

Tunable Supramolecular Ag+-Host Interactions in Pillar[n]arene[m]quinones and Ensuing Specific Binding to 1-Alkynes


Abstract
:1. Introduction
2. Results and Discussion
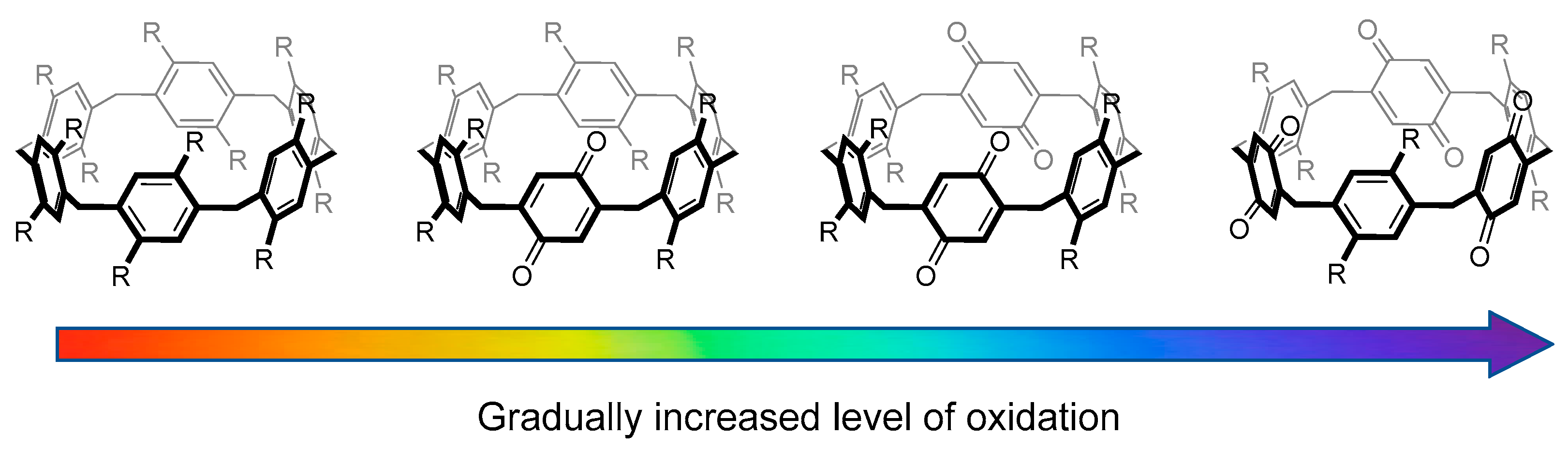
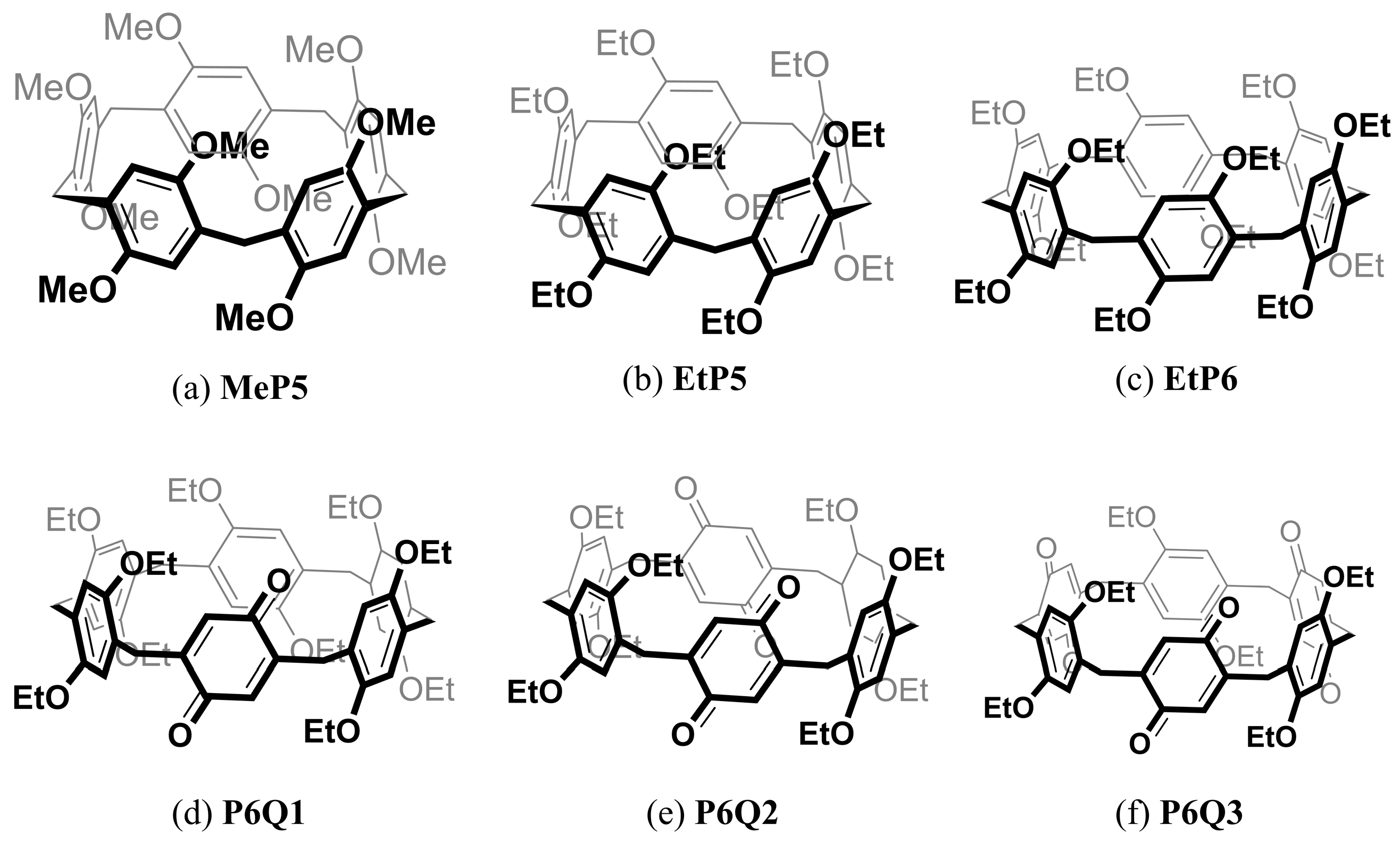
2.1. Synthesis of Different Pillararene Hosts
2.2. Determining Association Constants of Four Hosts with CF3COOAg via NMR Titration Experiments
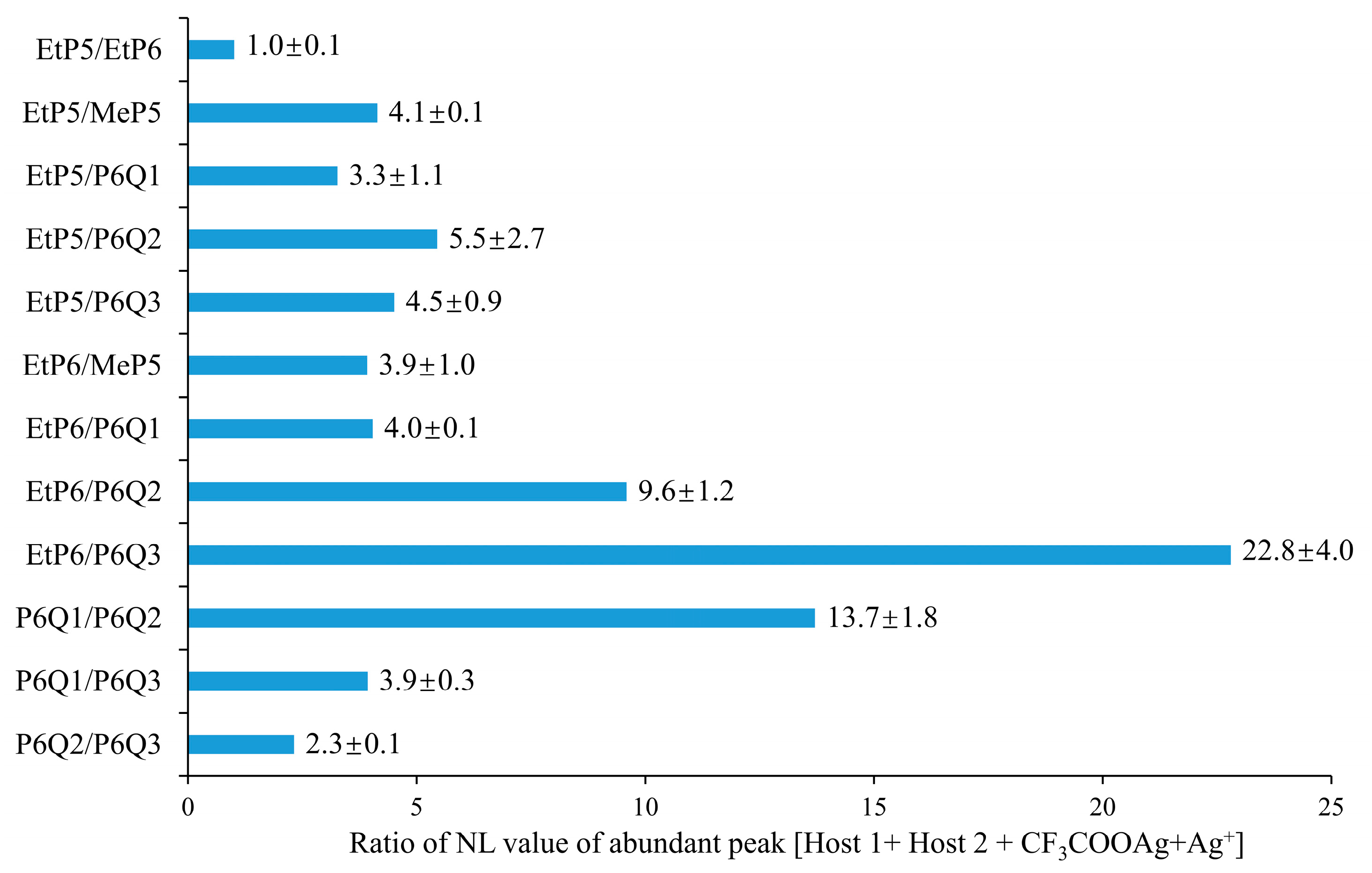
2.3. Complexation of the Host Molecule with Silver Trifluoroacetate by ESI-MS
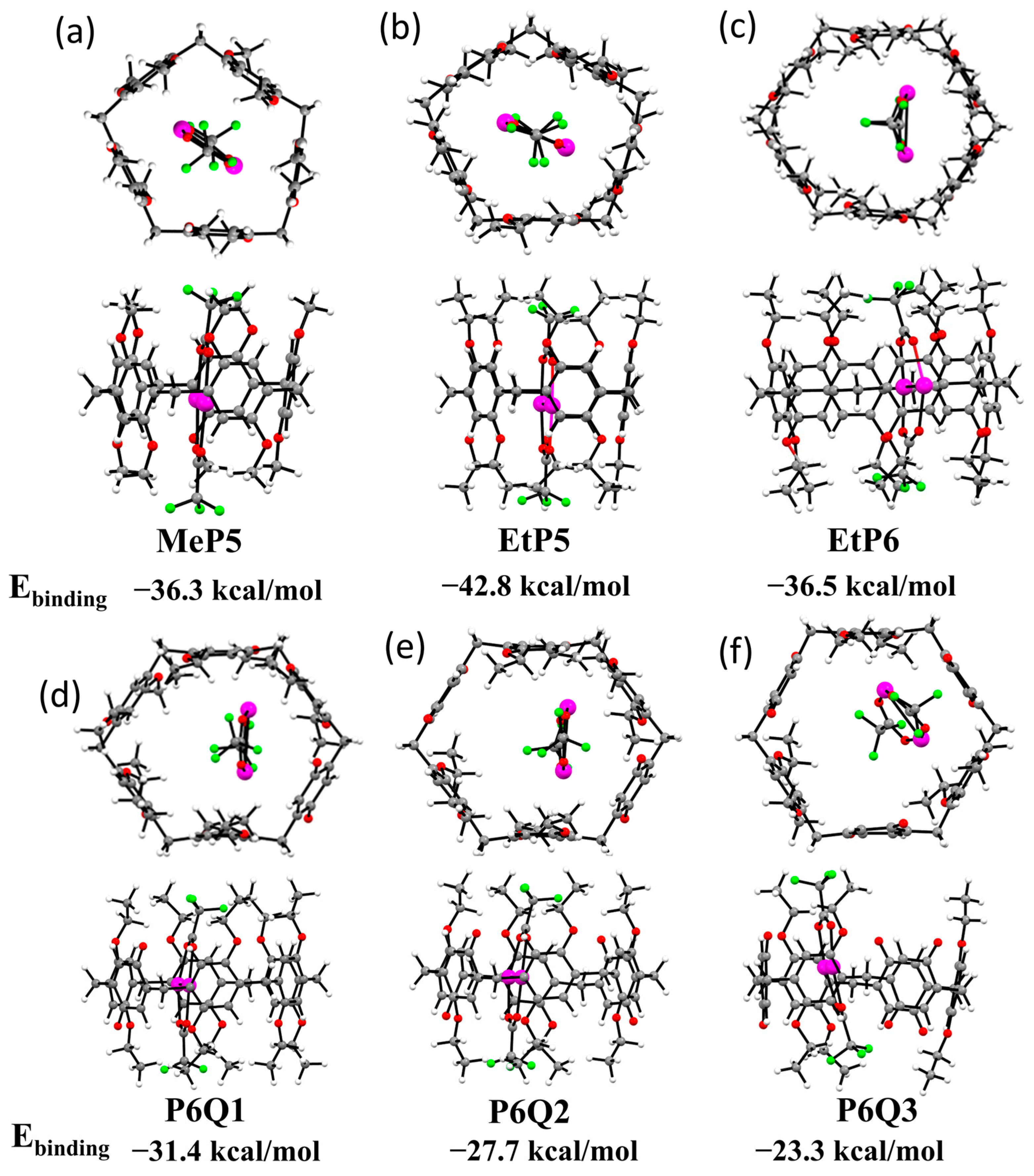
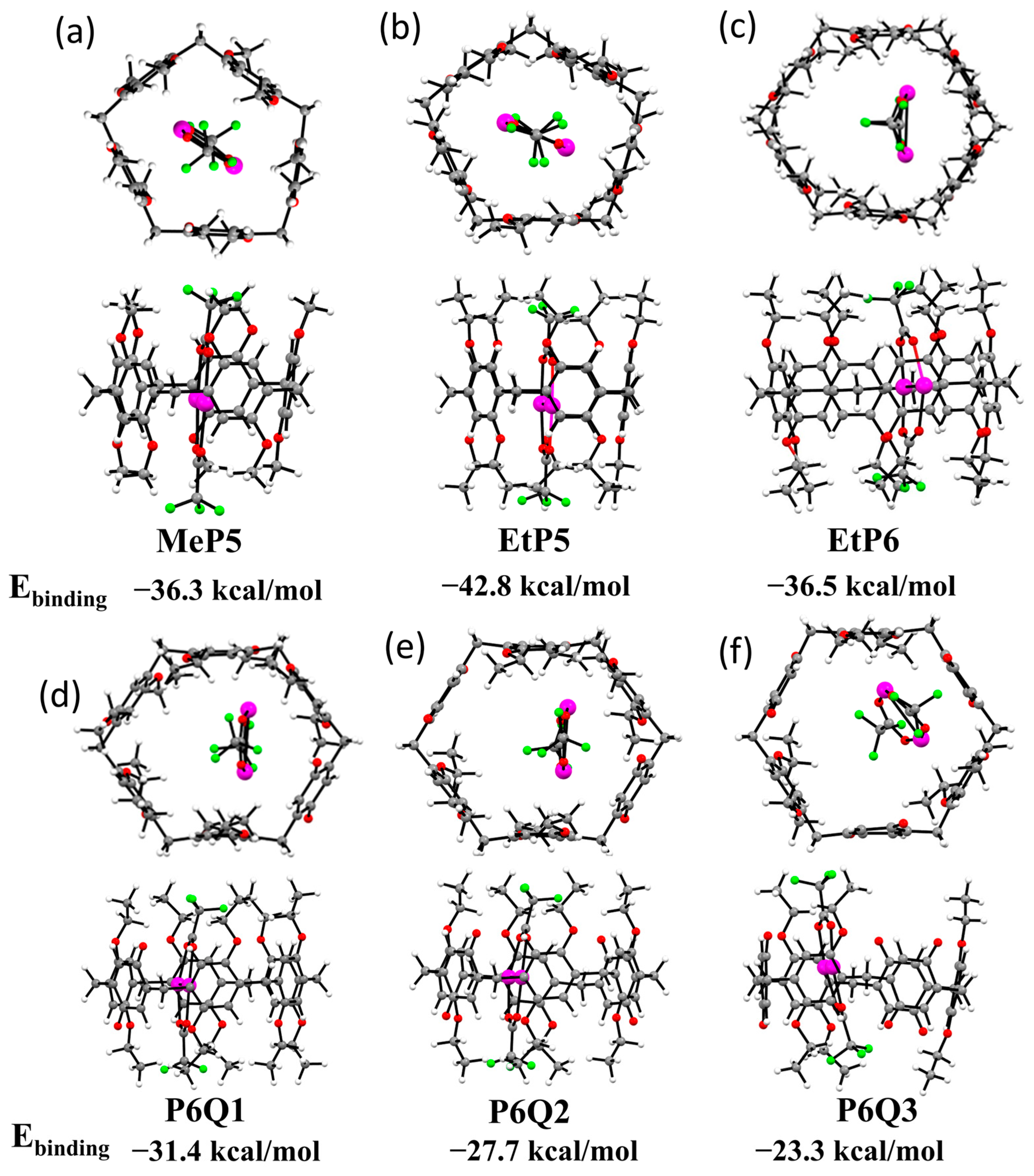
2.4. DFT-Optimized Structures for Six Different Pillararene-Ag2 Complexes
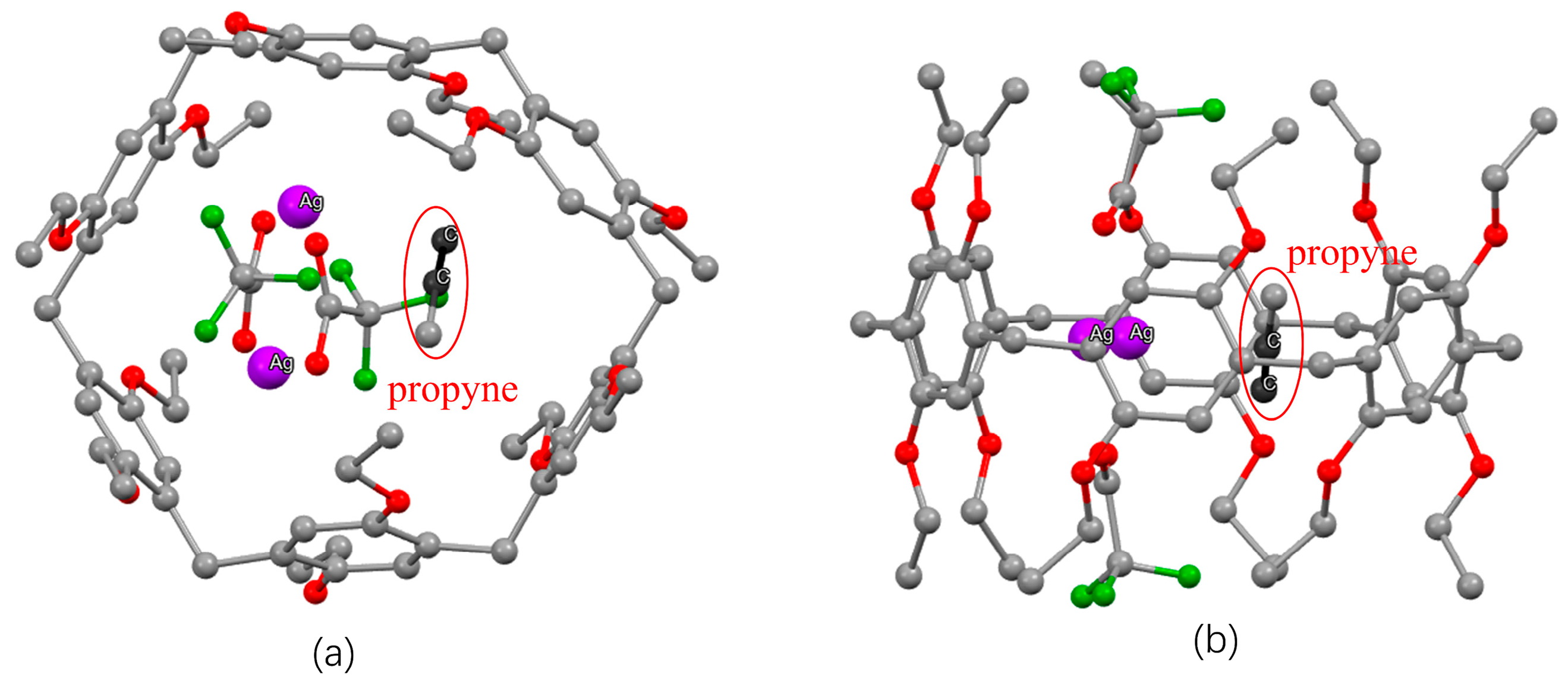
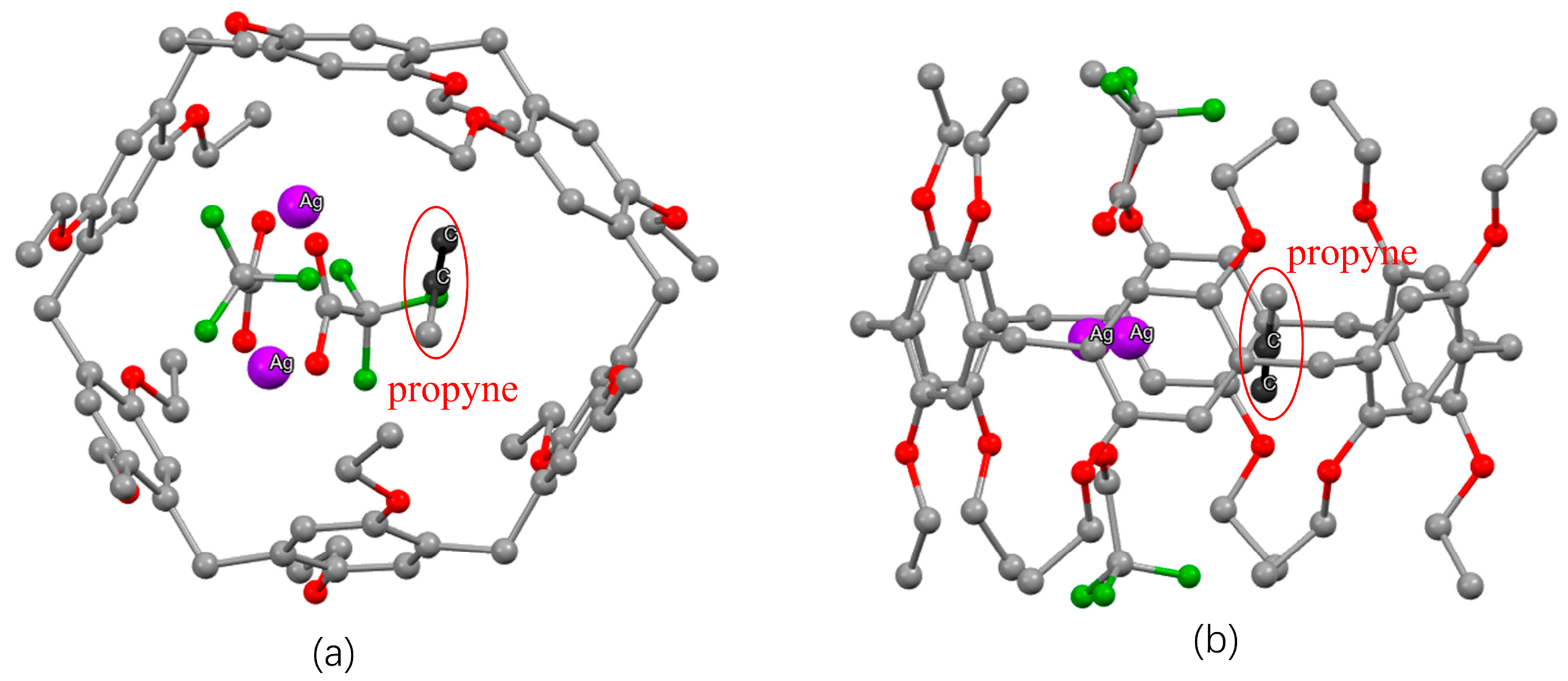
2.5. Specific Binding of 1-Alkynes to Silver-Loaded Pillararenes
3. Materials and Methods
3.1. Materials
General Procedures
3.2. Stoichiometry Determination
3.3. Binding Constants Determination
3.4. ESI-MS Competition Experiments
3.5. Optimized Geometry Structures of Pillararenes-Ag+ and Silver-Loaded-1-Alkyne Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, Y.; Ping, G.; Li, C. Efficient complexation between pillar[5]arenes and neutral guests: From host–guest chemistry to functional materials. Chem. Commun. 2016, 52, 9858–9872. