Abstract
The present study aims to report the design, synthesis, and biological activity of new ethacrynic acid (EA) analogs (6–10) obtained by the double modulation of the carboxylic acid moiety and the aromatic ring with the aim to increase the chemical reactivity of Michael acceptor of EA. All obtained compounds were characterized by 1H and 13C NMR, IR, and high-resolution mass spectrometry. The antiproliferative activity was evaluated in vitro using MMT test, in a first step, against HL60 cell line and in a second step, on a panel of human cancer cell lines such as HCT116, A549, MCF7, PC3, U87-MG, and SKOV3, and normal cell line MRC5 in comparison with positive control doxorubicin. Among all the tested compounds, the product 8 containing a propargyl and a hydroxyl groups, allowing an intramolecular hydrogen bond with the keto group of EA, exhibited a pronounced and selective activity in a nanomolar range against HL60, A549, PC3, and MCF7 with IC50 values of 15, 41.2, 68.7, and 61.5 nM, respectively. Compound 8 also showed a good selectivity index (SI) against HL60 and moderate SI against the other three human cancer cells (A549, PC3, and MCF7). The study of the structure-activity relationship showed that both modifications of the carboxylic group and the introduction of an intramolecular hydrogen bond are highly required to improve the antiproliferative activities. The molecular modeling studies of compound 8 revealed that it favorably binds to the glutathione S-transferase active site, which may explain its interesting anticancer activity. These new compounds have potential to be developed as novel therapeutic agents against various cancer types.
Keywords:
ethacrynic acid; anticancer; synthesis; antiproliferative; α; β-unsaturated ketone; Michael acceptor; MTS 1. Introduction
Currently, cancer is the leading cause of death in high and upper-middle income countries and the second most common cause of death worldwide [1]. In 2020, the World Health Organization (WHO) estimated that almost 10 million people died from cancer and 19.3 million new cases were diagnosed in the same year [2]. Hence, developing new small molecules with high specificity in order to combat this disease is a big challenge for researchers.
The α, β-unsaturated carbonyl groups are attractive in medicinal chemistry due to their chemical properties, biological activities, and therapeutic potential, especially in oncology [3].
Several natural and synthetic compounds, such as chalcones [4], ibrutinib [5,6], canertinib [7,8], afatinib [9,10], neratinib [11,12], hypothemycin [13], and their analogues, are known for their antitumor activities, thanks to their α, β-unsaturated carbonyl which acts as a Michael acceptor. This system can easily react with nucleophiles, such as thiol of cysteine under physiological conditions [14]. Therefore, the increase of the reactivity of the Michael acceptor group has currently attracted more attention [15]. This electrophile site is present in ethacrynic acid (EA), which is thought to be responsible for its excellent inhibitory properties of the glutathione S-transferase P1-1 (GSTP1-1) [16]. The GSTP1-1 is actively involved in the detoxification of cells and even in the elimination of many cytotoxic agents, which represents a major obstacle to anticancer chemotherapy. Additionally, it has anti-apoptotic activities based on protein–protein interactions with the N-terminal c-Jun kinase (JNK) by directly blocking the function of JNK [17,18]. In fact, most anticancer agents induce cell death by activating the mitogen-activated protein–kinase (MAPK) pathways, particularly those involving JNK and p38-MAPK [19]. There is sufficient evidence to suggest that GSTP1-1 inhibitors may be useful therapeutic agents for the treatment of cancer and other diseases associated with aberrant cell proliferation. EA analogs, alone or as adjuvants, are known to increase the activity of chemotherapeutic agents [20,21,22,23]. Based on this observation, we have considered improving the reactivity of the Michael acceptor group of EA to increase the inhibitory activity of GSTP1-1, on the one hand, and on the other hand to increase the antiproliferative activity via the apoptosis process through the activation of the caspase protease as we have demonstrated in our previous work [24,25].
Regarding all these above-mentioned factors, we have considered synthesizing new analogs of EA, by replacing the chlorine at the ortho position of the α, β-unsaturated carbonyl group with a hydroxyl group (to favorize the formation of an intramolecular hydrogen bond) and by removing the chlorine in the meta position. At the same time, the carboxylic acid group should be transformed on either amide (G1) or propoargylic group (G2) which have shown good activities in our previously published works [26,27,28,29]. Consequently, the reactivity of the Michael acceptor of EA could be enhanced by the establishment of an intramolecular hydrogen bond between the carbonyl and the hydroxyl group (Figure 1). All the prepared compounds were tested first toward HLC60 cell line, then 7 and 8 compounds have been selected for antiproliferative studies against a panel of tumor cell lines such as human adenocarcinomic epithelial cell line (A549), human breast cancer cell line (MCF7), human prostate cancer cell line (PC3), human glioblastoma (U87-MG), ovarian carcinoma cells (SKOV3), and human colon cancer cell line (HCT116).
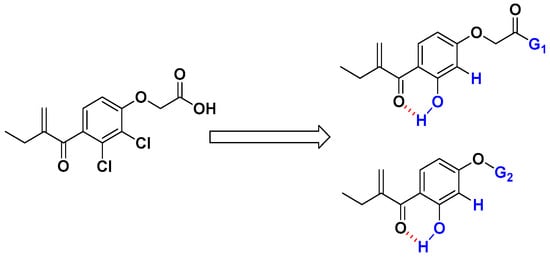
Figure 1.
Chemical modification to improve the electrophilic character of the Michael acceptor of EA analogs.
