The Tetrel Bonds of Hypervalent Halogen Compounds



Abstract
:1. Introduction
2. Results
2.1. Electrostatic Potentials of Monomers
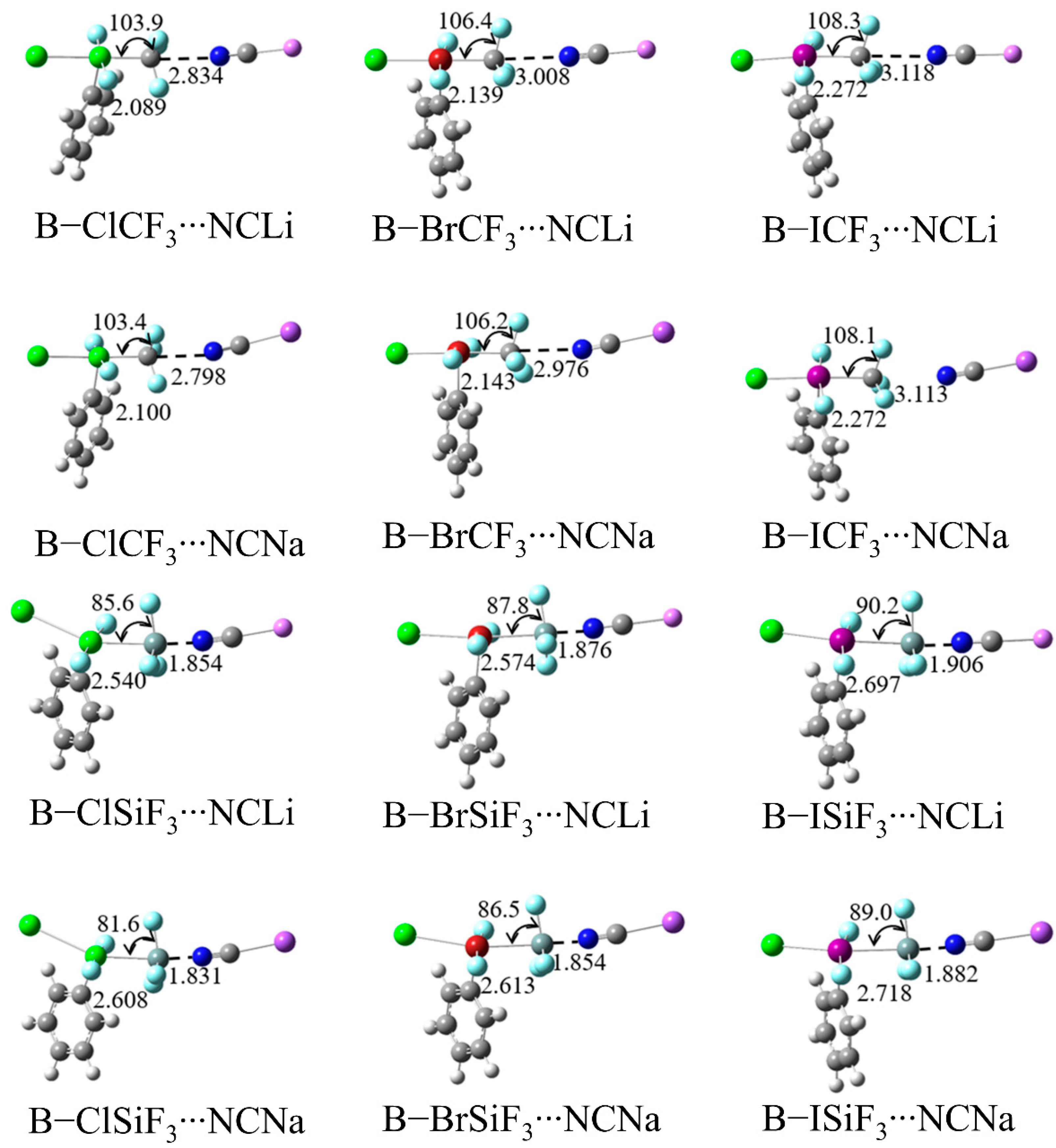
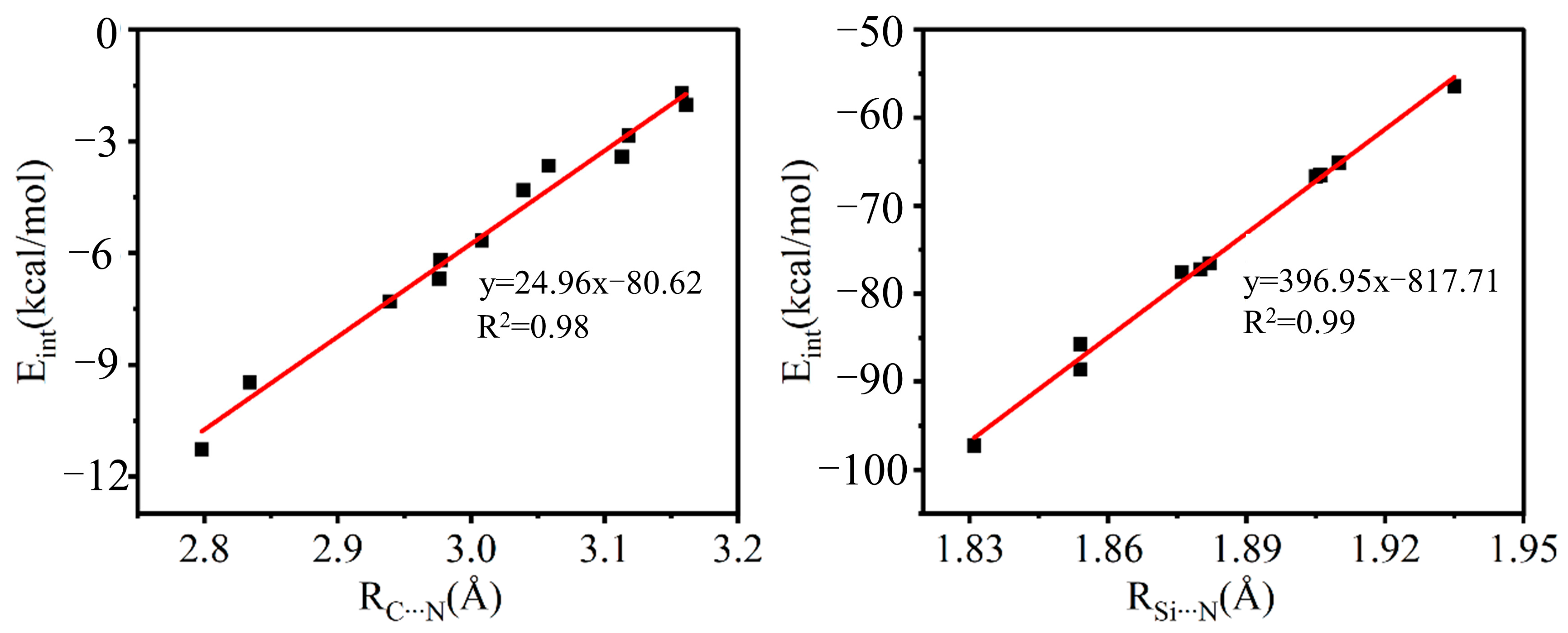
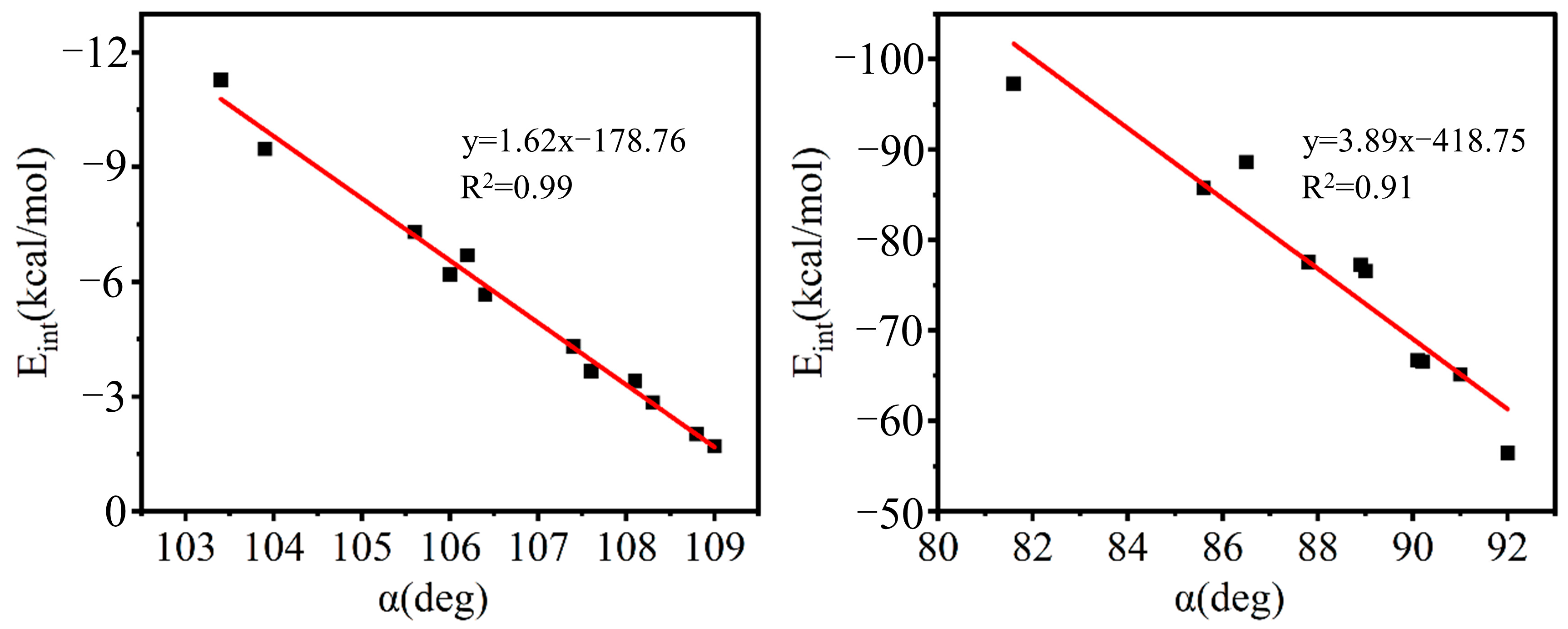
2.2. Geometric Structure and Interaction Energy
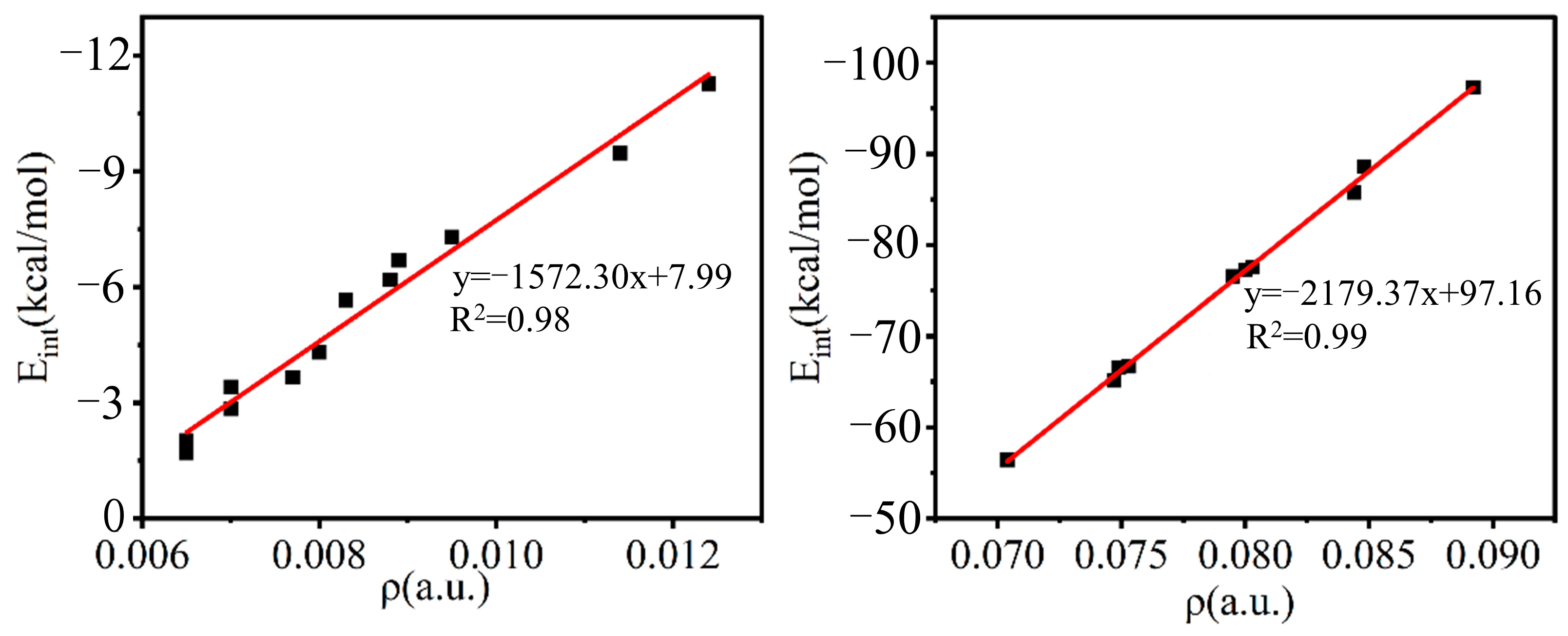
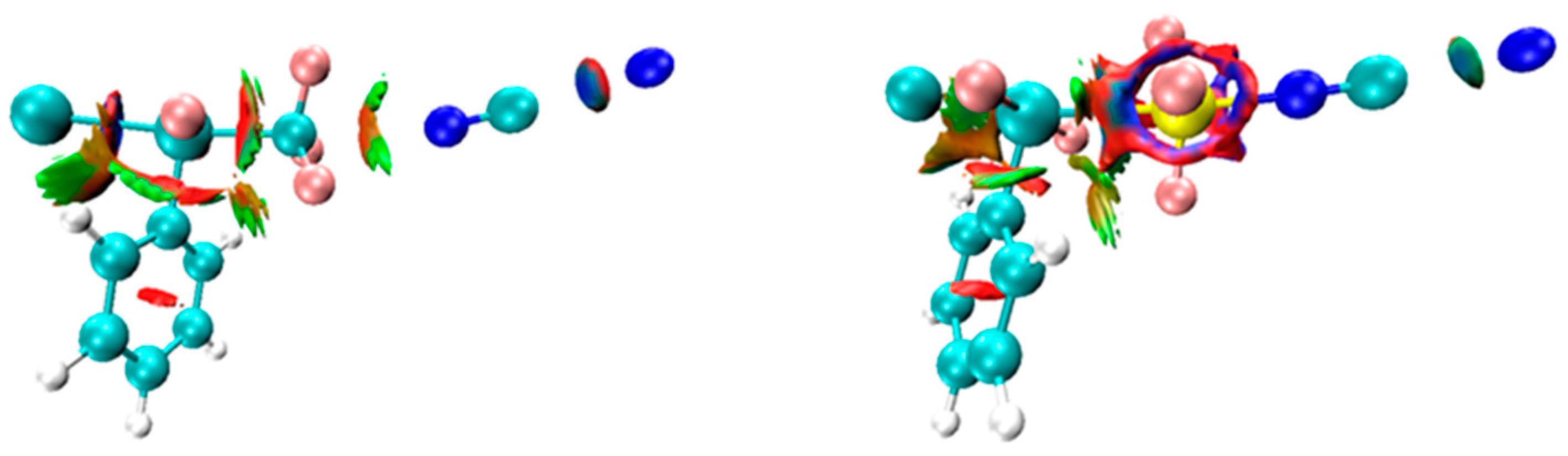
2.3. AIM Analysis
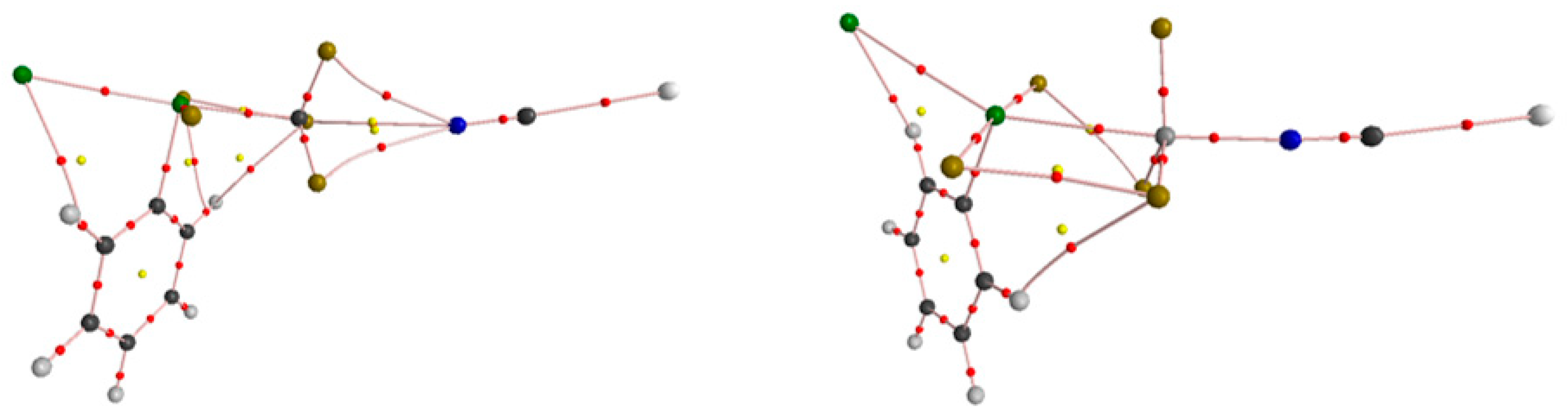
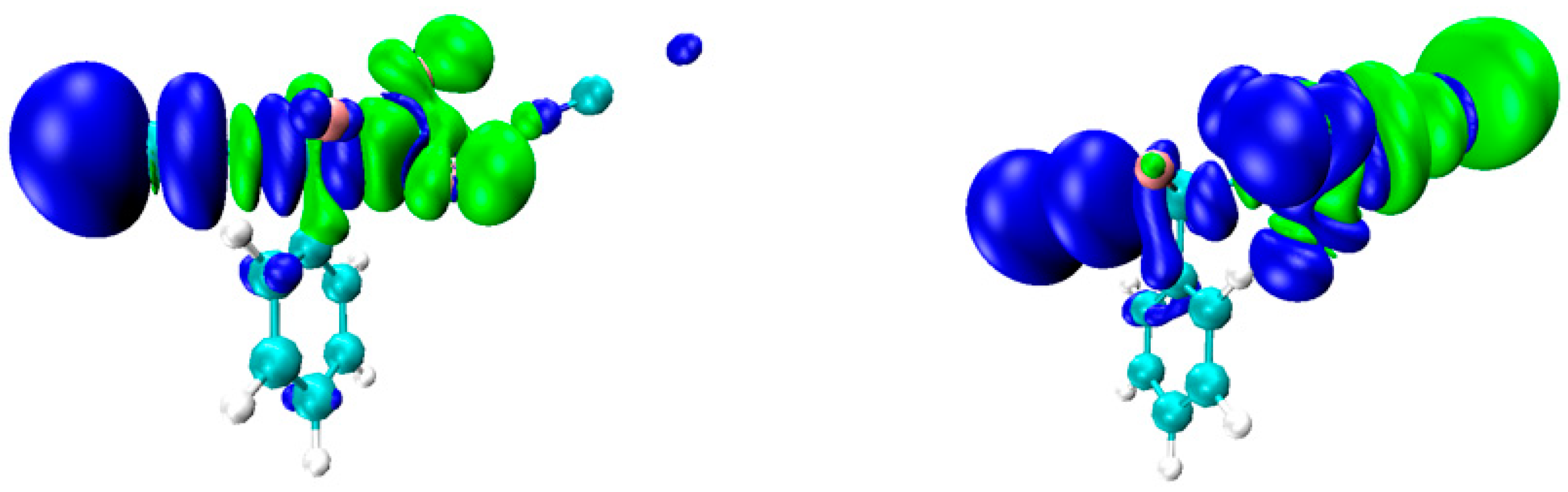
2.4. NOCV Analysis
2.5. Energy Decomposition Analysis
3. Computational Methodology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Bauzá, A.; Ramis, R.; Frontera, A. Computational study of anion recognition based on tetrel and hydrogen bonding interaction by calix [4] pyrrole derivatives. Comput. Theor. Chem. 2014, 1038, 67–70. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel bonding interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Daolio, A.; Scilabra, P.; Terraneo, G.; Resnati, G. C (sp3) atoms as tetrel bond donors: A crystallographic survey. Coord. Chem. Rev. 2020, 413, 213265. [Google Scholar] [CrossRef]
- Jena, S.; Dutta, J.; Tulsiyan, K.D.; Sahu, A.K.; Choudhury, S.S.; Biswal, H.S. Noncovalent interactions in proteins and nucleic acids: Beyond hydrogen bonding and π-stacking. Chem. Soc. Rev. 2022, 51, 4261–4286. [Google Scholar] [CrossRef] [PubMed]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent carbon-bonding interactions in proteins. Angew. Chem. Int. Ed. 2018, 57, 16496–16500. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Scheiner, S.; Nziko, V.P. Comparison of π-hole tetrel bonding with σ-hole halogen bonds in complexes of XCN (X= F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. The X–C∙∙∙Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef]
