
Calix[6]arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
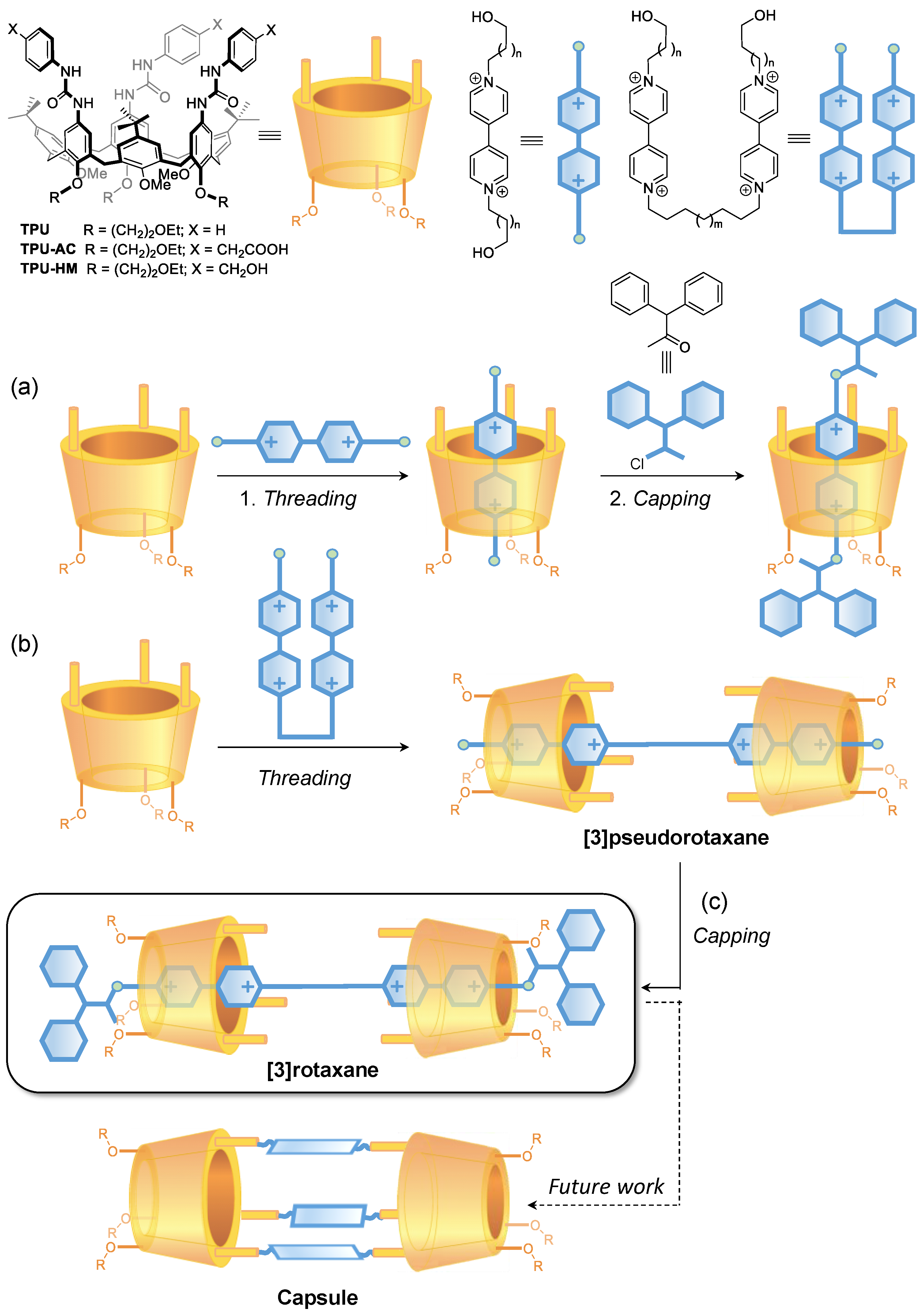
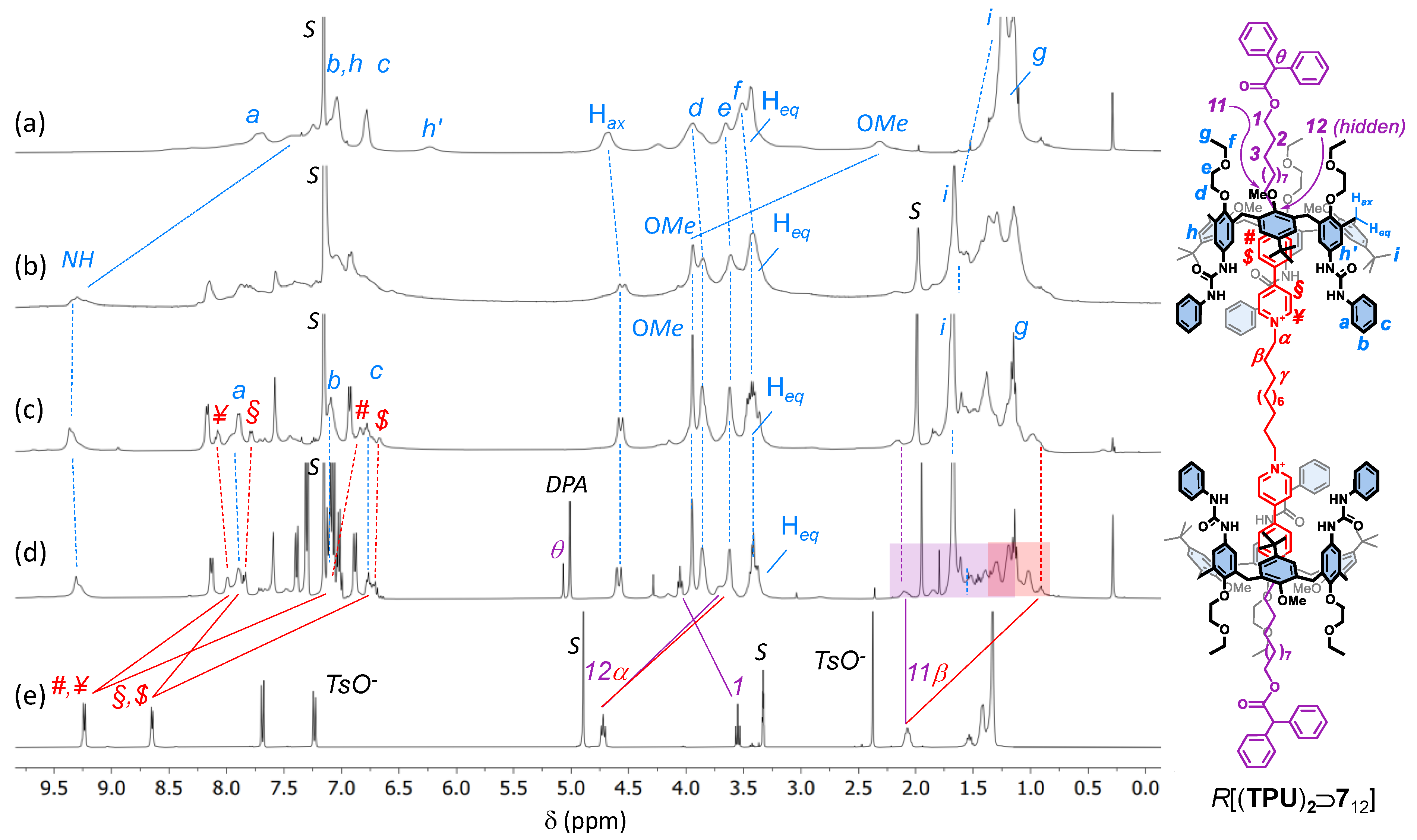
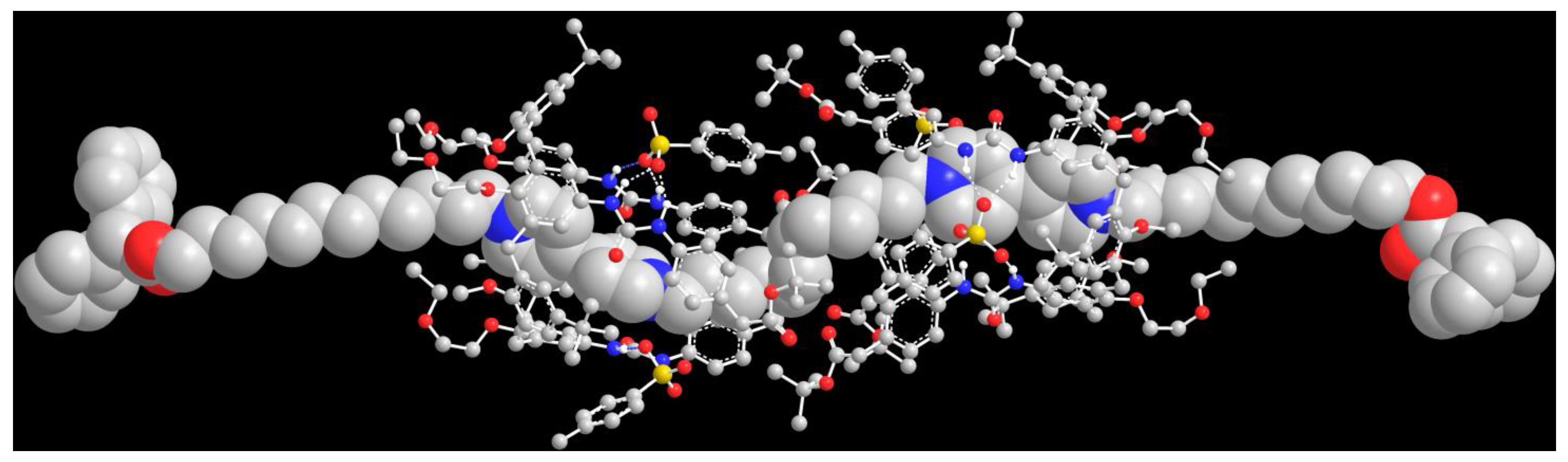
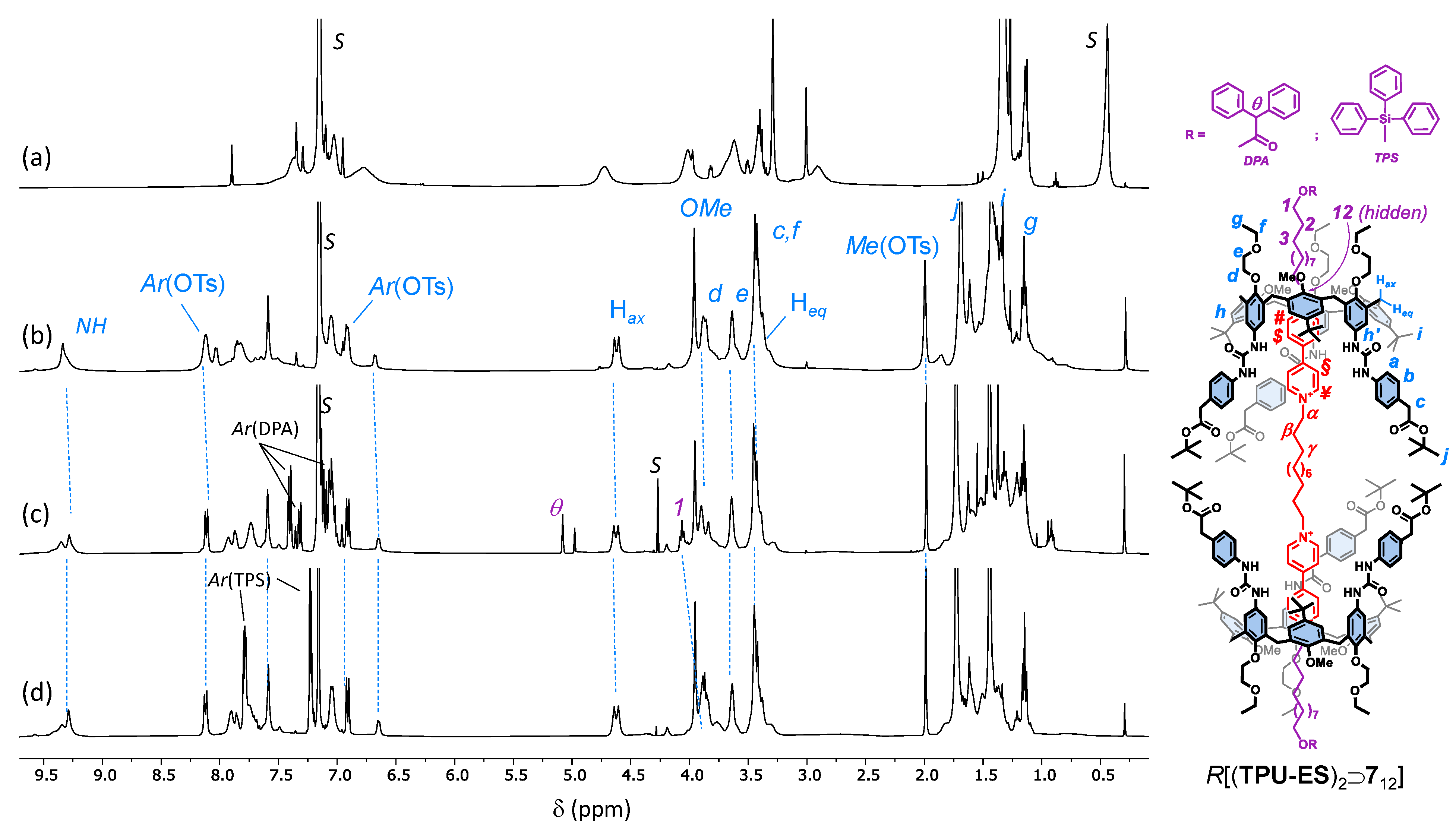
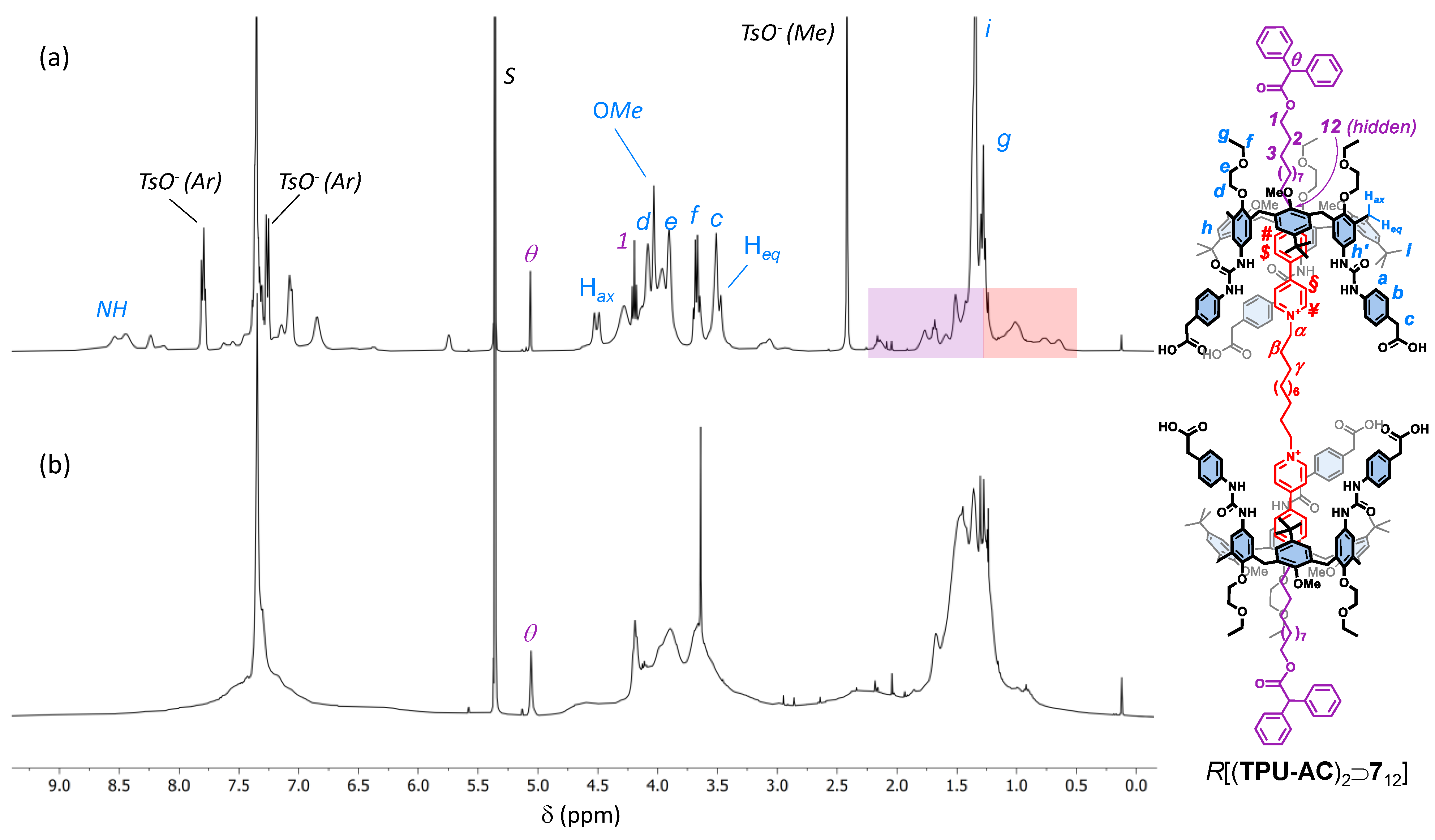
2. Results
3. Materials and Methods
3.1. General
3.2. Chemistry
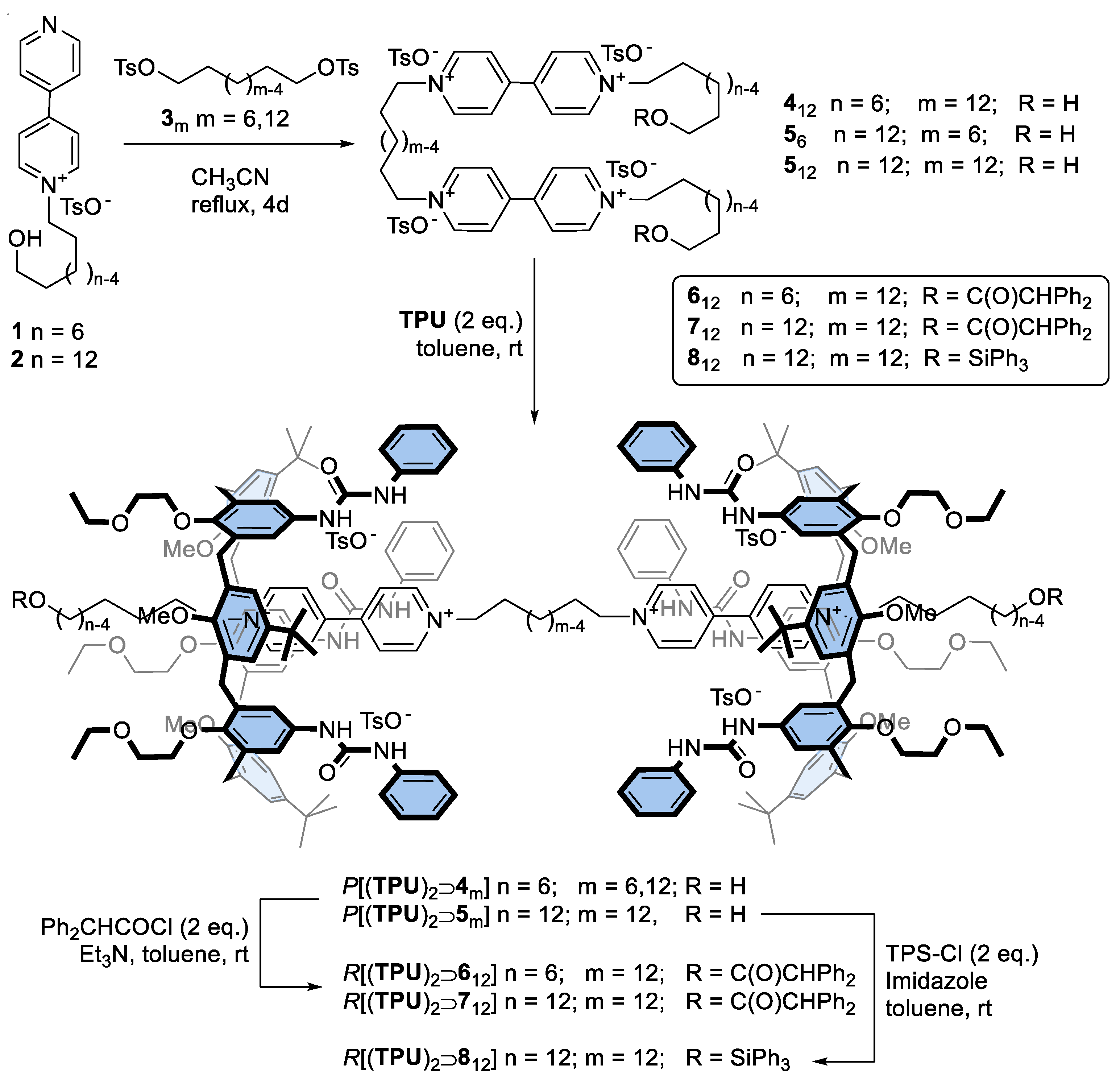
3.2.1. General Procedure for the Synthesis of the Bis-Viologen Axles 412 and 56–12
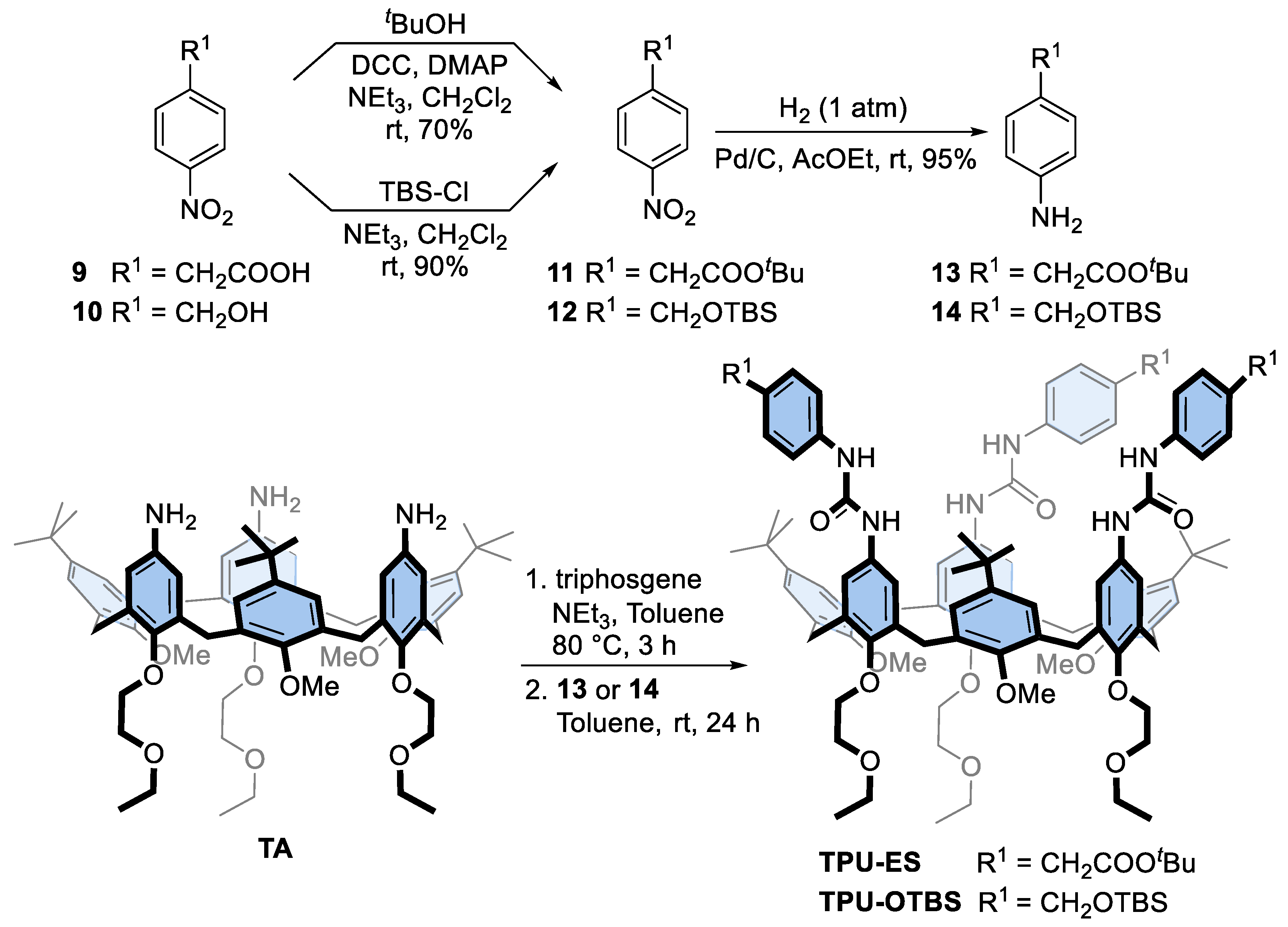
3.2.2. Synthesis of Tert-Butyl 2-(4-nitrophenyl)acetate (11)
3.2.3. Synthesis of Tert-Butyl 2-(4-aminophenyl)acetate (13)
3.2.4. General Procedure for the Synthesis of Calix[6]arenes TPU-ES and TPU-OTBS
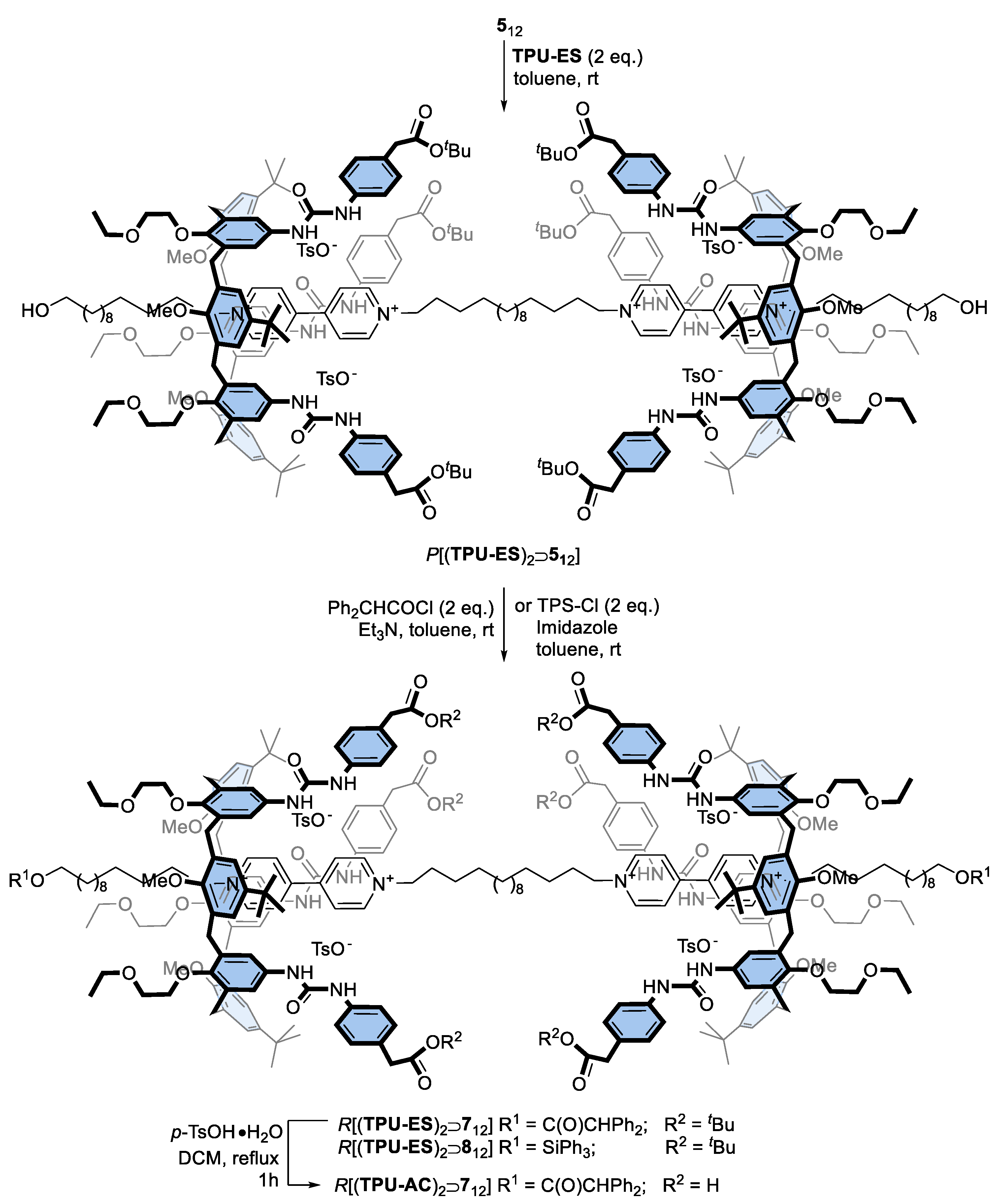
3.2.5. General Procedure for the Synthesis of [3]Rotaxanes R[(TPU)2⊃612], R[(TPU)2⊃712] and R[(TPU-ES)2⊃712]
3.2.6. General Procedure for the Synthesis of [3]Rotaxanes R[(TPU)2⊃812] and R[(TPU-ES)2⊃812]
3.2.7. Synthesis of [3]Rotaxane R[(TPU-AC)2⊃712]
3.3. Computational Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tapia, L.; Alfonso, I.; Solà, J. Molecular Cages for Biological Applications. Org. Biomol. Chem. 2021, 19, 9527–9540. [Google Scholar] [CrossRef]
- Acharyya, K.; Mukherjee, P.S. Organic Imine Cages: Molecular Marriage and Applications. Angew. Chem. Int. Ed. 2019, 58, 8640–8653. [Google Scholar] [CrossRef]