2. Results and Discussions
2.1. Chemistry
The synthetic route of the targeted EA analogue 6 is outlined in Scheme 1. In the first step, the compound 2 was prepared in 76% yield via a Friedel–Crafts reaction from 1 and butyryl chloride in the presence of titanium tetrachloride (TiCl4) as a Lewis acid in dichloromethane (DCM) [30]. Subsequently, the second step involves an aldol condensation reaction in the presence of formaldehyde and potassium carbonate in a mixture of ethanol/water (1/1) for 24 h to give compound 3 in 44% yield. Compound 4 as the main precursor for the synthesis of EA derivatives, was obtained under demethylation conditions using aluminum chloride (AlCl3) in anhydrous DCM in 78% yield. Next, the dihydroxyl compound 4 is involved in a Williamson-type selective nucleophilic substitution reaction in the presence of potassium iodide, tert-butyl-2-bromoacetate, and potassium tert-butoxide (t-BuOK) yielding compound 5 in 63%. It is very important to note that when ethyl ester and methyl ester were used substrates instead of tert-butyl ester 5, their hydrolysis under basic reaction conditions led to intramolecular cyclisation between the hydroxyl group and the Michael receptor. In the final step, the ester group of 5 is successfully converted into its corresponding carboxylic acid group using trifluoroacetic acid (TFA) in DCM at room temperature for 12 h to afford the target molecule 6 with a good yield (89%).
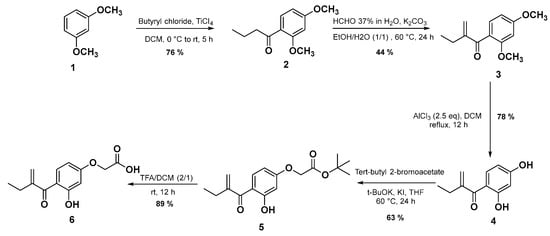
Scheme 1.
The synthetic route for the target compound 6.
After developing the synthesis of compound 6 as a new EA derivative, we were interested thereafter in carrying out two main structural modifications starting from the intermediate 4. Firstly, we have introduced the butyl acetamide function on the 4-hydroxy group to prepare an amide derivative of compound 6. In this case, the desired compound 7 was obtained via a nucleophilic substitution reaction (SN2) by reacting the compound 4 with the 2-bromo-N-butylacetamide, which was prepared and used without further purification according to the literature [31]. Secondly, following the same reactivity a propargylic group was introduced on the 4-hyrdroxy group by using propargyl bromide in the presence of t-BuOK and KI in THF at 60 °C for 24 h which afforded compound 8 in 41% yield.
In order to compare the effect of the substitution of the chlorine by the hydroxyl group on the antiproliferative activities, the analogs 9 and 10 were synthetized according to Scheme 2. Compound 9 was isolated in a 72% yield via an amidation reaction of EA with butan-1-amine in the presence of EDC and HOBt as activating agents through a mechanism similar to the previously reported in the literature [32]. Firstly, EA was deprotonated by EDC, then a nucleophilic attack of the protonated EDC gives a good leaving group which facilitates the nucleophilic attack of HOBt in the next step, giving EA derivative containing HOBt as the leaving group. Finally, the attack of the amine gave the desired product 9 and urea and HOBt as by-products. Compound 10 was prepared as previously reported [23] (Scheme 2). For more detail and data of the synthesized compounds, see the supplementary materials.
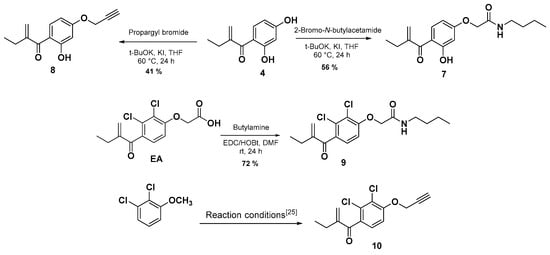
Scheme 2.
Synthesis of compounds 7–9 and 10 [25].
2.2. Biological Study
Based on a multiple target assay, all the prepared compounds (6–10) were first evaluated against HL60 leukemia cells to assess their in vitro antiproliferative activities. As mentioned in the Table 1, the obtained results were expressed as half-maximal inhibitory concentration (IC50) values with the average of at least three independent experiments and are compared to EA.

Table 1.
Antiproliferative activity of EA derivatives against cancer cell line HL60 expressed in terms of IC50 (μM).
From the screening results in Table 1, compound 6 exhibited weak antiproliferative activity with an IC50 value > 10 µM, showing that the modification of the aromatic ring without modifying the carboxylic group did not improve the antiproliferative activity of the EA. Effectively, once the carboxylic acid function of the compound 6 is modified by an amide function (compound 7), an interesting antiproliferative activity was observed with an IC50 = 1.7 μM. Interestingly, its analog (compound 9), which bears two chlorines on the aromatic ring, remains inactive. These results confirm the positive impact of the hydroxyl group on the biological activity and bode well with the development of new EA analogs containing a hydroxyl group in ortho position of the Michael acceptor. Interestingly, compound 8 exhibited a very potent antiproliferative activity with an IC50 = 0.015 µM, despite the absence of the amide function [22]. Furthermore, in agreement with the positive effect of the substitution of the chlorine atoms by a hydroxyl group, compound 8 shows markedly improved antiproliferative activity in comparison with compound 10 (IC50 value of 0.58 µM) (Figure 2) [25].