- Mani, D.; Arunan, E. Microwave spectroscopic and atoms in molecules theoretical investigations on the Ar∙∙∙propargyl alcohol complex: Ar∙∙∙H–O, Ar∙∙∙π, and Ar∙∙∙C interactions. ChemPhysChem 2013, 14, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Heywood, V.L.; Alford, T.P.; Roeleveld, J.J.; Deprez, S.J.L.; Verhoofstad, A.; Vlugt, J.V.; Domingos, S.R.; Schnell, M.; Davis, A.P.; Mooibroke, T.J. Observations of tetrel bonding between sp3-carbon and THF. Chem. Sci. 2020, 11, 5289–5293. [Google Scholar] [CrossRef]
- Scheiner, S. Comparison of CH···O, SH···O, chalcogen, and tetrel bonds formed by neutral and cationic sulfur-containing compounds. J. Phys. Chem. 2015, 119, 9189–9199. [Google Scholar] [CrossRef]
- Donald, K.J.; Tawfik, M. The weak helps the strong: Sigma-holes and the stability of MF4· base complexes. J. Phys. Chem. A 2013, 117, 14176–14183. [Google Scholar] [CrossRef]
- Grabowski, S.J. Tetrel bonds, penta-and hexa-coordinated tin and lead centres. Appl. Organomet. Chem. 2017, 31, e3727. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furanTF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic elucidation of factors that influence the strength of tetrel bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. π-Hole bonds: Boron and aluminum Lewis acid centers. ChemPhysChem 2015, 16, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Aman, Y.; Ji, X.; Mo, Y. Tetrel bonding interaction: An analysis with the block-localized wavefunction (BLW) approach. Phys. Chem. Chem. Phys. 2019, 21, 11776–11784. [Google Scholar] [CrossRef]
- Lin, H.; Meng, L.; Li, X.; Zeng, Y.; Zhang, X. Comparison of pnicogen and tetrel bonds in complexes containing CX2 carbenes (X = F, Cl, Br, OH, OMe, NH2, and NMe2). New J. Chem. 2019, 43, 15596–15604. [Google Scholar] [CrossRef]
- McDowell, S.A.; Wang, R.; Li, Q. Interactions in model ionic dyads and triads containing tetrel Aaoms. Molecules 2020, 25, 4197. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Steric crowding in tetrel bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef]
- Niu, Z.; McDowell, S.A.; Li, Q. Triel bonds with Au atoms as electron donors. ChemPhysChem 2023, 24, e202200748. [Google Scholar] [CrossRef] [PubMed]