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Kanai, S.; Fujinami, S.; Yamagishi, T.-A.; Nakamoto, Y. para-Bridged Symmetrical Pillar[5]arenes: Their Lewis Acid Catalyzed Synthesis and Host–Guest Property. J. Am. Chem. Soc. 2008, 130, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Zuilhof, H.; Sue, A.C.H.; Escorihuela, J. On the Stability and Formation of Pillar[n]arenes: A DFT Study. J. Org. Chem. 2021, 86, 14956–14963. [Google Scholar] [CrossRef] [PubMed]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Song, N.; Kakuta, T.; Yamagishi, T.-A.; Yang, Y.-W.; Ogoshi, T. Molecular-Scale Porous Materials Based on Pillar[n]arenes. Chem 2018, 4, 2029–2053. [Google Scholar] [CrossRef]
- Yang, K.; Pei, Y.; Wen, J.; Pei, Z. Recent advances in pillar[n]arenes: Synthesis and applications based on host–guest interactions. Chem. Commun. 2016, 52, 9316–9326. [Google Scholar] [CrossRef]
- Jia, Y.; Hu, J.-P.; Dang, L.-R.; Yao, H.; Shi, B.; Zhang, Y.-M.; Wei, T.-B.; Lin, Q. Rational Tuning of Binding Properties of Pillar[5]arene-Based Crystalline Material by Synergistic Effect and Its Application for Fluorescent Detection and Adsorption of 1,2-Ethylenediamine. ACS Sustain. Chem. Eng. 2021, 9, 16203–16209. [Google Scholar] [CrossRef]
