2.1. Simulation Setup
The current study is mainly focused on the effects of fCNT on the adsorption of saccharides on their external surfaces. Therefore, the starting point in modeling these compounds is the construction of the fCNT model. CNT are often purified using oxidative agents [
2,
4,
15].
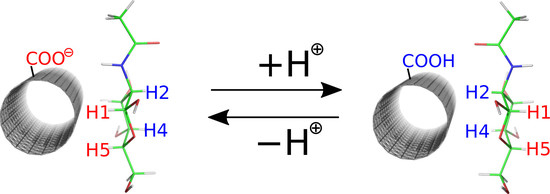
Thus, a common functional group present on the CNT surface is the carboxyl group. In aqueous dispersion, fCNT carboxyl groups partly dissociate depending on the pH leading to the carboxylic acid or carboxylate function. Thus, in order to fully understand the possible role of saccharides as agents tweaking the biocompatibility and hydrophilicity of fCNT surfaces, both the carboxylic acid and carboxylate forms of the carboxyl group have to be studied. In order to generate fCNT we assumed that the presence of carboxyl groups (either protonated or deprotonated) leads to the rehybridization (from sp
2 to sp
3) of the carbon atom to which the carboxyl group is bound. Also, the neighboring carbon atom needs similar rehybridization in order to keep the valence conserved. This is schematically shown in
Figure 1 together with the partial charges on atoms directly or indirectly involved in the functionalization scheme.
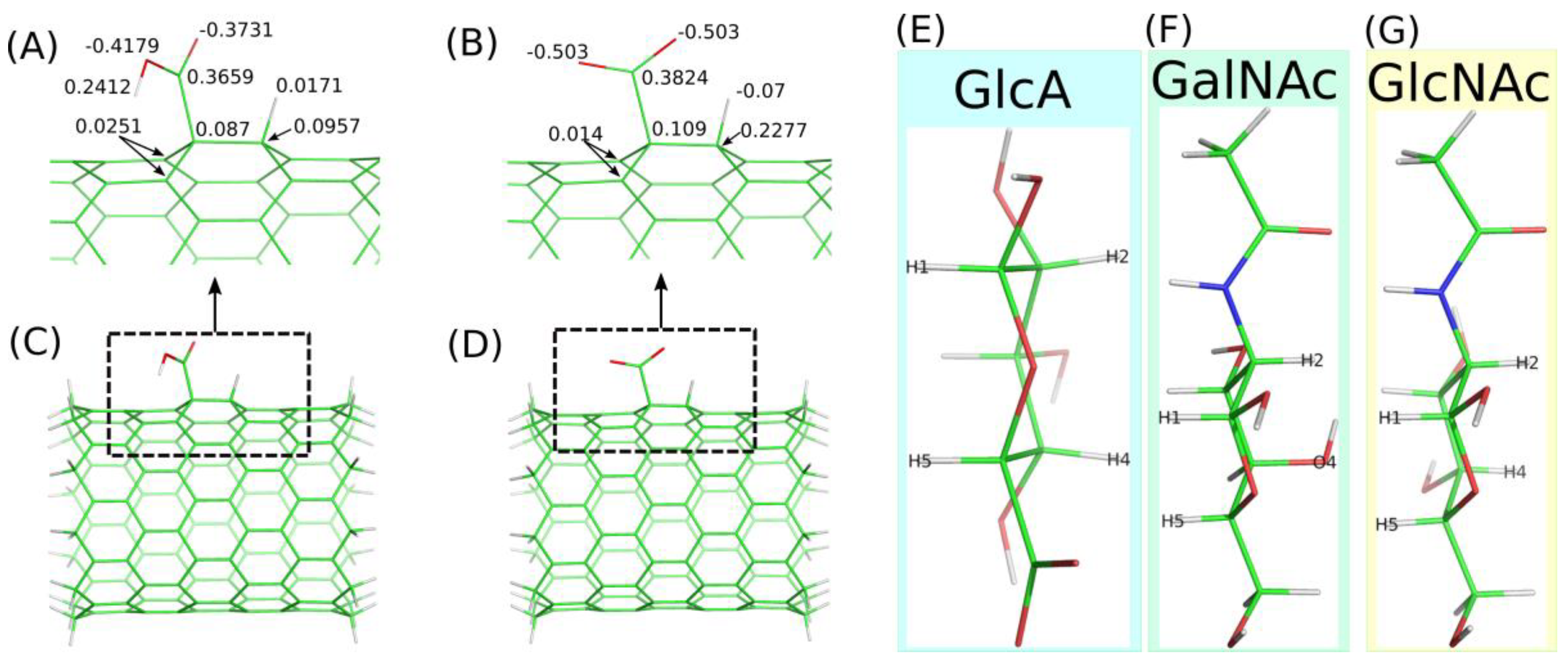
The detailed description of the empirical force field generation for fCNT is available in the Methods section. Here we need to mention several assumptions made during model building. One such assumption is that the presence of the functional groups leads to atomic charge values not equal to zero for these groups and for the neighboring CNT carbon atoms. The values of those charges are shown in
Figure 1A,B. These partial charges were derived using the RESP-X1 scheme for fitting quantum chemically determined electrostatic potentials into point charges [
16,
17]. Particular attention was paid to chemical and charge equivalencing for the fCNT atoms to derive equivalent charge values for chemically equivalent atoms. This was achieved by using the RED Server Development service which implements the methodology described by Vanquelef et al. [
18].