- Amgoune, A.; Bourissou, D. σ-Acceptor, Z-type ligands for transition metals. Chem. Commun. 2011, 47, 859–871. [Google Scholar] [CrossRef]
- Scheiner, S. Origins and properties of the tetrel bond. Phys. Chem. Chem. Phys. 2021, 23, 5702–5717. [Google Scholar] [CrossRef]
- Mikosch, J.; Trippel, S.; Eichhorn, C.; Otto, R.; Lourderaj, U.; Zhang, J.X.; Hase, W.L.; Weidemuller, M.; Wester, R. Imaging nucleophilic substitution dynamics. Science 2008, 319, 183–186. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Cheng, J.; Li, W.; Li, H. Tetrel bond of pseudohalide anions with XH3F (X = C, Si, Ge, and Sn) and its role in SN2 reaction. J. Chem. Phys. 2016, 145, 224310. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.; Schulz, N.; Andersson, H.; Nekoueishahraki, B.; Carlsson, A.; Sarabi, D.; Valkonen, A.; Rissanen, K.; Grafenstein, J.; Keller, S.; et al. Carbon’s three–center, four–electron tetrel bond, treated experimentally. J. Am. Chem. Soc. 2018, 140, 17571–17579. [Google Scholar] [CrossRef]
- Lim, D.W.; Sadakiyo, M.; Kitagawa, H. Proton transfer in hydrogen–bonded degenerate systems of water and ammonia in metal–organic frameworks. Chem. Sci. 2019, 10, 16–33. [Google Scholar] [CrossRef]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π–hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef]
- Liu, N.; Wu, Q.; Li, Q.; Scheiner, S. Promotion of TH3 (T = Si and Ge) group transfer within a tetrel bond by a cation–π interaction. Phys. Chem. Chem. Phys. 2022, 24, 1113–1119. [Google Scholar] [CrossRef]
- Zhdankin, V.V.; Stang, P.J. Chemistry of polyvalent iodine. Chem. Rev. 2008, 108, 5299–5358. [Google Scholar] [CrossRef]
- Akiba, K. Chemistry of Hypervalent Compounds; John Wiley & Sons: Hoboken, NJ, USA, 1998. [Google Scholar]
- Sikalov, A.A. Hypervalent halogen hydrides HalHn (Hal = Cl, Br, I; n = 3, 5, 7): DFT and ab initio stability prediction. Theor. Chem. Acc. 2020, 139, 8. [Google Scholar] [CrossRef]