- Jia, Y.; Guan, W.-L.; Liu, J.; Hu, J.-P.; Shi, B.; Yao, H.; Zhang, Y.-M.; Wei, T.-B.; Lin, Q. Novel conductive metallo-supramolecular polymer AIE gel for multi-channel highly sensitive detection of hydrazine hydrate. Chin. Chem. Lett. 2023, 34, 108082. [Google Scholar] [CrossRef]
- Guo, M.; Wang, X.; Zhan, C.; Demay-Drouhard, P.; Li, W.; Du, K.; Olson, M.A.; Zuilhof, H.; Sue, A.C.H. Rim-Differentiated C5-Symmetric Tiara-Pillar[5]arenes. J. Am. Chem. Soc. 2018, 140, 74–77. [Google Scholar] [CrossRef]
- Demay-Drouhard, P.; Du, K.; Samanta, K.; Wan, X.; Yang, W.; Srinivasan, R.; Sue, A.C.H.; Zuilhof, H. Functionalization at Will of Rim-Differentiated Pillar[5]arenes. Org. Lett. 2019, 21, 3976–3980. [Google Scholar] [CrossRef]
- Chao, Y.; Thikekar, T.U.; Fang, W.; Chang, R.; Xu, J.; Ouyang, N.; Xu, J.; Gao, Y.; Guo, M.; Zuilhof, H.; et al. “Rim-Differentiated” Pillar[6]arenes. Angew. Chem. Int. Ed. 2022, 61, e202204589. [Google Scholar] [CrossRef]
- Wang, H.; Yang, W.; Baldridge, K.K.; Zhan, C.-H.; Thikekar, T.U.; Sue, A.C.H. Spontaneous and induced chiral symmetry breaking of stereolabile pillar[5]arene derivatives upon crystallisation. Chem. Sci. 2021, 12, 10985–10989. [Google Scholar] [CrossRef]
- Wan, X.; Li, S.; Tian, Y.; Xu, J.; Shen, L.-C.; Zuilhof, H.; Zhang, M.; Sue, A.C.H. Twisted pentagonal prisms: AgnL2 metal-organic pillars. Chem 2022, 8, 2136–2147. [Google Scholar] [CrossRef]