- Bols, P.S.; Anderson, H.L. Template-Directed Synthesis of Molecular Nanorings and Cages. Acc. Chem. Res. 2018, 51, 2083–2092. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.-T.; Yuan, Y.-X.; Xiong, J.-B.; Zheng, Y.-S.; Tang, B.Z. Macrocycles and Cages Based on Tetraphenylethylene with Aggregation-Induced Emission Effect. Chem. Soc. Rev. 2018, 47, 7452–7476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Martinez, A.; Dutasta, J.-P. Emergence of Hemicryptophanes: From Synthesis to Applications for Recognition, Molecular Machines, and Supramolecular Catalysis. Chem. Rev. 2017, 117, 4900–4942. [Google Scholar] [CrossRef]
- Díaz-Moscoso, A.; Ballester, P. Light-Responsive Molecular Containers. Chem. Commun. 2017, 53, 4635–4652. [Google Scholar] [CrossRef]
- Hasell, T.; Cooper, A.I. Porous Organic Cages: Soluble, Modular and Molecular Pores. Nat. Rev. Mater. 2016, 1, 1–14. [Google Scholar] [CrossRef]
- Lorenzetto, T.; Fabris, F.; Scarso, A. A Resorcin[4]arene Hexameric Capsule as a Supramolecular Catalyst in Elimination and Isomerization Reactions. Beilstein J. Org. Chem. 2022, 18, 337–349. [Google Scholar] [CrossRef]
- La Manna, P.; Talotta, C.; Floresta, G.; De Rosa, M.; Soriente, A.; Rescifina, A.; Gaeta, C.; Neri, P. Mild Friedel–Crafts Reactions inside a Hexameric Resorcinarene Capsule: C−Cl Bond Activation through Hydrogen Bonding to Bridging Water Molecules. Angew. Chem. Int. Ed. 2018, 57, 5423–5428. [Google Scholar] [CrossRef]
- Ziegler, M.; Brumaghim, J.L.; Raymond, K.N. Stabilization of a Reactive Cationic Species by Supramolecular Encapsulation. Angew. Chem. Int. Ed. 2000, 39, 4119–4121. [Google Scholar] [CrossRef]