Small clusters of (10,0) CNT were used in these cases as models of carboxylated fCNT. These models are shown in
Figure 1C,D and contain 164 and 165 atoms for CNT-COO
⊖ and CNT-COOH, respectively. Nevertheless, the density-functional theory calculation for optimizing geometries and computing molecular electrostatic potentials was used; the combination of functional and basis set was B3LYP/6-31G(d).
The derived charge values (
Figure 1A,B) were used during the grafting of each carboxylic acid/carboxylate group to the CNT structure. The total number of these groups of atoms were assumed to represent 2% of the CNT carbon atoms. This is a rather common value reported in commercially available samples. We used (17,0) CNT with a length of 5.9 nm: the number of functional groups was 20 per CNT. The incorporation of these groups of atoms into the large CNT molecule needed charge adjustment for other atoms. This was achieved by dividing the total residual charge determined from each group by the number of carbon atoms in the CNT. Thus, each atom in the fCNT bears a charge value close to zero in order to compensate the charges computed for the functional groups.
The fCNT was treated as an infinite molecule by applying periodic replicas along its axis for bonds, angles, dihedrals and all other factors. The box size was adjusted to the length of the main (17,0) fCNT and the number of carbon atoms in that nanotube was 952. The fCNT was initially put in the middle of a simulation box, and saccharide molecules were placed in a way that prevents overlaps with fCNT or with other molecules. In this work we investigated the interaction of fCNT with monosacchrides such as β-D-glucopyranuronic acid (GlcA), N-acetyl-β-D-glucosamine (GlcNAc) and N-acetyl-β-D-galactosamine (GalNAc). Additionally, we also studied the interaction of two oligosaccharides: hyaluronic acid (HA) and chondroitin (Ch), both composed of monosaccharide decamers with a GlcA-β-1,3-GlcNAc- and GlcA-β-1,3-GalNAc- repetitive pattern, respectively.
Figure 1E–G show stick representation of monosaccharides utilized in the calculations and they also show atom name labels on hydrogen and oxygen atoms, which are used to analyze the carbohydrate orientation in reference to the fCNT surface (according to IUPAC nomenclature).
The combination of carboxylic acid and carboxylate fCNT (CNT-COOH and CNT-COO
⊖) and the considered monosaccharides and oligosaccharides lead to ten different systems. Additionally, we also studied four other molecular systems with four oligosaccharides interacting with a single fCNT and a single Ch chain system alone in water in order to estimate the influence of fCNT on the conformational properties of the glycan chain. In this context, the analogous data for the HA chain were taken from our previous paper [
11].