- Yang, W.; Wang, H.; Chang, R.; Feng, Z.; Zhu, Y.; Sue, A.C.H. Handcuff-like metallo-pseudorotaxanes consisting of tiara[5]arene wheels and dimeric silver trifluoroacetate axles. Chem. Commun. 2023, 59, 2457–2460. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Yamafuji, D.; Kotera, D.; Aoki, T.; Fujinami, S.; Yamagishi, T.-a. Clickable Di- and Tetrafunctionalized Pillar[n]arenes (n = 5, 6) by Oxidation–Reduction of Pillar[n]arene Units. J. Org. Chem. 2012, 77, 11146–11152. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Zhang, Z.; Yu, G.; Huang, F. Syntheses of a pillar[4]arene[1]quinone and a difunctionalized pillar[5]arene by partial oxidation. Chem. Commun. 2012, 48, 9876–9878. [Google Scholar] [CrossRef] [PubMed]
- Lao, K.U.; Yu, C.H. A computational study of unique properties of pillar[n]quinones: Self-assembly to tubular structures and potential applications as electron acceptors and anion recognizers. J. Comput. Chem. 2011, 32, 2716–2726. [Google Scholar] [CrossRef] [PubMed]
- Rashvand Avei, M.; Etezadi, S.; Captain, B.; Kaifer, A.E. Visualization and quantitation of electronic communication pathways in a series of redox-active pillar[6]arene-based macrocycles. Commun. Chem. 2020, 3, 117. [Google Scholar] [CrossRef]
