.-X. Coordination polymerization of polar vinyl monomers by single-site metal catalysts. Chem. Rev. 2009, 109, 5157–5214. [Google Scholar] [CrossRef]
- Drent, E.; van Dijk, R.; van Ginkel, R.; van Oort, B.; Pugh, R.I. Palladium catalysed copolymerisation of ethene with alkylacrylates: Polar comonomer built into the linear polymer chain. Chem. Commun. 2002, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.; Lief, G.R.; Shen, Z.; Jordan, R.F. Ethylene polymerization by palladium alkyl complexes containing bis (aryl) phosphino-toluenesulfonate ligands. Organometallics 2007, 26, 6624–6635. [Google Scholar] [CrossRef]
- Guironnet, D.; Roesle, P.; Rünzi, T.; Göttker-Schnetmann, I.; Mecking, S. Insertion polymerization of acrylate. J. Am. Chem. Soc. 2009, 131, 422–423. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Shen, Z.; Jordan, R.F. Copolymerization of ethylene and vinyl fluoride by (phosphine-sulfonate) Pd (Me)(py) catalysts. J. Am. Chem. Soc. 2007, 129, 15450–15451. [Google Scholar] [CrossRef]
- Wada, S.; Jordan, R.F. Olefin Insertion into a Pd–F Bond: Catalyst Reactivation Following β-F Elimination in Ethylene/Vinyl Fluoride Copolymerization. Angew. Chem. Int. Ed. 2017, 56, 1820–1824. [Google Scholar] [CrossRef]
- Lanzinger, D.; Giuman, M.M.; Anselment, T.M.; Rieger, B. Copolymerization of ethylene and 3, 3, 3-trifluoropropene using (phosphine-sulfonate) Pd (Me)(DMSO) as catalyst. ACS Macro Lett. 2014, 3, 931–934. [Google Scholar] [CrossRef]
- Borkar, S.; Newsham, D.K.; Sen, A. Copolymerization of Ethene with Styrene Derivatives, Vinyl Ketone, and Vinylcyclohexane Using a (Phosphine–sulfonate) palladium (II) System: Unusual Functionality and Solvent Tolerance. Organometallics 2008, 27, 3331–3334. [Google Scholar] [CrossRef]
- Rünzi, T.; Mecking, S. Saturated Polar-Substituted Polyethylene Elastomers from Insertion Polymerization. Adv. Funct. Mater. 2014, 24, 387–395. [Google Scholar] [CrossRef]
- Wucher, P.; Roesle, P.; Falivene, L.; Cavallo, L.; Caporaso, L.; Göttker-Schnetmann, I.; Mecking, S. Controlled acrylate insertion regioselectivity in diazaphospholidine-sulfonato palladium (II) complexes. Organometallics 2012, 31, 8505–8515. [Google Scholar] [CrossRef]
- Von Schenck, H.; Strömberg, S.; Zetterberg, K.; Ludwig, M.; Åkermark, B.; Svensson, M. Insertion Aptitudes and Insertion Regiochemistry of Various Alkenes Coordinated to Cationic (σ-R)(diimine) palladium (II)(R = –CH3,–C6H5). A Theoretical Study. Organometallics 2001, 20, 2813–2819. [Google Scholar] [CrossRef]
- Shen, Z.; Jordan, R.F. Copolymerization of ethylene and vinyl fluoride by (phosphine-bis (arenesulfonate)) PdMe (pyridine) catalysts: Insights into inhibition mechanisms. Macromolecules 2010, 43, 8706. [Google Scholar] [CrossRef]
- Wu, F.; Foley, S.R.; Burns, C.T.; Jordan, R.F. Acrylonitrile insertion reactions of cationic palladium alkyl complexes. J. Am. Chem. Soc. 2005, 127, 1841–1853. [Google Scholar] [CrossRef] [PubMed]
- Groux, L.F.; Weiss, T.; Reddy, D.N.; Chase, P.A.; Piers, W.E.; Ziegler, T.; Parvez, M.; Benet-Buchholz, J. Insertion of acrylonitrile into palladium methyl bonds in neutral and anionic Pd (II) complexes. J. Am. Chem. Soc. 2005, 127, 1854–1869. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.; Kang, X.; Luo, Y. Origin of different chain-end microstructures in ethylene/vinyl halide copolymerization catalysed by phosphine–sulfonate palladium complexes. New J. Chem. 2020, 44, 16941–16947. [Google Scholar] [CrossRef]