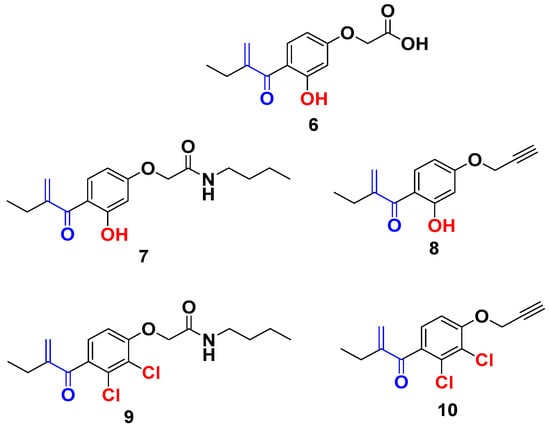
Figure 2.
New EA family selected for antiproliferative activity tests.
Based on these results, we have evaluated the antiproliferative activity of the two selected compounds 7 and 8 against a panel of cancer and non-cancer cell lines representative of diverse tissues/organ tumors: human adenocarcinomic epithelial cell line (A549), human breast cancer cell line (MCF7), human prostate cancer cell line (PC3), human glioblastoma (U87-MG), ovarian carcinoma cells (SKOV3), and human colon cancer cell line (HCT116) using cell survival assays. Moreover, in order to evaluate the safety index, their effect on the proliferation of human lung fibroblasts (MCR5) as a normal cell line was also studied. The cytotoxicities of compounds 7 and 8 (expressed as IC50 values) obtained after 72 h of exposure are summarized in Table 2.
Table 2.
Antiproliferative activity of compounds 7, 8, and doxorubicin upon various cancer human cells and normal cells expressed in terms of IC50 (nM).
The data shown in Table 2 indicates that for A549 and MCF7 cells, compound 8 was found to be more active than both compound 7 and doxorubicin, used as a reference, with IC50 values of 41.2 and 61.5 nM, respectively. We have also noticed in the case of PC3 and U87-MG cells that compound 8 was more active than compound 7 but less active than doxorubicin with IC50 values of 68.7, 369, and 2.09 nM, respectively. Conversely, we have observed in the case of HCT116 that compound 7 is slightly more active than compound 8, but both were less active than doxorubicin with IC50 values of 920, 1260, and 90 nM, respectively.
Based on these very encouraging results, the selectivity index (SI) of compounds 7 and 8 was calculated for each cancer cell lines (SI, defined as the ratio of the IC50s of non-cancer cells/IC50s of cancer cells) (Table 3) [33,34]. The results have corroborated the well-known poor selectivity of doxorubicin and have emphasized the selectivity of our compounds, especially compound 8 which exhibits a good SI against HL60 (SI = 23.33) and moderate ones on the other three cancer cells (A549, MCF7, and PC3). Unfortunately, compound 7 is less selective compared to compound 8 on all cancer cells. Next, the classical physicochemical parameter such as the membrane–water partition coefficient (LogP) was evaluated, using Marvin application ChemAxon for the two compounds 7 and 8 in order to correlate it with their antiproliferative activities. The lipophilicity, which combines both biological and physicochemical properties, is a crucial property for drug absorption, distribution, potency, and elimination. The compounds 7 and 8 showed almost the same LogP with the values of 3.44 and 2.67, respectively, suitable for the future development of drug-like compounds [35].
Table 3.
Selectivity index (SI) of compounds 7, 8, and doxorubicin.
Then, we decided to achieve docking molecular studies using the best anticancer agent 8 and the glutathione S-transferase as target enzyme. With compound 8, we obtained the nine different conformations from docking analysis, from which the eighth conformer showed the best results with a total of 10 bonds, the first three being the closest to the Glutathione S-transferase active site (1.84 Å, 3.05 Å, and 3.57 Å respectively). The one at 3.05 Å from the active site showed the presence of a hydrogen bond with tyrosine at the position 108 of the pocket, as well as a weak hydrogen bond with glycine at the position 205. The rest are hydrophobic alkyl bonds, with a pi-pi stacked hydrophobic bond with phenylalanine at position 8 of the active site. The docking analysis shows that compound 8 favorably binds to the glutathione S-transferase and further explains the interesting anticancer results, especially, against the HL60 cancer cell line (Figure 3).
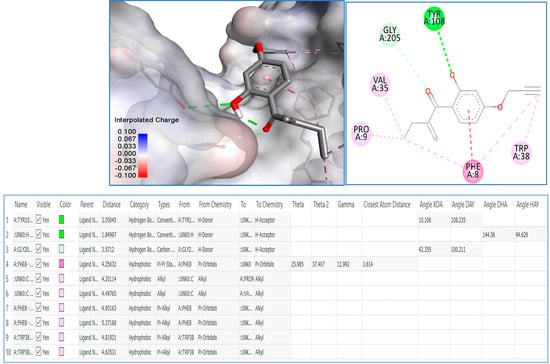
Figure 3.
Molecular modeling of compound 8 with glutathione S-transferase active site.
3. Materials and Methods
3.1. General Procedures
All reagent-grade chemicals and solvents commercially available were used without further purification. The reactions were monitored by thin-layer chromatography (TLC) using aluminum (Kiesel gel 60F254, Merck, Darmstadt, Germany) and visualized using ultraviolet light (λ = 254 nm or 365 nm). Flash column chromatography was performed on silica gel 60 (230–400 mesh, 0.040–0.063 mm). Melting points (mp (°C)) were carried out by open capillary tubes and are uncorrected using Thermo scientific digital melting point IA9200. IR absorption spectra were obtained on a Thermo Scientific Nicolet iS10, and the values are reported in inverse centimeters. 1H NMR and 13C NMR spectra were recorded on a Bruker AC300 (300 MHz for 1H and 75 MHz for 13C) or on a Bruker 400 (400 MHz for 1H and 100.6 MHz for 13C) spectrometer at room temperature, and the samples dissolved in an appropriate deuterated solvent. The peak patterns are indicated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; q, quartette. The coupling constants, J, are reported in Hertz (Hz). HRMS spectra were acquired in positive mode with an ESI source on a Q-TOF mass spectrometer with an accuracy tolerance of 2 ppm.