- Hirohata, T.; Shida, N.; Uekusa, H.; Yasuda, N.; Nishihara, H.; Ogoshi, T.; Tomita, I.; Inagi, S. Pillar[6]quinone: Facile synthesis, crystal structures and electrochemical properties. Chem. Commun. 2021, 57, 6360–6363. [Google Scholar] [CrossRef]
- Huang, S.; Claassen, F.W.; van Beek, T.A.; Chen, B.; Zeng, J.; Zuilhof, H.; Salentijn, G.I.J. Rapid Distinction and Semiquantitative Analysis of THC and CBD by Silver-Impregnated Paper Spray Mass Spectrometry. Anal. Chem. 2021, 93, 3794–3802. [Google Scholar] [CrossRef]
- Kaneti, J.; de Smet, L.C.P.M.; Boom, R.; Zuilhof, H.; Sudhölter, E.J.R. Computational Probes into the Basis of Silver Ion Chromatography. II. Silver(I)−Olefin Complexes. J. Phys. Chem. A 2002, 106, 11197–11204. [Google Scholar] [CrossRef]
- Hua, B.; Shao, L.; Zhang, Z.; Liu, J.; Huang, F. Cooperative Silver Ion-Pair Recognition by Peralkylated Pillar[5]arenes. J. Am. Chem. Soc. 2019, 141, 15008–15012. [Google Scholar] [CrossRef]
- Boinski, T.; Szumna, A. A facile, moisture-insensitive method for synthesis of pillar[5]arenes—The solvent templation by halogen bonds. Tetrahedron 2012, 68, 9419–9422. [Google Scholar] [CrossRef]
- Cao, J.; Shang, Y.; Qi, B.; Sun, X.; Zhang, L.; Liu, H.; Zhang, H.; Zhou, X. Synthesis of pillar[n]arenes (n = 5 and 6) with deep eutectic solvent choline chloride 2FeCl3. RSC Adv. 2015, 5, 9993–9996. [Google Scholar] [CrossRef]