- Tian, W.K.; Miao, Q.; Li, Q.Z.; Li, W.; Cheng, J. Superalkali Li3M (M = Cl, Br, I) as a Lewis base in halogen bonding: A heavier halogen is a stronger Lewis base than a lighter halogen. Comput. Theor. Chem. 2013, 1012, 41–46. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 09, Revision, A.02; Gaussian, Inc.: Wallingford, UK, 2009. [Google Scholar]
- Yang, F.L.; Yang, X.; Wu, R.Z.; Yan, C.X.; Yang, F.; Ye, W.; Zhang, L.W.; Zhou, P.P. Intermolecular interactions between σ- and π-holes of bromopentafluorobenzene and pyridine: Computational and experimental investigations. Phys. Chem. Chem. Phys. 2018, 20, 11386–11395. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Q.; Yang, F.L.; Lu, K.; Yian, C.X.; Dou, W.; Yang, L.; Zhou, P.P. Competition and cooperativity of hydrogen-bonding and tetrel-bonding interactions involving triethylene diamine (DABCO), H2O and CO2 in air. New J. Chem. 2020, 44, 2328–2338. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 2002, 19, 553–566. [Google Scholar] [CrossRef]
- Bulat, F.A.; Toro-Labbé, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital. Chem. Rev. 1998, 88, 899–926. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Mitoraj, M.P.; Michalak, A.; Ziegler, T. A combined charge and energy decomposition scheme for bond analysis. J. Chem. Theory Comput. 2009, 5, 962–975. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Li, H. Energy decomposition analysis of covalent bonds and intermolecular interactions. J. Chem. Phys. 2009, 131, 014102. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Non–covalent sp3 carbon bonding with ArCF3 is analogous to CH–π interactions. Chem. Commun. 2014, 50, 12626–12629. [Google Scholar] [CrossRef]
- Liu, M.; Yang, L.; Li, Q.; Li, W.; Cheng, J.; Xiao, B.; Yu, X. Modulating the strength of tetrel bonding through beryllium bonding. J. Mol. Model. 2016, 22, 192. [Google Scholar] [CrossRef]
- Solimannejad, M.; Orojloo, M.; Amani, S. Effect of cooperativity in lithium bonding on the strength of halogen bonding and tetrel bonding: (LiCN)n∙∙∙ClYF3 and (LiCN)n∙∙∙YF3Cl (Y= C, Si and n= 1–5) complexes as a working model. J. Mol. Model. 2015, 21, 183. [Google Scholar] [CrossRef]