2.2. Adsorption of Monosaccharides on fCNT
The study of the interaction of monosaccharides GlcA, GalNAc and GlcNAc with CNT-COOH and CNT-COO
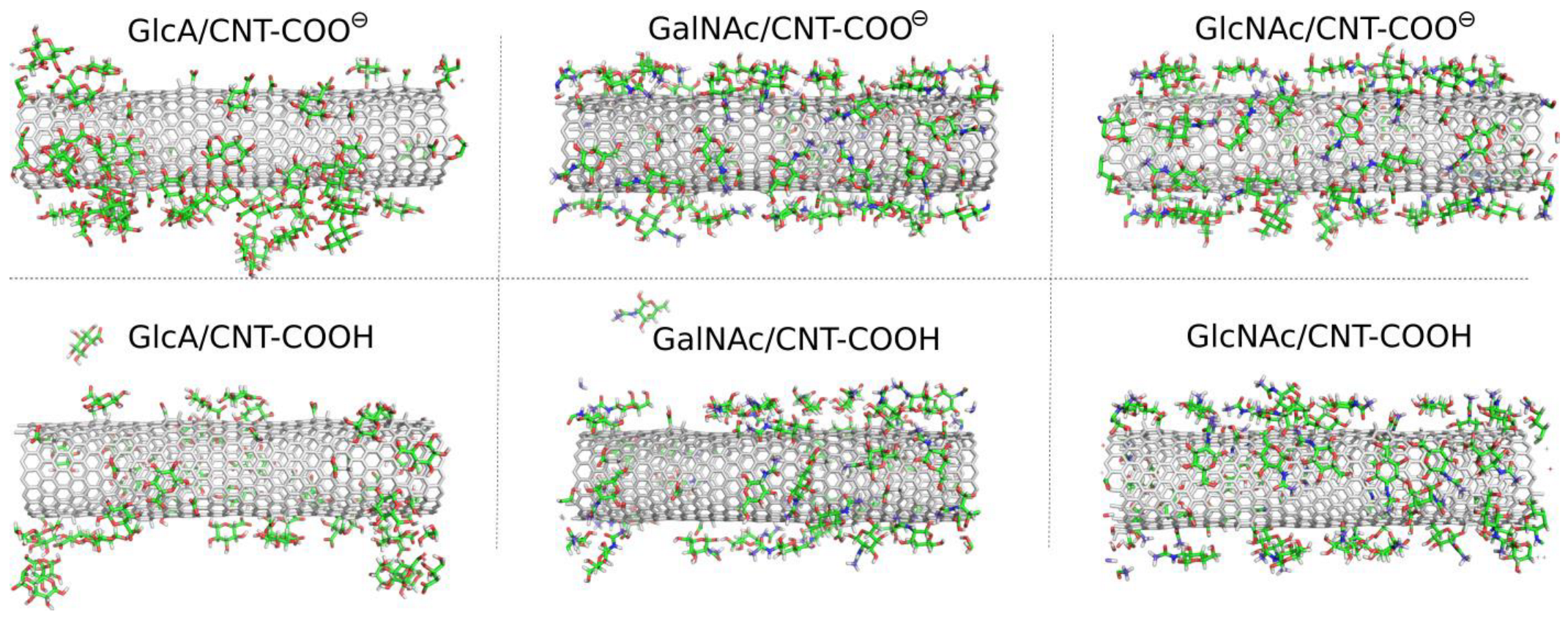
⊖ using molecular dynamics (MD) simulations in a condensed phase led to significant qualitative differences between the considered cases. Visual inspection of the MD snapshots is useful for drawing the first qualitative conclusions about the properties of systems under investigation. Thus,
Figure 2 shows the MD snapshots for monosaccharide adsorption taken at the last simulation frames, i.e., after 600 ns of the production runs.
As described in
Figure 2 the monosaccharides attach to the surfaces of the fCNT, but in some cases individual molecules were found also in the bulk water. In the GlcA/CNT-COOH and GalNAc/CNT-COOH molecular systems the adsorption is considered as unstable. This is because in a possible experimental situation, the re-adsorption of temporarily desorbed molecules is unlikely to be observed. This is due to the much larger volume of the aqueous phase in a possible experimental system. During MD simulations the re-adsorption occurs relatively quickly due to the rather small distance necessary to be passed by the desorbed molecule and to interact with a fCNT image in the periodic conditions.
Thus, individual events of spontaneous detachments seen in MD simulations mean that in experimental conditions the adsorption/desorption events will be occurring simultaneously (reversible adsorption). The effective surface coverage will be strongly dependent on the bulk concentration of monosaccharides and temperature.
A more quantitative analysis of the adsorption of monosaccharides on the fCNT surface was based on the determination of pair-interaction energies between fCNT and adsorbed molecules and also depends on the determination of the radial density profiles (RDF) of adsorbed molecules in reference to the fCNT axis. These results are collected in
Table 1 and
Figure 2 and
Figure 3. Thus,
Table 1 shows the determined mean-pair-interaction energies for the different studied molecular systems. These energies are split into the Lennard–Jones and electrostatic contributions.
Figure 2 in turn shows the total pair interaction energies as bar charts for the molecular systems.
The qualitative conclusion drawn from the MD simulation snapshots are supported by the analysis of pair-interaction energies between adsorbed carbohydrates and fCNT. Thus, the strongest interaction is observed for GalNAc and GlcNAc and deprotonated fCNT. This is quite surprising since these molecules are neutral. Moreover, a significant contribution from the electrostatic interactions to the total energy (−26 kJ mol
−1 as seen in
Table 1) was not expected. The CNT-COOH protonated molecular systems actually reveal a similar range of dispersion interactions, with a significantly smaller contribution for the Coulomb energies. Intuitively, the electrostatic energy values for negatively charged fCNT with neutral monosaccharides are not expected to be large. However, the observations are clear: the contribution of the electrostatic interactions for the studied monosaccharides is significant and leads to a qualitative change in properties for the analyzed systems.