- Da Pian, M.; De Lucchi, O.; Strukul, G.; Fabris, F.; Scarso, A. Cation templated improved synthesis of pillar[6]arenes. RSC Adv. 2016, 6, 48272–48275. [Google Scholar] [CrossRef]
- Mirzaei, S.; Wang, D.; Lindeman, S.V.; Sem, C.M.; Rathore, R. Highly Selective Synthesis of Pillar[n]arene (n = 5, 6). Org. Lett. 2018, 20, 6583–6586. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Cao, D.; Liu, L.; Kou, Y.; Wang, L.; Meier, H. Synthesis and host-guest properties of pillar[6]arenes. Sci. China Chem. 2012, 55, 223–228. [Google Scholar] [CrossRef]
- Ogoshi, T.; Ueshima, N.; Akutsu, T.; Yamafuji, D.; Furuta, T.; Sakakibara, F.; Yamagishi, T.-A. The template effect of solvents on high yield synthesis, co-cyclization of pillar[6]arenes and interconversion between pillar[5]- and pillar[6]arenes. Chem. Commun. 2014, 50, 5774–5777. [Google Scholar] [CrossRef] [PubMed]
- Santra, S.; Kovalev, I.S.; Kopchuk, D.S.; Zyryanov, G.V.; Majee, A.; Charushin, V.N.; Chupakhin, O.N. Role of polar solvents for the synthesis of pillar[6]arenes. RSC Adv. 2015, 5, 104284–104288. [Google Scholar] [CrossRef]
- Santra, S.; Kopchuk, D.S.; Kovalev, I.S.; Zyryanov, G.V.; Majee, A.; Charushin, V.N.; Chupakhin, O.N. Solvent-free synthesis of pillar[6]arenes. Green Chem. 2016, 18, 423–426. [Google Scholar] [CrossRef]