- Scheiner, S. The ditetrel bond: Noncovalent bond between neutral tetrel atoms. Phys. Chem. Chem. Phys. 2020, 22, 16606–16614. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, H.; Cheng, J.; Li, W.; Li, Q. Prominent enhancing effects of substituents on the strength of π···σ–hole tetrel bond. Int. J. Quantum Chem. 2017, 117, e25448. [Google Scholar] [CrossRef]
- Wei, Y.; Cheng, J.; Li, W.; Li, Q. Regulation of coin metal substituents and cooperativity on the strength and nature of tetrel bonds. RSC Adv. 2017, 7, 46321–46328. [Google Scholar] [CrossRef]
- Wu, Q.; Xie, X.; Li, Q.; Scheiner, S. Enhancement of tetrel bond involving tetrazole–TtR3 (Tt = C., Si; R. = H., F). Promotion of SiR3 transfer by a triel bond. Phys. Chem. Chem. Phys. 2022, 24, 25895–25903. [Google Scholar] [CrossRef]
- Niu, Z.H.; Wu, Q.Z.; Li, Q.Z.; Scheiner, S. C∙∙∙O and Si∙∙∙O tetrel bonds: Substituent effects and transfer of the SiF3 group. Int. J. Mol. Sci. 2023, 24, 11884. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eint | R1 | R2 | R1–R2 | ΔR2 | γ1 a | γ2 a | α | |
|---|---|---|---|---|---|---|---|---|
| A–ClCF3⋯NCLi | −6.19 | 2.977 | 2.012 | 0.965 | 0.034 | 0.750 | 0.490 | 106.0 |
| A–ClCF3⋯NCNa | −7.30 | 2.939 | 2.017 | 0.922 | 0.039 | 0.740 | 0.491 | 105.6 |
| A–BrCF3⋯NCLi | −3.66 | 3.058 | 2.097 | 0.961 | 0.012 | 0.770 | 0.492 | 107.6 |
| A–BrCF3⋯NCNa | −4.31 | 3.039 | 2.099 | 0.940 | 0.014 | 0.765 | 0.493 | 107.4 |
| A–ICF3⋯NCLi | −1.70 | 3.158 | 2.253 | 0.905 | 0.000 | 0.795 | 0.512 | 109.0 |
| A–ICF3⋯NCNa | −2.02 | 3.161 | 2.254 | 0.907 | 0.001 | 0.796 | 0.512 | 108.8 |
| B–ClCF3⋯NCLi | −9.47 | 2.834 | 2.089 | 0.745 | 0.057 | 0.714 | 0.508 | 103.9 |
| B–ClCF3⋯NCNa | −11.27 | 2.798 | 2.100 | 0.698 | 0.068 | 0.705 | 0.511 | 103.4 |
| B–BrCF3⋯NCLi | −5.66 | 3.008 | 2.139 | 0.869 | 0.02 | 0.758 | 0.502 | 106.4 |