- Cram, D.J.; Tanner, M.E.; Thomas, R. The Taming of Cyclobutadiene. Angew. Chem. Int. Ed. Eng. 1991, 30, 1024–1027. [Google Scholar] [CrossRef]
- Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J.R. White Phosphorus Is Air-Stable Within a Self-Assembled Tetrahedral Capsule. Science 2009. [Google Scholar] [CrossRef]
- Bolliger, J.L. Self-Assembled Coordination Cages and Organic Capsules as Catalytic Supramolecular Reaction Vessels. In Effects of Nanoconfinement on Catalysis; Poli, R., Ed.; Fundamental and Applied Catalysis; Springer International Publishing: Cham, Switzerland, 2017; pp. 17–48. ISBN 978-3-319-50207-6. [Google Scholar]
- Voloshin, Y.; Belaya, I.; Krämer, R. The Encapsulation Phenomenon: Synthesis, Reactivity and Applications of Caged Ions and Molecules; Springer: Cham, Switzerland, 2016; ISBN 978-3-319-27738-7. [Google Scholar]
- Hof, F.; Rebek, J. Molecules within Molecules: Recognition through Self-Assembly. Proc. Natl. Acad. Sci. USA 2002, 99, 4775–4777. [Google Scholar] [CrossRef] [PubMed]
- Hof, F.; Craig, S.L.; Nuckolls, C.; Rebek, J., Jr. Molecular Encapsulation. Angew. Chem. Int. Ed. 2002, 41, 1488–1508. [Google Scholar] [CrossRef]
- Yamanaka, M.; Kobayashi, K. Capsular Assemblies of Calix[4]resorcinarene-Based Cavitands. Asian J. Org. Chem. 2013, 2, 276–289. [Google Scholar] [CrossRef]
- Puchnin, K.; Zaikin, P.; Cheshkov, D.; Vatsouro, I.; Kovalev, V. Calix[4]tubes: An Approach to Functionalization. Chem. Eur. J. 2012, 18, 10954–10968. [Google Scholar] [CrossRef]