3.2. Synthesis and Characterization
The compounds 2 and 10 were prepared according to the methodology described in the literature [25,30,36].
1-(2,4-Dimethoxyphenyl)-2-methylenebutan-1-one (3). To a mixed solution of water and ethanol (17 mL/10 mL), compound 2 (1.1 g, 5 mmol) in ethanol (34 mL), 30% solution of formaldehyde (0.5 mL, 5 mmol), and potassium carbonate (0.67 g, 5 mmol) were added. The mixture was stirred for 24 h at 60 °C. To the reaction mixture, a hydrochloric acid solution (a mixture of 2 mL of concentrated hydrochloric acid and 100 mL of water) was added; then, the medium was decanted, extracted with (3 × 10 mL) ethyl acetate, dried over magnesium sulfate, filtered, and concentrated to dryness. The crude residue was purified by chromatography on a silica gel column using hexane/EtOAc, 7: 3 (v/v) as eluent, to obtain the expected product 3 under form of a white solid with a yield of 44%. Mp: 109–113 °C. 1H NMR (400 MHz, CDCl3), δ 7.30 (d, 3JHH = 8.3 Hz, 1H), 6.56-6.45 (m, 2H), 5.72 (s, 1H), 5.56 (s, 1H), 3.86 (s, 3H), 3.79 (s, 3H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.13 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 197.7, 162.7, 159.3, 151.7, 131.3, 124.2, 122.1, 104.1, 98.8, 55.6, 55.4, 24.1, 12.5. IR: ν (cm−1): = 1661 (C=O Ketone). HRMS (+ESI) m/z: [M+H]+ calculated for C13H17O3: 221.1171, found, 221.1172.
1-(2,4-Dihydroxyphenyl)-2-methylenebutan-1-one (4). A solution of anisole 3 (5 g, 22.5 mmol) in DCM (120 mL) was cooled to −78 °C, then an amount of AlCl3 (1 M in DCM, 149 mL, 149 mmol) was added dropwise over 10 min. The reaction mixture was stirred at 40 °C for 24 h. After total consumption of the limiting reagent, the reaction was stopped by the addition of methanol (60 mL). The reaction medium was concentrated to dryness then taken up in ethyl acetate and washed several times with an aqueous solution of HCl (1 M). The organic phases were dried over magnesium sulfate, filtered, and concentrated to dryness. The crude residue was purified by chromatography on a silica gel column using hexane/EtOAc, 7: 3 (v/v) as eluent, to obtain the expected product 4 as liquid form with a yield of 78%. 1H NMR (400 MHz, CDCl3), δ 12.64 (s, 1H), 7.68 (d, 3JHH = 8.8 Hz, 1H), 6.92 (br, 1H), 6.45 (d, 3JHH = 2.4 Hz, 1H), 6.41 (dd, 3JHH = 2.4, 8.8 Hz, 1H), 5.58 (s, 1H), 5.32 (s, 1H), 2.47 (t, 3JHH = 7.4 Hz, 2H), 1.12 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 203.1, 165.9, 163.6, 148.4, 135.6, 119.0, 113.1, 107.8, 103.6, 26.5, 12.1. IR: ν (cm−1): = 3333 (OH), 1669 (C=O Ketone). HRMS (+ESI) m/z: [M+H]+ calculated for C11H13O3: 193.0856, found: 193.0859.
Tert-butyl 2-(3-hydroxy-4-(2-methylenebutanoyl)phenoxy)acetate (5). To a solution of phenol 4 (0.92 g, 3 mmol) in THF, potassium tert-butoxide (0.33 g, 5 mmol), a catalytic amount of KI (0.05 g, 0.3 mmol), and tert-butyl bromoacetate (0.58 g, 3 mmol) were added, and the whole mixture was stirred at 60 °C for 24 h. After cooling to room temperature, a solution of concentrated HCl was added to pH = 1, and then the mixture was extracted three times with ethyl acetate. The organic phases were washed with brine, dried over magnesium sulphate, filtered, and then concentrated to dryness. The crude residue was purified by chromatography on a silica gel column using DCM/EtOAc, 4: 1 (v/v) as eluent, to obtain the expected product 5 as a white solid with a yield of 63%. Mp. 125–126 °C. 1H NMR (400 MHz, CDCl3),), δ 12.59 (s, 1H), 7.70 (d, 3JHH = 8.9 Hz, 1H), 6.47 (dd, 3JHH = 8.9, 4JHH = 2.5 Hz, 1H), 6.41 (d, 3JHH = 2.5 Hz, 1H), 5.59 (s, 1H), 5.32 (s, 1H), 4.57 (s, 2H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.52 (s, 9H), 1.13 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 202.6, 167.0, 166.0, 164.4, 148.5, 134.7, 119.0, 113.6, 107.3, 101.8, 82.9, 65.4, 28.0 (3C), 26.4, 12.1. IR: ν (cm−1): = 3312 (OH), 1651 (C=O Ketone), 1586 (C=O Ester). HRMS (+ESI) m/z: [M+H]+ calculated for C17H23O5: 307.1538, found: 307.1500.