| B–BrCF3⋯NCNa | −6.69 | 2.976 | 2.143 | 0.833 | 0.024 | 0.750 | 0.503 | 106.2 |
| B–ICF3⋯NCLi | −2.84 | 3.118 | 2.272 | 0.846 | 0.004 | 0.785 | 0.503 | 108.3 |
| B–ICF3⋯NCNa | −3.41 | 3.113 | 2.272 | 0.841 | 0.004 | 0.784 | 0.516 | 108.1 |
| A–BrSiF3⋯NCLi | −66.69 | 1.905 | 2.508 | −0.603 | -- | 0.451 | 0.556 | 90.1 |
| A–BrSiF3⋯NCNa | −77.25 | 1.880 | 2.535 | −0.655 | -- | 0.445 | 0.562 | 88.9 |
| A–ISiF3⋯NCLi | −56.43 | 1.935 | 2.656 | −0.721 | 0.09 | 0.459 | 0.571 | 92.0 |
| A–ISiF3⋯NCNa | −65.13 | 1.910 | 2.673 | −0.763 | 0.107 | 0.453 | 0.575 | 91.0 |
| B–ClSiF3⋯NCLi | −85.74 | 1.854 | 2.540 | −0.686 | -- | 0.439 | 0.583 | 85.6 |
| B–ClSiF3⋯NCNa | −97.27 | 1.831 | 2.608 | −0.777 | -- | 0.434 | 0.598 | 81.6 |
| B–BrSiF3⋯NCLi | −77.57 | 1.876 | 2.574 | −0.698 | 0.199 | 0.445 | 0.571 | 87.8 |
| B–BrSiF3⋯NCNa | −88.60 | 1.854 | 2.613 | −0.759 | 0.238 | 0.439 | 0.579 | 86.5 |
| B–ISiF3⋯NCLi | −66.53 | 1.906 | 2.697 | −0.791 | 0.11 | 0.452 | 0.580 | 90.2 |
| B–ISiF3⋯NCNa | −76.56 | 1.882 | 2.718 | −0.836 | 0.131 | 0.446 | 0.585 | 89.0 |
| T⋯N a | X–T | |||||
|---|---|---|---|---|---|---|
| ρ | ∇2ρ | H | ρ | ∇2ρ | H | |
| A–ClCF3⋯NCLi | 0.0088 | 0.0456 | 0.0022 | 0.1393 | −0.1276 | −0.0735 |
| A–ClCF3⋯NCNa | 0.0095 | 0.0491 | 0.0023 | 0.1372 | −0.1207 | −0.0715 |
| A–BrCF3⋯NCLi | 0.0077 | 0.0409 | 0.0021 | 0.1387 | −0.1689 | −0.0784 |
| A–BrCF3⋯NCNa | 0.0080 | 0.0420 | 0.0021 | 0.1381 | −0.1673 | −0.0777 |
| A–ICF3⋯NCLi | 0.0065 | 0.0347 | 0.0018 | 0.1171 | −0.0795 | −0.0614 |
| A–ICF3⋯NCNa | 0.0065 | 0.0344 | 0.0018 | 0.1172 | −0.0818 | −0.0615 |
| B–ClCF3⋯NCLi | 0.0114 | 0.0575 | 0.0025 | 0.1142 | −0.0480 | −0.0509 |
| B–ClCF3⋯NCNa | 0.0124 | 0.0608 | 0.0024 | 0.1108 | −0.0388 | −0.0481 |
| B–BrCF3⋯NCLi | 0.0083 | 0.0437 | 0.0022 | 0.1252 | −0.1196 | −0.0641 |
| B–BrCF3⋯NCNa | 0.0089 | 0.0464 | 0.0022 | 0.1241 | −0.1163 | −0.0629 |
| B–ICF3⋯NCLi | 0.0070 | 0.0372 | 0.0019 | 0.1132 | −0.0801 | −0.0568 |
| B–ICF3⋯NCNa | 0.0070 | 0.0381 | 0.0021 | 0.1134 | −0.0823 | −0.0570 |