- Rincón, A.M.; Prados, P.; de Mendoza, J. A Calix[6]arene Dimer Linked Through Amino Acid Hydrogen Bond Interactions. Eur. J. Org. Chem. 2002, 2002, 640–644. [Google Scholar] [CrossRef]
- Le Gac, S.; Zeng, X.; Reinaud, O.; Jabin, I. Synthesis and Conformational Study of the First Triply Bridged Calix[6]azatubes. J. Org. Chem. 2005, 70, 1204–1210. [Google Scholar] [CrossRef]
- Moerkerke, S.; Ménand, M.; Jabin, I. Calix[6]arene-Based Cascade Complexes of Organic Ion Triplets Stable in a Protic Solvent. Chem. Eur. J. 2010, 16, 11712–11719. [Google Scholar] [CrossRef]
- Arduini, A.; Ferdani, R.; Pochini, A.; Secchi, A.; Ugozzoli, F.; Sheldrick, G.M.; Prados, P.; González, J.J.; de Mendoza, J. Non-Bonded Water Molecules Confined Into a Self-Assembled Calixarene Cage. J. Supramol. Chem. 2002, 2, 85–88. [Google Scholar] [CrossRef]
- Arduini, A.; Domiano, L.; Ogliosi, L.; Pochini, A.; Secchi, A.; Ungaro, R. Self-Assembled Hydrogen-Bonded Molecular Cages of Calix[6]arenetricarboxylic Acid Derivatives. J. Org. Chem. 1997, 62, 7866–7868. [Google Scholar] [CrossRef]
- González, J.J.; Ferdani, R.; Albertini, E.; Blasco, J.M.; Arduini, A.; Pochini, A.; Prados, P.; de Mendoza, J. Dimeric Capsules by the Self-Assembly of Triureidocalix[6]arenes through Hydrogen Bonds. Chem. Eur. J. 2000, 6, 73–80. [Google Scholar] [CrossRef]
- Arduini, A.; Ferdani, R.; Pochini, A.; Secchi, A. Synthesis of Upper Rim Covalently Linked Double Calix[6]arenes. Tetrahedron 2000, 56, 8573–8577. [Google Scholar] [CrossRef]