2-(3-Hydroxy-4-(2-methylenebutanoyl)phenoxy)acetic acid (6). To a solution of ester 5 (0.08 g, 0.261 mmol) in DCM (10 mL), trifluoroacetic acid (6 mL) was added, and the mixture was stirred at room temperature for 12 h. The resulting mixture was concentrated, and the residue was dissolved in DCM (1 mL), then the desired product 6 was precipitated by the technique of co-evaporation using diethyl ether. The expected product 6 was obtained as a white solid with a yield of 89%. Mp. 62–164 °C. 1H NMR (400 MHz, CDCl3), δ 12.58 (br s, 1H), 10.72 (br s, 1H), 7.72 (d, 3JHH = 8.9 Hz, 1H), 6.49 (dd, 3JHH = 8.9, 4JHH = 2.5 Hz, 1H), 6.45 (d, 3JHH = 2.5 Hz, 1H), 5.60 (s, 1H), 5.33 (s, 1H), 4.74 (s, 2H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.12 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 202.7, 173.1, 165.9, 163.8, 148.4, 134.9, 119.2, 113.9, 107.1, 101.9, 64.5, 26.3, 12.1. IR: ν (cm−1): = 3402 (OH), 1652 (C=O Ketone), 1622 (C=O Acid). HRMS (+ESI) m/z: [M+H]+ calculated for C13H15O5: 251.0910, found: 251.0914.
N-Butyl-2-(3-hydroxy-4-(2-methylenebutanoyl)phenoxy)acetamide (7). To a solution of phenol 4 (0.57, 3 mmol) in tetrahydrofuran, potassium tert-butoxide (0.33 g, 5 mmol), a catalytic amount of KI (0.05 g, 0.3 mmol), and 2-bromo-N-butylacetamide (0.58, 3 mmol) were added. The reaction mixture was heated at 60 °C for 24 h. After cooling to room temperature, a solution of concentrated HCl was added to pH = 1, and then the mixture was extracted three times with ethyl acetate. The organic phases were combined, washed with brine, dried over magnesium sulphate, filtered, and concentrated to dryness. The crude residue was purified by chromatography on a silica gel column using DCM/EtOAc, 4: 2 (v/v) as eluent, to obtain the expected product 7 as a white solid with a yield of 56%. Mp. 133–134 °C. 1H NMR (400 MHz, CDCl3), δ 12.52 (s, 1H), 7.73 (d, 3JHH = 8.9 Hz, 1H), 6.57–6.43 (m, 3H), 5.61 (s, 1H), 5.33 (s, 1H), 4.54 (s, 2H), 3.37 (q, 3JHH = 7.4 Hz, 2H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.59–1.53 (m, 2H), 1.42–1.32 (m, 2H), 1.13 (t, 3JHH = 7.4 Hz, 3H), 0.95 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 202.7, 167.0, 165.9, 163.3, 148.5, 135.0, 119.3, 114.1, 106.4, 102.7, 67.2, 38.9, 31.6, 26.3, 20.0, 13.7, 12.1. IR: ν (cm−1): = 3350 (OH), 1663 (C=O Ketone), 1602 (C=O Amide). HRMS (+ESI) m/z: [M+H]+ calculated for C17H24NO4: 306.1700, found: 306.1699.
1-(2-Hydroxy-4-(prop-2-yn-1-yloxy)phenyl)-2-methylenebutan-1-one (8). To a solution of phenol 4 (0.69 g, 3 mmol) in tetrahydrofuran, potassium tert-butoxide (0.33 g, 5 mmol), a catalytic amount of KI (0.05 g, 0.3 mmol) and propargyl bromide (0.35 g, 3 mmol) were added. The reaction mixture was heated at 60 °C for 24 h. After cooling to room temperature, a solution of concentrated HCl was added to pH = 1 and then the mixture was extracted three times with ethyl acetate. The organic phases were combined, washed with brine, dried over magnesium sulphate, filtered, and concentrated to dryness. The crude residue was purified by chromatography on a silica gel column using Hexane/DCM, 1: 1 (v/v) as eluent, to obtain the expected product 8 as liquid form with a yield of 41%. 1H NMR (300 MHz, CDCl3), δ 12.6 (s, 1H), 7.71 (d, 3JHH = 8.8 Hz, 1H), 6.57 (d, 3JHH = 2.4 Hz, 1H), 6.49 (dd, 3JHH = 2.4, 8.8 Hz, 1H), 5.99 (s, 1H), 5.32 (s, 1H), 4.75 (d, 4JHH = 2.4 Hz, 2H), 2.59 (t, 4JHH = 2.4 Hz, 1H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.12 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3), δ 202.7, 165.9, 164.0, 148.6, 134.7, 119.0, 113.6, 107.4, 102.4, 76.4, 56.0, 56.0, 26.4, 12.1. IR: ν (cm−1): = 3289 (OH), 2122 (C≡C Alkyne), 1678 (C=O Ketone). HRMS (+ESI) m/z: [M+H]+ calculated for C14H15O3: 231.0975, found: 231.0977.