| A–BrSiF3⋯NCLi | 0.0753 | 0.3637 | −0.0148 | 0.0589 | 0.0156 | −0.0300 |
| A–BrSiF3⋯NCNa | 0.0800 | 0.4014 | −0.0156 | 0.0554 | 0.0124 | −0.0276 |
| A–ISiF3⋯NCLi | 0.0704 | 0.3240 | −0.0141 | 0.0664 | −0.0435 | −0.0384 |
| A–ISiF3⋯NCNa | 0.0747 | 0.3576 | −0.0147 | 0.0638 | −0.0404 | −0.0364 |
| B–ClSiF3⋯NCLi | 0.0844 | 0.4452 | −0.0156 | 0.0384 | 0.0280 | −0.0145 |
| B–ClSiF3⋯NCNa | 0.0892 | 0.4855 | −0.0165 | 0.0333 | 0.0270 | −0.0110 |
| B–BrSiF3⋯NCLi | 0.0803 | 0.4080 | −0.0151 | 0.0498 | 0.0085 | −0.0237 |
| B–BrSiF3⋯NCNa | 0.0848 | 0.4444 | −0.0160 | 0.0458 | 0.0066 | −0.0208 |
| B–ISiF3⋯NCLi | 0.0749 | 0.3636 | −0.0144 | 0.0598 | −0.0391 | −0.0330 |
| B–ISiF3⋯NCNa | 0.0795 | 0.3995 | −0.0151 | 0.0568 | −0.0355 | −0.0305 |
| CT | E | Δq | |
|---|---|---|---|
| A–ClCF3⋯NCLi | 0.0049 | −0.54 | −0.1121 |
| A–ClCF3⋯NCNa | 0.0049 | −0.65 | −0.1289 |
| A–BrCF3⋯NCLi | 0.0064 | −0.44 | −0.0844 |
| A–BrCF3⋯NCNa | 0.0065 | −0.52 | −0.0950 |
| A–ICF3⋯NCLi | 0.0069 | −0.39 | −0.0656 |
| A–ICF3⋯NCNa | 0.0069 | −0.46 | −0.0743 |
| B–ClCF3⋯NCLi | 0.0033 | −0.67 | −0.1630 |
| B–ClCF3⋯NCNa | 0.0023 | −0.81 | −0.1890 |
| B–BrCF3⋯NCLi | 0.0058 | −0.55 | −0.1093 |
| B–BrCF3⋯NCNa | 0.0058 | −0.67 | −0.1242 |
| B–ICF3⋯NCLi | 0.0067 | −0.46 | −0.0819 |
| B–ICF3⋯NCNa | 0.0068 | −0.55 | −0.0919 |
| A–BrSiF3⋯NCLi | 0.2038 | −59.35 | -- |
| A–BrSiF3⋯NCNa | 0.2167 | −64.21 | -- |
| A–ISiF3⋯NCLi | 0.1890 | −54.86 | −0.3051 |
| A–ISiF3⋯NCNa | 0.2017 | −59.38 | −0.3316 |
| B–ClSiF3⋯NCLi | 0.2268 | −73.27 | -- |
| B–ClSiF3⋯NCNa | 0.2379 | −82.60 | -- |
| B–BrSiF3⋯NCLi | 0.2167 | −65.42 | −0.3282 |
| B–BrSiF3⋯NCNa | 0.2287 | −71.74 | −0.3612 |
| B–ISiF3⋯NCLi | 0.2026 | −59.63 | −0.3553 |
| B–ISiF3⋯NCNa | 0.2148 | −64.52 | −0.3882 |
| Ees | Eex | Erep | Epol | Edisp | Etotal | |
|---|---|---|---|---|---|---|
| A–ClCF3⋯NCLi | −8.36 | −4.17 | 13.73 | −2.03 | −5.34 | −6.17 |
| A–ClCF3⋯NCNa | −9.82 | −4.90 | 15.69 | −2.44 | −5.77 | −7.23 |
| A–BrCF3⋯NCLi | −5.50 | −3.62 | 12.06 | −1.77 | −4.90 | −3.73 |