The properties of negatively charged GlcA with fCNT is more intuitive: the neutral CNT-COOH interacts more strongly with GlcA molecules than the negatively charged CNT-COO⊖. However, the difference is not that large when looking at the total energies. Nevertheless, these two molecular systems are definitely the weakest in terms of the adsorption strengths when compared to GlcNAc and GalNAc.
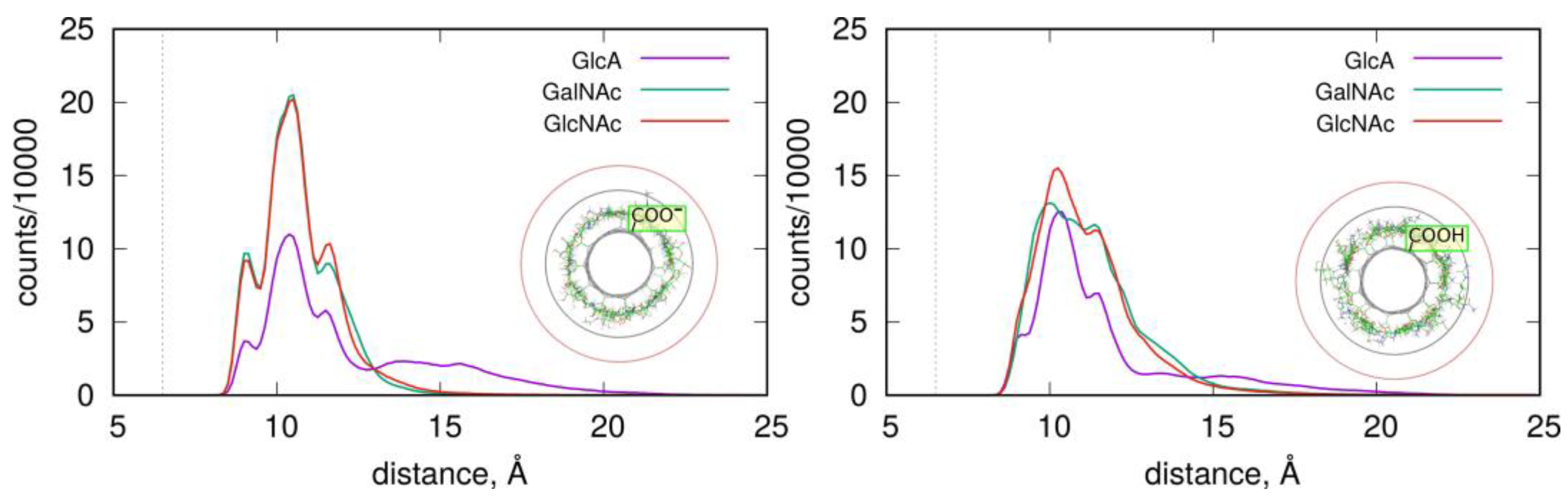
The structures and stabilities of the adsorbed layers are more quantitatively described using the RDF functions of the monosaccharides in reference to the fCNT axis.
Figure 4 shows the corresponding data for the studied systems.
As described in
Figure 4, the adsorbed layers are the most compact in the case of GalNAc and GlcNAc adsorbed at the CNT-COO
⊖ surface. This is in agreement with the pair-interaction energies reported in
Figure 3 and
Table 1. In these cases, all monosaccharide atoms fit within a distance smaller than 15 Å from the nanotube axis. As observed in insets in
Figure 4, this distance corresponds to the monolayer coverage of the nanotube surface. However, in all cases, the densities decay is almost at zero within the distance 20 Å from the fCNT axis. The non-zero density within the 15 Å–20 Å means that the adsorbed layers are more diffuse or that transient detachments of the adsorbed molecules occur. This happens to the GlcA for both fCNT types as well as GlcNAc and GalNAc in the case of protonated fCNT.
The shapes of the density profiles differ significantly depending on the protonation state of the fCNT surface in the cases of GlcNAc and GalNAc. It seems that fCNT-carboxylate groups lead to the formation of stiffer monosaccharide–fCNT complexes and well-defined peaks on the distributions. In the cases of fCNT carboxylic acid groups, the adsorbed layers are more loosely defined and the density profiles are more diffuse.