- Mehmood, A.; Mahmood, A.; Xu, X.; Raza, W.; Ahmed, S.; Ullah, N.; Luo, Y.; Tian, X. Mechanistic study to reveal steric and electronic aspects involved in the formation of microstructures during Pd-catalyzed olefin/divinyl formal copolymerization: Reactivity to catalyst choice. Phys. Chem. Chem. Phys. 2023, 25, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, A.; Xu, X.; Raza, W.; Kukkar, D.; Kim, K.-H.; Luo, Y. Computational study of the copolymerization mechanism of ethylene with methyl 2-acetamidoacrylate catalyzed by phosphine-sulfonate palladium complexes. New J. Chem. 2021, 45, 16670–16678. [Google Scholar] [CrossRef]
- Mehmood, A.; Xu, X.; Raza, W.; Kim, K.-H.; Luo, Y. Mechanistic studies for palladium catalyzed copolymerization of ethylene with vinyl ethers. Polymers 2020, 12, 2401. [Google Scholar] [CrossRef]
- Xu, X.; Luo, G.; Mehmood, A.; Zhao, Y.; Zhou, G.; Hou, Z.; Luo, Y. Theoretical mechanistic studies on redox-switchable polymerization of trimethylene carbonate catalyzed by an indium complex bearing a ferrocene-based ligand. Organometallics 2018, 37, 4599–4607. [Google Scholar] [CrossRef]
- Piche, L.; Daigle, J.C.; Rehse, G.; Claverie, J.P. Structure–Activity Relationship of Palladium Phosphanesulfonates: Toward Highly Active Palladium-Based Polymerization Catalysts. Chem. A Eur. J. 2012, 18, 3277–3285. [Google Scholar] [CrossRef]
- Skupov, K.M.; Marella, P.R.; Simard, M.; Yap, G.P.; Allen, N.; Conner, D.; Goodall, B.L.; Claverie, J.P. Palladium aryl sulfonate phosphine catalysts for the copolymerization of acrylates with ethene. Macromol. Rapid Commun. 2007, 28, 2033–2038. [Google Scholar] [CrossRef]
- Shen, Z.; Jordan, R.F. Self-assembled tetranuclear palladium catalysts that produce high molecular weight linear polyethylene. J. Am. Chem. Soc. 2010, 132, 52–53. [Google Scholar] [CrossRef]
- Rünzi, T.; Fröhlich, D.; Mecking, S. Direct synthesis of ethylene− acrylic acid copolymers by Insertion polymerization. J. Am. Chem. Soc. 2010, 132, 17690–17691. [Google Scholar] [CrossRef] [PubMed]
- Nakano, R.; Chung, L.W.; Watanabe, Y.; Okuno, Y.; Okumura, Y.; Ito, S.; Morokuma, K.; Nozaki, K. Elucidating the key role of phosphine–sulfonate ligands in palladium-catalyzed ethylene polymerization: Effect of ligand structure on the molecular weight and linearity of polyethylene. ACS Catal. 2016, 6, 6101–6113. [Google Scholar] [CrossRef]
- Rezabal, E.; Ugalde, J.M.; Frenking, G. The trans Effect in Palladium Phosphine Sulfonate Complexes. J. Phys. Chem. A 2017, 121, 7709–7716. [Google Scholar] [CrossRef]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Huang, X.; Meggers, E.; Houk, K. Origins of Enantioselectivity in Asymmetric Radical Additions to Octahedral Chiral-at-Rhodium Enolates: A Computational Study. J. Am. Chem. Soc. 2017, 139, 17902–17907. [Google Scholar] [CrossRef] [PubMed]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing reaction rates with the distortion/interaction-activation strain model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef]