| A–BrCF3⋯NCNa | −6.25 | −4.03 | 13.11 | −2.07 | −5.12 | −4.35 |
| A–ICF3⋯NCLi | −2.93 | −7.04 | 11.85 | −0.93 | −2.66 | −1.71 |
| A–ICF3⋯NCNa | −3.15 | −2.89 | 10.17 | −1.78 | −4.38 | −2.03 |
| B–ClCF3⋯NCLi | −12.72 | −12.45 | 22.10 | −4.69 | −1.56 | −9.33 |
| B–ClCF3⋯NCNa | −14.88 | −6.66 | 20.85 | −3.62 | −6.72 | −11.03 |
| B–BrCF3⋯NCLi | −7.51 | −3.93 | 13.00 | −2.18 | −5.03 | −5.65 |
| B–BrCF3⋯NCNa | −8.74 | −4.54 | 14.63 | −2.59 | −5.37 | −6.62 |
| B–ICF3⋯NCLi | −4.18 | −2.99 | 10.61 | −1.78 | −4.50 | −2.85 |
| B–ICF3⋯NCNa | −4.67 | −3.19 | 11.10 | −2.07 | −4.59 | −3.42 |
| A–BrSiF3⋯NCLi | −94.92 | −56.18 | 171.44 | −70.13 | −20.35 | −70.14 |
| A–BrSiF3⋯NCNa | −104.78 | −59.61 | 181.87 | −77.47 | −20.76 | −80.75 |
| A–ISiF3⋯NCLi | −87.57 | −55.70 | 169.53 | −61.97 | −20.73 | −56.44 |
| A–ISiF3⋯NCNa | −96.58 | −59.36 | 180.35 | −68.34 | −21.20 | −65.13 |
| B–ClSiF3⋯NCLi | −116.07 | −71.79 | 212.37 | −94.69 | −19.52 | −89.70 |
| B–ClSiF3⋯NCNa | −130.90 | −83.04 | 241.07 | −108.94 | −19.57 | −101.38 |
| B–BrSiF3⋯NCLi | −104.62 | −61.62 | 186.40 | −81.40 | −19.93 | −81.16 |
| B–BrSiF3⋯NCNa | −116.54 | −68.45 | 204.53 | −91.58 | −20.12 | −92.16 |
| B–ISiF3⋯NCLi | −95.17 | −57.62 | 176.40 | −69.50 | −20.62 | −66.52 |
| B–ISiF3⋯NCNa | −105.16 | −61.62 | 188.03 | −76.80 | −21.01 | −76.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Z.; McDowell, S.A.C.; Li, Q. The Tetrel Bonds of Hypervalent Halogen Compounds. Molecules 2023, 28, 7087. https://doi.org/10.3390/molecules28207087
Niu Z, McDowell SAC, Li Q. The Tetrel Bonds of Hypervalent Halogen Compounds. Molecules. 2023; 28(20):7087. https://doi.org/10.3390/molecules28207087
Chicago/Turabian StyleNiu, Zhihao, Sean A. C. McDowell, and Qingzhong Li. 2023. "The Tetrel Bonds of Hypervalent Halogen Compounds" Molecules 28, no. 20: 7087. https://doi.org/10.3390/molecules28207087
APA StyleNiu, Z., McDowell, S. A. C., & Li, Q. (2023). The Tetrel Bonds of Hypervalent Halogen Compounds. Molecules, 28(20), 7087. https://doi.org/10.3390/molecules28207087