Interestingly, both GlcNAc and GalNAc monosaccharides display nearly the same density profiles and interaction energies, independently of the fCNT type. This suggests that the glucose to galactose epimerization at the C4 atom is not of primary relevance for the structural features of adsorbed layer, nor for the energetic properties of adsorption.
More detailed information about the structures of adsorbed layers comes from the analysis of the orientations of the GlcA, GalNAc and GlcNAc monosaccharide at/on the fCNT surfaces. These orientations can be defined by the density profiles of some selected atoms, which are shown in
Figure 1. Each monosaccharide has a distinct “side” defined by the arrangement of H1–H5 and H2–H4 (or O4) atoms, as described in
Figure 1E–G. Thus, determination of the density profiles of the H1–H5 and H2–H4(O4) pairs gives direct information about the preferred orientation of a monosaccharide on the fCNT surface. The obtained results are shown in
Figure 5.
As shown, each monosaccharide has a preferred orientation towards the fCNT surface. A similar observation was reported in the analysis of HA oligosaccharide adsorption on the surfaces of bare unfunctionalized CNT in a previous work [
11]. It was found that GlcA and GlcNAc prefer to be adsorbed via the H1-H5 and H2-H4 side, respectively. The current study provides even more striking results since we conclude that the protonation state of the fCNT affects the preferred orientation of GlcNAc and GalNAc. In this new work, GlcA still reveals the preferred H1-H5 orientation no matter what the protonation state of the fCNT is. Moreover, GlcNAc and GalNAc display a preferred orientation with their H1-H5 side towards the CNT-COO
⊖ surface, and on the other side towards the CNT-COOH surface. This means that these changes can be externally controlled by the changes in the pH of the aqueous solution. Thus, this new property of the monosaccharides/fCNT systems can be utilized in practical realizations/experimental trials of molecular nanodevices and sensors.
Finally, the clear preferences of monosaccharides to interact with CNT via the face-to-face contact of their aliphatic patches with the CNT surface is a clear indicator of CH-π-attractive interactions. Such interactions were unambiguously demonstrated in our previous study [
11] as being responsible for the adsorption of HA onto the surface of bare unfunctionalized CNT. Similar interactions are responsible for the binding of carbohydrates to proteins [
13,
19] where the role of π donor is played by aromatic amino acids.
2.3. Adsorption of Oligosaccharides on fCNTs
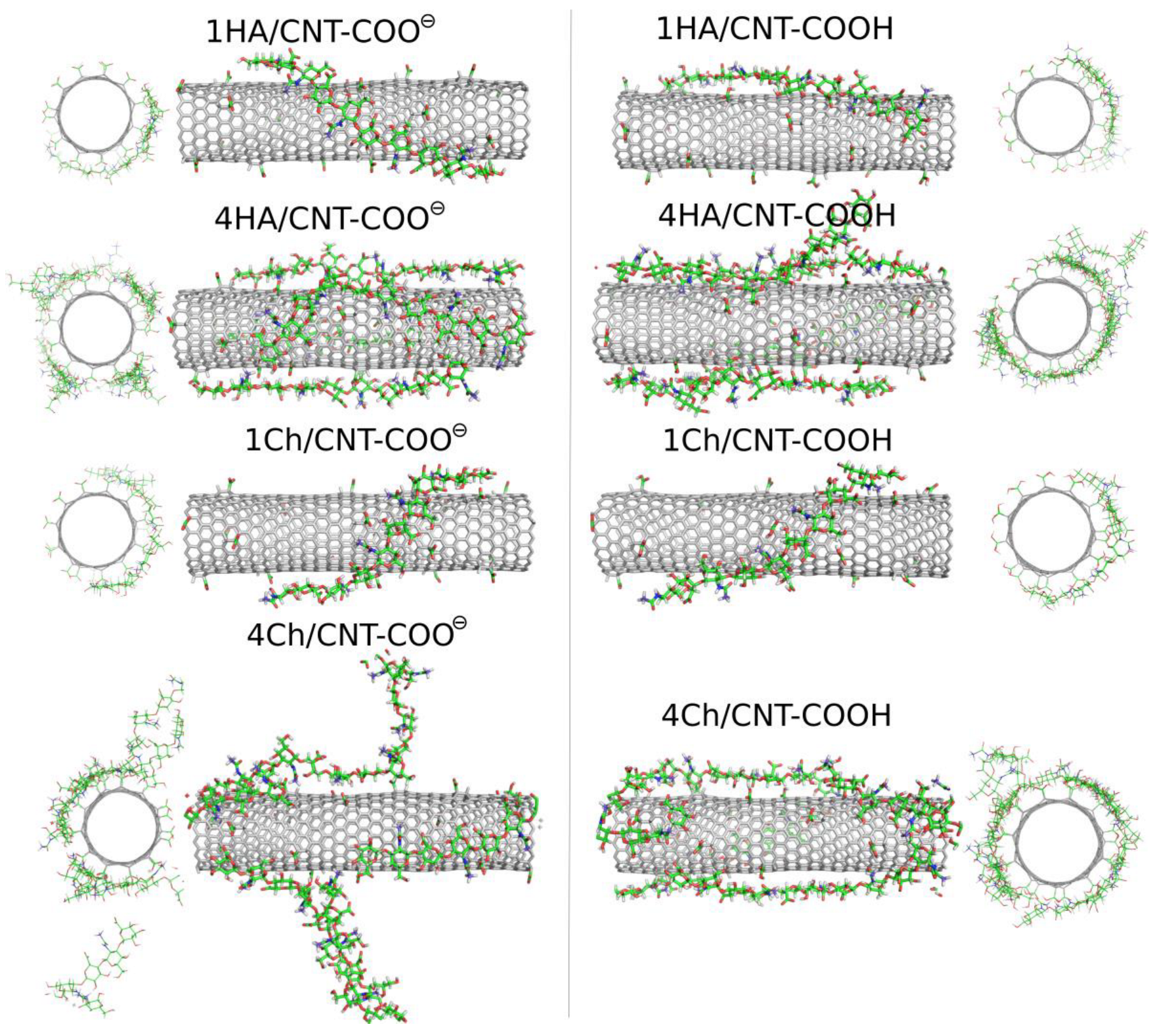
Oligosaccharides can potentially be more useful as CNT surface-modifying agents due to their larger molar mass and their stronger binding to the surface. In this study, we analyzed the adsorption behavior of decameric HA and decameric Ch chains. We start the analysis of the results from the visual inspection of the simulation boxes at the final step, i.e., after 600 ns of productive MD simulations.