- Arduini, A.; Credi, A.; Faimani, C.; Massera, C.; Pochini, A.; Secchi, A.; Semeraro, M.; Silvi, S.; Ugozzoli, F. Self-Assembly of a Double Calix[6]arene Pseudorotaxane in Oriented Channels. Chem. Eur. J. 2008, 14, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Scelle, J.; Vervoitte, H.; Bouteiller, L.; Chamoreau, L.-M.; Sollogoub, M.; Vives, G.; Hasenknopf, B. Size-Dependent Compression of Threaded Alkyldiphosphate in Head to Head Cyclodextrin [3]Pseudorotaxanes. Chem. Sci. 2022, 13, 2218–2225. [Google Scholar] [CrossRef]
- Yamashina, M.; Kusaba, S.; Akita, M.; Kikuchi, T.; Yoshizawa, M. Cramming versus Threading of Long Amphiphilic Oligomers into a Polyaromatic Capsule. Nat. Commun. 2018, 9, 4227. [Google Scholar] [CrossRef]
- Tuncel, D.; Ünal, Ö.; Artar, M. Supramolecular Assemblies Constructed by Cucurbituril-Catalyzed Click Reaction. Isr. J. Chem. 2011, 51, 525–532. [Google Scholar] [CrossRef]
- Cera, G.; Arduini, A.; Secchi, A.; Credi, A.; Silvi, S. Heteroditopic Calix[6]arene Based Intervowen and Interlocked Molecular Devices. Chem. Rec. 2021, 21, 1161–1181. [Google Scholar] [CrossRef]
- Orlandini, G.; Casimiro, L.; Bazzoni, M.; Cogliati, B.; Credi, A.; Lucarini, M.; Silvi, S.; Arduini, A.; Secchi, A. Synthesis and Properties of a Redox-Switchable Calix[6]arene-Based Molecular Lasso. Org. Chem. Front. 2020, 7, 648–659. [Google Scholar] [CrossRef]