N-Butyl-2-(2,3-dichloro-4-(2-methylenebutanoyl)phenoxy)acetamide (9). To a solution of EDC (0.56 g, 3.63 mmol), HOBt (0.55 g, 3.63 mmol), DMAP (0.02 g, 0.16 mmol), and ethacrynic acid (0.10 g, 3.30 mmol) in DMF anhydrous (5 mL), butan-1-amine (0.24 g, 3.30 mmol) was added at 0 °C, and the whole mixture was stirred overnight at room temperature. The mixture was extracted with ethyl acetate, and the combined organic phases were washed with brine, dried over magnesium sulfate, and then concentrated under pressure. The crude residue was purified by chromatography on a silica gel column using DCM/EtOAc, 4: 1 (v/v) as eluent, to obtain the expected product 9 as a white solid with a yield of 72%. Mp. 98–100 °C. 1H NMR (400 MHz, CDCl3), δ 7.19 (d, 3JHH = 8.5 Hz, 1H), 6.87 (d, 3JHH = 8.5 Hz, 1H), 6.78 (br s, 1H), 5.96 (s, 1H), 5.59 (s, 1H), 4.57 (s, 2H), 3.39 (q, 3JHH = 7.4 Hz, 2H), 2.47 (q, 3JHH = 7.4 Hz, 2H), 1.63–1.53 (m, 2H), 1.47–1.32 (m, 2H), 1.15 (t, 3JHH = 7.4 Hz, 3H), 0.95 (t, 3JHH = 7.4 Hz, 3H). 13C NMR (101 MHz, CDCl3), δ 195.5, 166.5, 154.5, 150.2, 134.1, 131.4, 128.7, 127.2, 122.9, 110.9, 68.2, 38.9, 31.5, 23.4, 20.0, 13.7, 12.4. IR: ν (cm−1): 1660 (C=O Ketone), 1599 (C=O Amide). HRMS (+ESI) m/z: [M+H]+ calculated for C17H22Cl2NO3: 358.0973, found: 358.0971.
3.3. Biological Evaluation
All cancer cell lines (HCT116, A549, MCF7, PC3, U87-MG, SKOV3, and MRC5) were obtained from the European Collection of Cell Culture (ECACC, Salisbury, UK) or from the American Type Culture Collection (Rockville, MD, USA) and were cultured according to the supplier’s instructions. Human HCT-116 colorectal carcinoma, MCF7 breast adenocarcinoma, PC3 prostate adenocarcinoma, A549 lung carcinoma, and SK-OV-3 ovary carcinoma were grown in Gibco McCoy’s 5A supplemented with 10% fetal calf serum (FCS) and 1% glutamine. HL60 myelogenous leukemia cells were grown in RPMI 1640 supplemented with 10% fetal calf serum (FCS) and 1% glutamine. U87-MG glioblastoma and human MRC-5 cells were grown in Gibco medium DMEM supplemented with 10% fetal calf serum (FCS) and 1% glutamine. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. Cell growth inhibition was determined by an MTS assay according to the manufacturer’s instructions (Promega, Madison, WI, USA). Briefly, the cells were seeded in 96-well plates (2.5 × 103 cells/well) containing 100 μL of growth medium. After 24 h of culture, the cells were treated with the tested compounds at 10 different final concentrations. After incubation for 72 h, 20 µL of Cell Titer 96®AQueous. One Solution Reagent was added for 2 h before recording absorbance at 490nm with a spectrophotometric plate reader. The concentration–response curves were generated with Graph Prism software and the 50% inhibition concentration values (IC50) were calculated from polynomial curves (four or five-parameter logistic equations).
3.4. Molecular Modeling
Molecular docking was performed using the Autodock Vina [37,38]. AutoDockTools was required to prepare the PDBQT file for Glutathione S-transferase and to set the size and the center of the grid box. Kollman charges, Gasteiger charges, AD4 type atom, and polar hydrogen atoms were included in the Glutathione S-transferase structure. The grid size was set at 30 × 30 × 30 (x, y, and z) points, and the grid center was designated at x, y, and z dimensions of 19.951167, −0.126697, and 20.556417, respectively. Post-docking analyses were visualized using Discovery Studio Biovia 2021, which showed the sizes and locations of binding sites, hydrogen-bond interactions, hydrophobic interactions, and bonding distances at 5 Å from the position of the docked ligand; compound being docked to the active site of Glutathione S-transferase. Subsequently, binding poses of each ligand were observed and their interactions with the protein were characterized, and the best conformations of each ligand were selected.
4. Conclusions
In summary, new EA analogues have been synthesized by modifying two parts of the EA, namely the carboxylic acid part and the aromatic ring part. All the synthesized compounds were evaluated and screened for their in vitro antiproliferative activities first against HL60. Based on their activities, two selected compounds, 7 and 8, were tested on a panel of human cancer cell lines and on a normal cell line. Interestingly, compound 8 shows very potent anti-proliferative activity against the four human cancer cells HL60, A549, PC3, and MCF7 with IC50 values of 15 nM, 41.2 nM, 68.7, and 61.5 nM, respectively. Compound 8 also shows a good SI against HL60 and moderate SI on the other three cancer cells (A549, PC3, and MCF7). This study reports the crucial and positive impact of an intramolecular hydrogen bond to increase the chemical reactivity of the Michael acceptor of the EA analogs, which correlates with the improvement of the antiproliferative activities.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28020910/s1, Spectral Data of All the Synthesized Products, 1H and 13C NMR Spectra of All the Products.
Author Contributions
Conceptualization, G.G., F.S. and S.E.K.; methodology, G.G., F.S. and S.E.K.; validation, A.E.A., N.E.B. and M.-A.H.; formal analysis, A.E.A., N.E.B., H.G., E.E.F. and J.B.; investigation, S.E.K., G.G. and F.S.; resources, G.G., F.S. and S.E.K.; data curation, A.E.A., S.E.K., H.G., E.E.F. and G.G.; writing—Original draft preparation, A.E.A., S.E.K. and G.G.; writing—Review and editing, J.B., F.S., E.E.F. and G.G.; visualization, S.E.K., A.E.A. and G.G.; supervision, F.S., G.G. and S.E.K.; project administration, F.S., G.G. and S.E.K.; funding acquisition, S.E.K., F.S. and G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Euromed University of Fes, CNRST of Morocco and the University of Orleans.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data supporting this research are availible Supplementary Materials.