- Hristova, Y.R.; Smulders, M.M.J.; Clegg, J.K.; Breiner, B.; Nitschke, J.R. Selective anion binding by a “Chameleon” capsule with a dynamically reconfigurable exterior. Chem. Sci. 2011, 2, 638–641. [Google Scholar] [CrossRef]
- Smulders, M.M.J.; Zarra, S.; Nitschke, J.R. Quantitative Understanding of Guest Binding Enables the Design of Complex Host–Guest Behavior. J. Am. Chem. Soc. 2013, 135, 7039–7046. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. PCCP 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Xie, Y.-M.; Fan, Y.-Y.; Lin, F.-L.; Hu, T.; Liu, J.; Lu, C.-Z. Luminescent silver(I) tert-butylethynide compounds with nicotinic/isonicotinic acid as ligands. J. Mol. Struct. 2017, 1150, 335–339. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Thordarson, P. Determining association constants from titration experiments in supramolecular chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Brynn Hibbert, D.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef] [PubMed]
- Ulatowski, F.; Dąbrowa, K.; Bałakier, T.; Jurczak, J. Recognizing the Limited Applicability of Job Plots in Studying Host–Guest Interactions in Supramolecular Chemistry. J. Org. Chem. 2016, 81, 1746–1756. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Main Product | Solvent | Equiv. Oxidant | Reaction Time | Isolated Yield |
|---|---|---|---|---|
| P6Q1 | DCM/THF (9:1) | 2.2 | 30 min | 50% |