Figure 6 shows these snapshots for the studied systems involving oligosaccharides.
The qualitative behavior of HA and Ch oligosaccharides on fCNT surfaces can be summarized as follows. The single decamers tightly wrap the fCNT no matter what the considered oligosaccharides and fCNT are. However, at a higher concentration of oligosaccharides represented by four decamers with a fCNT, destabilization of the adsorption is observed. Particularly, in the four Ch/CNT-COO
⊖ system, Ch oligosaccharides show a predilection for partial detachments from the fCNT surface and tend to stand perpendicularly to the fCNT axis. Such behavior is not observed in the case of HA or Ch adsorbed on CNT-COOH and is therefore not an effect of the crowded conditions. Elsewhere, the free energy component coming from the interaction between Ch molecules and fCNT is comparable to that for the interaction with water. As seen in
Table 1 and
Figure 3, increasing the number of the adsorbed molecules reduces the effective pair interaction per single molecule. Thus, the balance between free energy components from fCNT and water becomes shifted towards water as the number of Ch molecules increases. This leads to the unstable adsorption of Ch on fCNT with carboxylate groups. We can further conclude that changing the pH so that the surface carboxyl groups switch from the protonated to the deprotonated state (or vice versa) should lead to the reversible adsorption and desorption of Ch molecules on fCNT. This effect is not observed for HA molecules, at least in the considered conditions, i.e., up to four HA molecules on the (17,0) 5.9 nm long fCNT.
These conclusions are also supported by the RDF, which contain information from the whole-productive MD simulations (as for energy values in
Table 1).
Figure 7 shows such profiles, which are analogous to those in
Figure 4. The density of four Ch oligosaccharides at the CNT-COO
⊖ surface do not decay to zero. This means that perpendicularly standing Ch oligosaccharides are observed during the whole-productive MD simulations. In the case of CNT-COOH, the adsorbed layer of the four Ch decamers terminates at 20 Å, which demonstrate the presence of a single layer and a stable adsorption. Interestingly, the four HA/CNT-COOH system reveals a long tail on the RDF indicating similar behavior to the Ch/CNT-COOH system, but with a much weaker intensity.
Single-molecule adsorption is in both cases strong and limited strictly to the single-layer occupation without any transient displacement from the fCNT surface. This conclusion comes from either the pair-interaction energies shown in
Table 1 and
Figure 3 or from the fact that RDF never exceed 15 Å, as seen in
Figure 7. Increasing the concentration of oligosaccharides leads to the reduction of the pair-interaction energy with fCNT, and to significant displacements of oligosaccharides from the fCNT surfaces.