Acknowledgments
This work was supported by Euromed University of Fes and Campus France and partially supported by Orléans University, CNRST, CNRS, Labex SynOrg (ANR-11-LABX-0029), and Labex IRON (ANR-11-LABX-0018-01).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 3–9 are available from the authors.
References
- Ur Rashid, H.; Xu, Y.; Muhammad, Y.; Wang, L.; Jiang, J. Research advances on anticancer activities of matrine and its derivatives: An updated overview. Eur. J. Med. Chem. 2019, 161, 205–238. [Google Scholar] [CrossRef] [PubMed]
- Globocan (The Global Cancer Observatory). All Cancers; International Agency for Research on Cancer—WHO: Lyon, France, 2020; Volume 419, pp. 199–200. Available online: https://gco.iarc.fr/today/home (accessed on 10 July 2022).
- Bukhari, S.N.A.; Jantan, I.; Unsal Tan, O.; Sher, M.; Naeem-ul-Hassan, M.; Qin, H.-L. Biological activity and molecular docking studies of curcumin-related α, β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J. Agric. Food Chem. 2014, 62, 5538–5547. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.M.; Mustafa, R.Z.; Gyamfi, J.A.; Pittaluga, S.; Chang, S.; Chang, B.; Farooqui, M.; Wiestner, A. Ibrutinib inhibits BCR and NF-ΚB signaling and reduces tumor proliferation in tissue-resident cells of patients with CLL. Blood J. Am. Soc. Hematol. 2014, 123, 3286–3295. [Google Scholar] [CrossRef] [PubMed]
- Saleh, L.M.; Wang, W.; Herman, S.E.M.; Saba, N.S.; Anastas, V.; Barber, E.; Corrigan-Cummins, M.; Farooqui, M.; Sun, C.; Sarasua, S.M. Ibrutinib downregulates a subset of MiRNA leading to upregulation of tumor suppressors and inhibition of cell proliferation in chronic lymphocytic leukemia. Leukemia 2017, 31, 340–349. [Google Scholar] [CrossRef]
- Hightower, M.; Belani, C.P.; Jain, V.K. Erlotinib (OSI-774, TarcevaTM), a Selective epidermal growth factor receptor tyrosine kinase inhibitor, in combination with chemotherapy for advanced non–small-cell lung Cancer. Clin. Lung Cancer 2003, 4, 336–338. [Google Scholar] [CrossRef]
- Severinsson, E.A.D.; Trinks, C.; Gréen, H.; Abdiu, A.; Hallbeck, A.-L.; Stål, O.; Walz, T.M. The Pan-ErbB Receptor tyrosine kinase inhibitor canertinib promotes apoptosis of malignant melanoma in vitro and displays anti-tumor activity in vivo. Biochem. Biophys. Res. Commun. 2011, 414, 563–568. [Google Scholar] [CrossRef]
- Hurvitz, S.A.; Shatsky, R.; Harbeck, N. Afatinib in the treatment of breast cancer. Expert Opin. Investig. Drugs 2014, 23, 1039–1047. [Google Scholar] [CrossRef]
- D’Arcangelo, M.; Hirsch, F.R. Clinical and comparative utility of afatinib in non-small cell lung cancer. Biol. Targets Ther. 2014, 8, 183–192. [Google Scholar] [CrossRef]
- Bose, P.; Ozer, H. Neratinib: An Oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin. Investig. Drugs 2009, 18, 1735–1751. [Google Scholar] [CrossRef]
- Burstein, H.J.; Sun, Y.; Dirix, L.Y.; Jiang, Z.; Paridaens, R.; Tan, A.R.; Awada, A.; Ranade, A.; Jiao, S.; Schwartz, G. Neratinib, an irreversible ErbB receptor tyrosine kinase inhibitor, in patients with advanced ErbB2-positive breast cancer. J. Clin. Oncol. 2010, 28, 1301–1307. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Nishida, K.; Sugita, K.; Yoshioka, T. Antitumor efficacy of hypothemycin, a new ras-signaling inhibitor. Jpn. J. Cancer Res. 1999, 90, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.H.; Khalaila, I.; Allardyce, C.S.; Juillerat-Jeanneret, L.; Dyson, P.J. Rational design of platinum (iv) compounds to overcome glutathione-s-transferase mediated drug resistance. J. Am. Chem. Soc. 2005, 127, 1382–1383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Neumann, H.; Beller, M. Synthesis of α, β-unsaturated carbonyl compounds by carbonylation reactions. Chem. Soc. Rev. 2020, 49, 3187–3210. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.; Dutta, S.; Tew, K.D. Inhibitors of glutathione s-transferases as therapeutic agents. Adv. Drug Deliv. Rev. 1997, 26, 91–104. [Google Scholar] [CrossRef]
- Wang, T.; Arifoglu, P.; Ronai, Z.; Tew, K.D. Glutathione S-transferase P1–1 (GSTP1–1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J. Biol. Chem. 2001, 276, 20999–21003. [Google Scholar] [CrossRef]
- Zhou, L.; Jing, Y.; Styblo, M.; Chen, Z.; Waxman, S. Glutathione-S-transferase π inhibits As2O3-induced apoptosis in lymphoma cells: Involvement of hydrogen peroxide catabolism. Blood 2005, 105, 1198–1203. [Google Scholar] [CrossRef]
- Yin, Z.; Ivanov, V.N.; Habelhah, H.; Tew, K.; Ronai, Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000, 60, 4053–4057. [Google Scholar]