- Bazzoni, M.; Zanichelli, V.; Casimiro, L.; Massera, C.; Credi, A.; Secchi, A.; Silvi, S.; Arduini, A. New Geometries for Calix[6]arene-based Rotaxanes. Eur. J. Org. Chem. 2019, 2019, 3513–3524. [Google Scholar] [CrossRef]
- Zanichelli, V.; Dallacasagrande, L.; Arduini, A.; Secchi, A.; Ragazzon, G.; Silvi, S.; Credi, A. Electrochemically Triggered Co-Conformational Switching in a [2]Catenane Comprising a Non-Symmetric Calix[6]arene Wheel and a Two-Station Oriented Macrocycle. Molecules 2018, 23, 1156–1168. [Google Scholar] [CrossRef]
- Zanichelli, V.; Bazzoni, M.; Arduini, A.; Franchi, P.; Lucarini, M.; Ragazzon, G.; Secchi, A.; Silvi, S. Redox-Switchable Calix[6]arene-Based Isomeric Rotaxanes. Chem. Eur. J. 2018, 24, 12370–12382. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Bussolati, R.; Credi, A.; Pochini, A.; Secchi, A.; Silvi, S.; Venturi, M. Rotaxanes with a Calix[6]arene Wheel and Axles of Different Length. Synthesis, Characterisation, and Photophysical and Electrochemical Properties. Tetrahedron 2008, 64, 8279–8286. [Google Scholar] [CrossRef]
- Arduini, A.; Calzavacca, F.; Pochini, A.; Secchi, A. Unidirectional Threading of Triphenylureidocalix[6]arene-Based Wheels: Oriented Pseudorotaxane Synthesis. Chem. Eur. J. 2003, 9, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Arduini, A.; Ciesa, F.; Fragassi, M.; Pochini, A.; Secchi, A. Selective Synthesis of Two Constitutionally Isomeric Oriented Calix[6]arene-Based Rotaxanes. Angew. Chem. Int. Ed. 2005, 44, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, M.; Andreoni, L.; Silvi, S.; Credi, A.; Cera, G.; Secchi, A.; Arduini, A. Selective Access to Constitutionally Identical, Orientationally Isomeric Calix[6]arene-Based [3]Rotaxanes by an Active Template Approach. Chem. Sci. 2021, 12, 6419–6428. [Google Scholar] [CrossRef]
- Arduini, A.; Bussolati, R.; Credi, A.; Secchi, A.; Silvi, S.; Semeraro, M.; Venturi, M. Toward Directionally Controlled Molecular Motions and Kinetic Intra- and Intermolecular Self-Sorting: Threading Processes of Nonsymmetric Wheel and Axle Components. J. Am. Chem. Soc. 2013, 135, 9924–9930. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. MOPAC2016 2016.
- Arduini, A.; Bussolati, R.; Credi, A.; Faimani, G.; Garaudée, S.; Pochini, A.; Secchi, A.; Semeraro, M.; Silvi, S.; Venturi, M. Towards Controlling the Threading Direction of a Calix[6]arene Wheel by Using Nonsymmetric Axles. Chem. Eur. J. 2009, 15, 3230–3242. [Google Scholar] [CrossRef]
- Burns, D.H.; Chan, H.; Miller, J.D.; Jayne, C.L.; Eichhorn, D.M. Synthesis, Modification, and Characterisation of a Family of Homologues of Exo-Calix[4]arene: Exo-[n.m.n.m]Metacyclophanes, n,m ≥ 3. J. Org. Chem. 2000, 65, 5185–5196. [Google Scholar] [CrossRef]
- Walczak, R.M.; Cowart, J.S.; Reynolds, J.R. Tethered PProDOTs: Conformationally Restricted 3,4-Propylenedioxythiophene Based Electroactive Polymers. J. Mater. Chem. 2007, 17, 254–260. [Google Scholar] [CrossRef]