2.5. Influence of Glycan Adsorption on Its Conformation
As the CNT molecule displays a significant degree of rigidity, we did not analyze their potential structural alterations induced by the presence of adsorbed saccharides. On the contrary, carbohydrate molecules are conformationally heterogeneous and deserving of a systematic analysis. We studied how the conformation of HA and Ch decamers is affected by CNT, when the complex is formed. Thus, free energy maps were recovered from MD trajectories by using the main conformational descriptors, illustrating the three-dimensional shape of the glycosaminoglycan backbone, i.e., the ϕ and ψ glycosidic linkage torsion angles as defined in the Methods section.
Figure 8 shows the free energy maps generated for the HA decamer in the glycosidic dihedral angle ϕ, ψ coordinates. The most notable observed effect of HA adsorption on fCNT is the reduction of its conformational space in comparison to that of the free glycan molecule. This is due to the influence of external forces originating from the presence of CNT and the limited mobility of the adsorbed oligosaccharide. When adsorbed onto CNT-COO
⊖, the HA conformational space shows significant alterations. Namely, the secondary glycosidic-linkage-type of geometry shapes appears. Based on the production MD simulations, the presence of this alternative conformation is associated with the existence of conformationally locked ‘kinks’ in the adsorbed HA decamer, while the rate of conformational exchanges involving reorientation of linkages is too slow to fully recover the conformational, dynamic equilibria.
The discussed changes in backbone geometry take place in the β(1–4) and β(1–3) glycosidic linkage present in HA, but the first one seems to be affected to a larger extent in comparison to the second one, as the populations of
syn- and
anti- conformers [
21] are nearly equal.
Our previous work [
11], which focused on the HA/unfunctionalized–CNT interaction resulted in similar findings, showing that the adsorption of HA is associated with conformational alterations of the HA backbone. This occurs via the rotation of the ψ dihedral angle. In spite of the qualitatively distinct behaviors observed in the cases of CNT-COO
⊖ and CNT-COOH (conformational alteration of backbone for the ψ dihedral angle vs. no alteration, respectively), we refrain from drawing any ultimate conclusion about the influence of CNT on the dynamics and the conformational feature of adsorbed glycan decamers. This is because the production MD simulation time is not sufficiently long enough to observe multiple binding/unbinding events and, consequently, may not correctly render the dynamics of the ϕ and ψ dihedral angles.
Nevertheless, it is clear that the fCNT–HA interactions are strong enough to perturb the intrinsic dynamic equilibrium related to the conformation of the glycosidic linkage within the HA oligosaccharide.