- Tew, K.D. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 1994, 54, 4313–4320. [Google Scholar] [CrossRef] [PubMed]
- Ploemen, J.; Van Schanke, A.; Van Ommen, B.; Van Bladeren, P.J. Reversible conjugation of ethacrynic acid with glutathione and human glutathione S-transferase P1-1. Cancer Res. 1994, 54, 915–919. [Google Scholar]
- Oakley, A.J.; Rossjohn, J.; Lo Bello, M.; Caccuri, A.M.; Federici, G.; Parker, M.W. The Three-dimensional structure of the human Pi class glutathione transferase P1-1 in complex with the inhibitor ethacrynic acid and its glutathione conjugate. Biochemistry 1997, 36, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Gast, S.-M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G.H. In vivo efficacy of the diuretic agent ethacrynic acid against multiple myeloma. Leuk. Res. 2012, 36, 598–600. [Google Scholar] [CrossRef]
- Mignani, S.; El Brahmi, N.; El Kazzouli, S.; Eloy, L.; Courilleau, D.; Caron, J.; Bousmina, M.M.; Caminade, A.M.; Cresteil, T.; Majoral, J.P. A Novel class of ethacrynic acid derivatives as promising drug-like potent generation of anticancer agents with established mechanism of action. Eur. J. Med. Chem. 2016, 122, 656–673. [Google Scholar] [CrossRef] [PubMed]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Synthesis and evaluation of a novel class of ethacrynic acid derivatives containing triazoles as potent anticancer agents. Bioorg. Chem. 2021, 115, 105293. [Google Scholar] [CrossRef]
- El Abbouchi, A.; El Brahmi, N.; Hiebel, M.-A.; Bignon, J.; Guillaumet, G.; Suzenet, F.; El Kazzouli, S. Synthesis and biological evaluation of ethacrynic acid derivatives bearing sulfonamides as potent anti-cancer agents. Bioorg. Med. Chem. Lett. 2020, 30, 127426. [Google Scholar] [CrossRef] [PubMed]
- El Brahmi, N.; Mignani, S.M.; Caron, J.; El Kazzouli, S.; Bousmina, M.M.; Caminade, A.-M.; Cresteil, T.; Majoral, J.-P. Investigations on dendrimer space reveal solid and liquid tumor growth-inhibition by original phosphorus-based dendrimers and the corresponding monomers and dendrons with ethacrynic acid motifs. Nanoscale 2015, 7, 3915–3922. [Google Scholar] [CrossRef]
- El Brahmi, N.; El Kazzouli, S.; Mignani, S.; Laurent, R.; Ladeira, S.; Caminade, A.M.; Bousmina, M.; Majoral, J.P. Symmetrical and unsymmetrical incorporation of active biological monomers on the surface of phosphorus dendrimers. Tetrahedron 2017, 73, 1331–1341. [Google Scholar] [CrossRef]
- El Kazzouli, S.; Zyad, A.; El Brahmi, N.; El Abbouchi, A.; Boujdi, K.; Bousmina, M.; Ait Mouse, H.; Tilaoui, M. Anticancer activities of a novel family of ethacrynic acid derivatives. U.S. Patent Application No. 17/297,252, 27 January 2022. [Google Scholar]
- Kaeppler, U.; Stiefl, N.; Schiller, M.; Vicik, R.; Breuning, A.; Schmitz, W.; Rupprecht, D.; Schmuck, C.; Baumann, K.; Ziebuhr, J. A New lead for nonpeptidic active-site-directed inhibitors of the severe acute respiratory syndrome coronavirus main protease discovered by a combination of screening and docking methods. J. Med. Chem. 2005, 48, 6832–6842. [Google Scholar] [CrossRef]
- Swyka, R.A.; Shuler, W.G.; Spinello, B.J.; Zhang, W.; Lan, C.; Krische, M.J. Conversion of aldehydes to branched or linear ketones via regiodivergent rhodium-catalyzed vinyl bromide reductive coupling–redox isomerization mediated by formate. J. Am. Chem. Soc. 2019, 141, 6864–6868. [Google Scholar] [CrossRef]
- Hawash, M.; Kahraman, D.C.; Ergun, S.G.; Cetin-Atalay, R.; Baytas, S.N. Synthesis of novel indole-isoxazole hybrids and evaluation of their cytotoxic activities on hepatocellular carcinoma cell lines. BMC Chem. 2021, 15, 66. [Google Scholar] [CrossRef]
- Hawash, M.; Jaradat, N.; Eid, A.M.; Abubaker, A.; Mufleh, O.; Al-Hroub, Q.; Sobuh, S. Synthesis of novel isoxazole–carboxamide derivatives as promising agents for melanoma and targeted nano-emulgel conjugate for improved cellular permeability. BMC Chem. 2022, 16, 47. [Google Scholar] [CrossRef] [PubMed]
- Weerapreeyakul, N.; Nonpunya, A.; Barusrux, S.; Thitimetharoch, T.; Sripanidkulchai, B. Evaluation of the anticancer potential of six herbs against a hepatoma cell line. Chin. Med. 2012, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Sakaeda, T.; Okamura, N.; Nagata, S.; Yagami, T.; Horinouchi, M.; Okumura, K.; Yamashita, F.; Hashida, M. Molecular and pharmacokinetic properties of 222 commercially available oral drugs in humans. Biol. Pharm. Bull. 2001, 24, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).