| P6Q2 | DCM/THF (8:2) | 4.4 | 3 h | 45% |
| P6Q3 | DCM/THF (7:3) | 6.6 | 3 h | 30% |
| Host | Association Constant/M−1 |
|---|---|
| MeP5 | (1.41 ± 0.04) × 103 |
| EtP5 | (2.24 ± 0.25) × 103 |
| EtP6 | (2.04 ± 0.41) × 103 |
| P6Q1 | (0.66 ± 0.08) × 103 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Escorihuela, J.; Wang, H.; Sue, A.C.-H.; Zuilhof, H. Tunable Supramolecular Ag+-Host Interactions in Pillar[n]arene[m]quinones and Ensuing Specific Binding to 1-Alkynes. Molecules 2023, 28, 7009. https://doi.org/10.3390/molecules28207009
Zhu Y, Escorihuela J, Wang H, Sue AC-H, Zuilhof H. Tunable Supramolecular Ag+-Host Interactions in Pillar[n]arene[m]quinones and Ensuing Specific Binding to 1-Alkynes. Molecules. 2023; 28(20):7009. https://doi.org/10.3390/molecules28207009
Chicago/Turabian StyleZhu, Yumei, Jorge Escorihuela, Haiying Wang, Andrew C.-H. Sue, and Han Zuilhof. 2023. "Tunable Supramolecular Ag+-Host Interactions in Pillar[n]arene[m]quinones and Ensuing Specific Binding to 1-Alkynes" Molecules 28, no. 20: 7009. https://doi.org/10.3390/molecules28207009
APA StyleZhu, Y., Escorihuela, J., Wang, H., Sue, A. C.-H., & Zuilhof, H. (2023). Tunable Supramolecular Ag+-Host Interactions in Pillar[n]arene[m]quinones and Ensuing Specific Binding to 1-Alkynes. Molecules, 28(20), 7009. https://doi.org/10.3390/molecules28207009