- Zou, C.; Pang, W.; Chen, C. Influence of chelate ring size on the properties of phosphine-sulfonate palladium catalysts. Sci. China Chem. 2018, 61, 1175–1178. [Google Scholar] [CrossRef]
- Tan, C.; Zou, C.; Chen, C. Material Properties of Functional Polyethylenes from Transition-Metal-Catalyzed Ethylene–Polar Monomer Copolymerization. Macromolecules 2022, 55, 1910–1922. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16 Rev. C. 01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Avcı, D.; Bahçeli, S.; Tamer, Ö.; Atalay, Y. Comparative study of DFT/B3LYP, B3PW91, and HSEH1PBE methods applied to molecular structures and spectroscopic and electronic properties of flufenpyr and amipizone. Can. J. Chem. 2015, 93, 1147–1156. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Beachy, M.D.; Ringnalda, M.N.; Pollard, W.T.; Dunietz, B.D.; Cao, Y. Correlated ab initio electronic structure calculations for large molecules. J. Phys. Chem. A 1999, 103, 1913–1928. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Martin, J.M.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Steinbrenner, U.; Bergner, A.; Dolg, M.; Stoll, H. On the transferability of energy adjusted pseudopotentiais: A calibration study for XH4 (X = C, Si, Ge, Sn, Pb). Mol. Phys. 1994, 82, 3–11. [Google Scholar] [CrossRef]
- Kaupp, M.; Schleyer, P.v.R.; Stoll, H.; Preuss, H. Pseudopotential approaches to Ca, Sr, and Ba hydrides. Why are some alkaline earth MX2 compounds bent? J. Chem. Phys. 1991, 94, 1360–1366. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Kozuch, S. A refinement of everyday thinking: The energetic span model for kinetic assessment of catalytic cycles. WIREs Comput. Mol. Sci. 2012, 2, 795–815. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef]
- Zhurko, G.; Zhurko, D. Chemcraft—Graphical Software for Visualization of Quantum Chemistry Computations. Version 1.8, 2009, Build 654. Available online: https://www.chemcraftprog.com (accessed on 10 October 2023).


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, A.; Mahmood, A.; AlMasoud, N.; Hassan, A.; Alomar, T.S.; El-Bahy, Z.M.; Raza, N.; Tian, X.; Ullah, N. Mechanistic Study on Steric Activity Interplay of Olefin/Polar Monomers for Industrially Selective Late Transition Metal Catalytic Reactions. Molecules 2023, 28, 7148. https://doi.org/10.3390/molecules28207148
Mehmood A, Mahmood A, AlMasoud N, Hassan A, Alomar TS, El-Bahy ZM, Raza N, Tian X, Ullah N. Mechanistic Study on Steric Activity Interplay of Olefin/Polar Monomers for Industrially Selective Late Transition Metal Catalytic Reactions. Molecules. 2023; 28(20):7148. https://doi.org/10.3390/molecules28207148
Chicago/Turabian StyleMehmood, Andleeb, Ayyaz Mahmood, Najla AlMasoud, Arzoo Hassan, Taghrid S. Alomar, Zeinhom M. El-Bahy, Nadeem Raza, Xiaoqing Tian, and Naeem Ullah. 2023. "Mechanistic Study on Steric Activity Interplay of Olefin/Polar Monomers for Industrially Selective Late Transition Metal Catalytic Reactions" Molecules 28, no. 20: 7148. https://doi.org/10.3390/molecules28207148
APA StyleMehmood, A., Mahmood, A., AlMasoud, N., Hassan, A., Alomar, T. S., El-Bahy, Z. M., Raza, N., Tian, X., & Ullah, N. (2023). Mechanistic Study on Steric Activity Interplay of Olefin/Polar Monomers for Industrially Selective Late Transition Metal Catalytic Reactions. Molecules, 28(20), 7148. https://doi.org/10.3390/molecules28207148