- Liu, P.; Xu, J.; Yan, D.; Zhang, P.; Zeng, F.; Li, B.; Wu, S. A DT-Diaphorase Responsive Theranostic Prodrug for Diagnosis, Drug Release Monitoring and Therapy. Chem. Commun. 2015, 51, 9567–9570. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck Molecular Force Field. V. Extension of MMFF94 Using Experimental Data, Additional Computational Data, and Empirical Rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeerschd, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cester Bonati, F.; Bazzoni, M.; Baccini, C.; Zanichelli, V.; Orlandini, G.; Arduini, A.; Cera, G.; Secchi, A. Calix[6]arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules. Molecules 2023, 28, 595. https://doi.org/10.3390/molecules28020595
Cester Bonati F, Bazzoni M, Baccini C, Zanichelli V, Orlandini G, Arduini A, Cera G, Secchi A. Calix[6]arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules. Molecules. 2023; 28(2):595. https://doi.org/10.3390/molecules28020595
Chicago/Turabian StyleCester Bonati, Federica, Margherita Bazzoni, Caterina Baccini, Valeria Zanichelli, Guido Orlandini, Arturo Arduini, Gianpiero Cera, and Andrea Secchi. 2023. "Calix[6]arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules" Molecules 28, no. 2: 595. https://doi.org/10.3390/molecules28020595
APA StyleCester Bonati, F., Bazzoni, M., Baccini, C., Zanichelli, V., Orlandini, G., Arduini, A., Cera, G., & Secchi, A. (2023). Calix[6]arene-Based [3]Rotaxanes as Prototypes for the Template Synthesis of Molecular Capsules. Molecules, 28(2), 595. https://doi.org/10.3390/molecules28020595