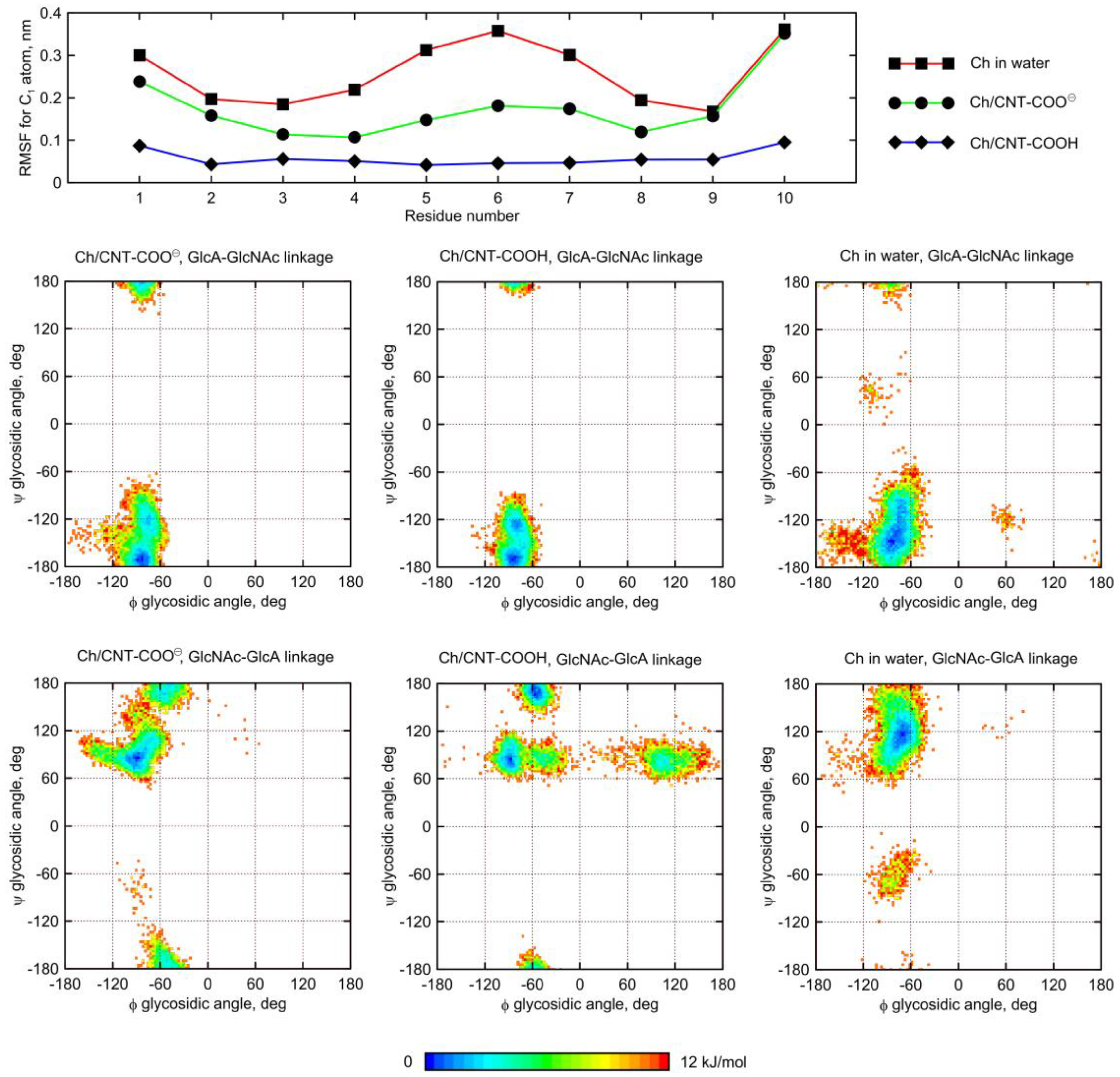
A similar behavior was observed for the Ch oligosaccharide (
Figure 9). However, the largest deformation of the glycosidic linkage is observed as a result of adsorption on CNT-COOH, whereas interactions of Ch with CNT-COO
⊖ display less significant differences. Interestingly, instead of expected
anti-ψ conformations, the glycosidic bonds of the adsorbed Ch chain now explore the quasi
trans-ψ conformation. This unusual behavior is observed in the case of both β(1–4) and β(1–3) linkages, and for CNT-COO
⊖ and CNT-COOH. Moreover, the deformation along the ϕ dihedral angle is observed, but only in the case of CNT-COO
⊖ and the β(1–4) linkage. Such changes in the ϕ value act against the exo-anomeric effect [
22] and, again, speaks for strong, attractive forces acting between glycans and fCNT. Apart from that, the expected reduction in the ϕ, ψ conformational space upon adsorption onto CNT is observed as well.
The dynamic geometry of glycan oligosaccharides affected by the CNT presence is also illustrated by the changes in root-mean-square-fluctuation (RMSF) parameter values (
Figure 8 and
Figure 9). Large decreases of the RMSF values calculated for the anomeric carbon atoms and adsorbed glycan chains speak for reduced mobilities of saccharides adsorbed on CNT and agree with both the monitored alterations of conformational properties and the expectations. The reduction of RMSF values upon adsorption on CNT-COO
⊖ is similar to that observed for HA-unfunctionalized–CNT interactions [
11] and equal to ca. 50%. However much larger reduction (even up to 80%) is observed in the case of both HA and Ch interacting with CNT-COOH. This is in line with pair-interaction energies determined for oligosaccharides (
Figure 3) and visual inspection of the behavior of oligosaccharides adsorbed on fCNT.
Finally, in the case of both HA and Ch, we observed several events of pyranose ring distortion, changing the ring geometry from
4C
1 to boat and skew-boat shapes. This effect concerns mainly GlcA units, in which the carboxylate group is forced to adopt the axial or quasi-axial conformation, and is located further from the CNT surface. Again, this observation can be interpreted in terms of strong saccharide-CNT affinity, which results in forces that emerge on the ring substituent and shift the dynamic equilibria of the ring conformation [
23].
Summarizing, the conformation of glycosidic linkages in the adsorbed saccharides may undergo significant alterations in order to facilitate the favorable CNT–glycan interactions and increase the area of the glycan-fCNT contact. The observed kinks in glycosidic linkages as well as the presence of alternative ring shapes are conformationally locked and are indicators of strong, attractive interactions between rings of adsorbed saccharides and fCNT.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}