
Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
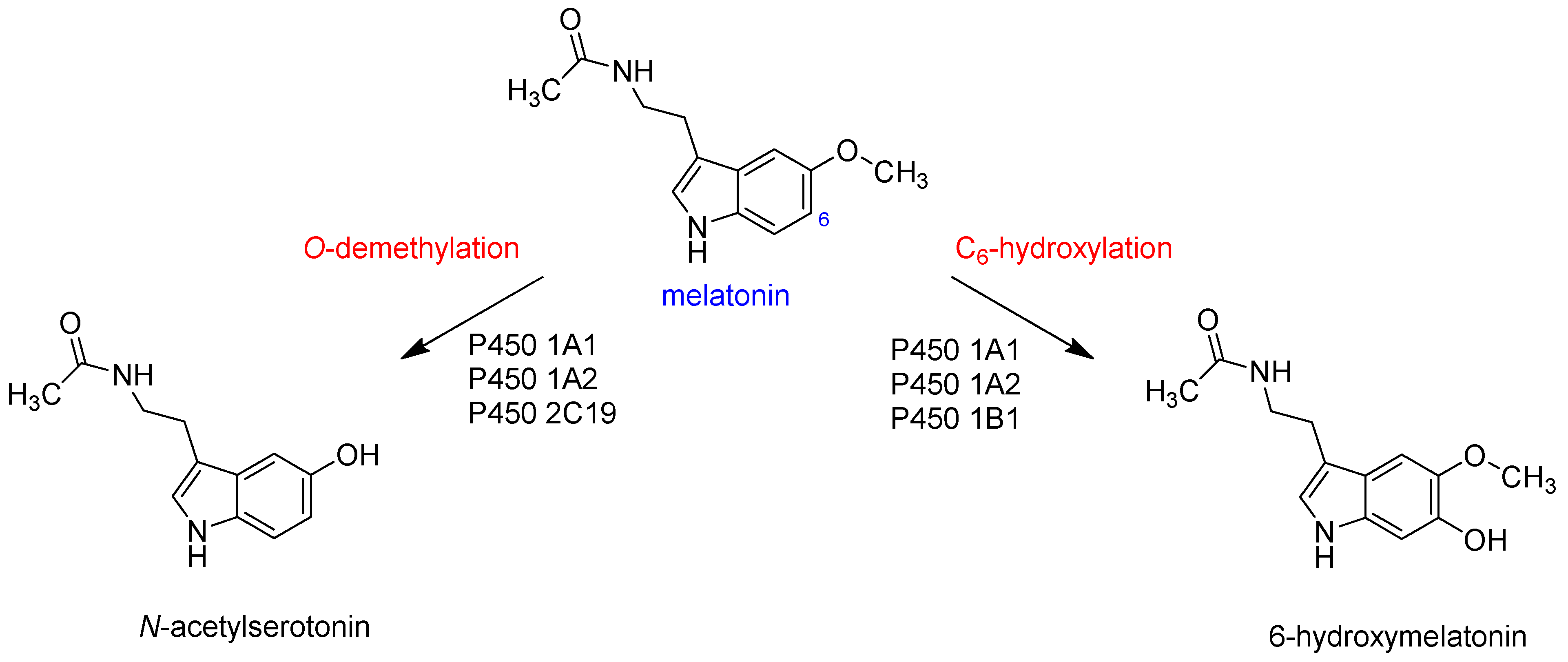
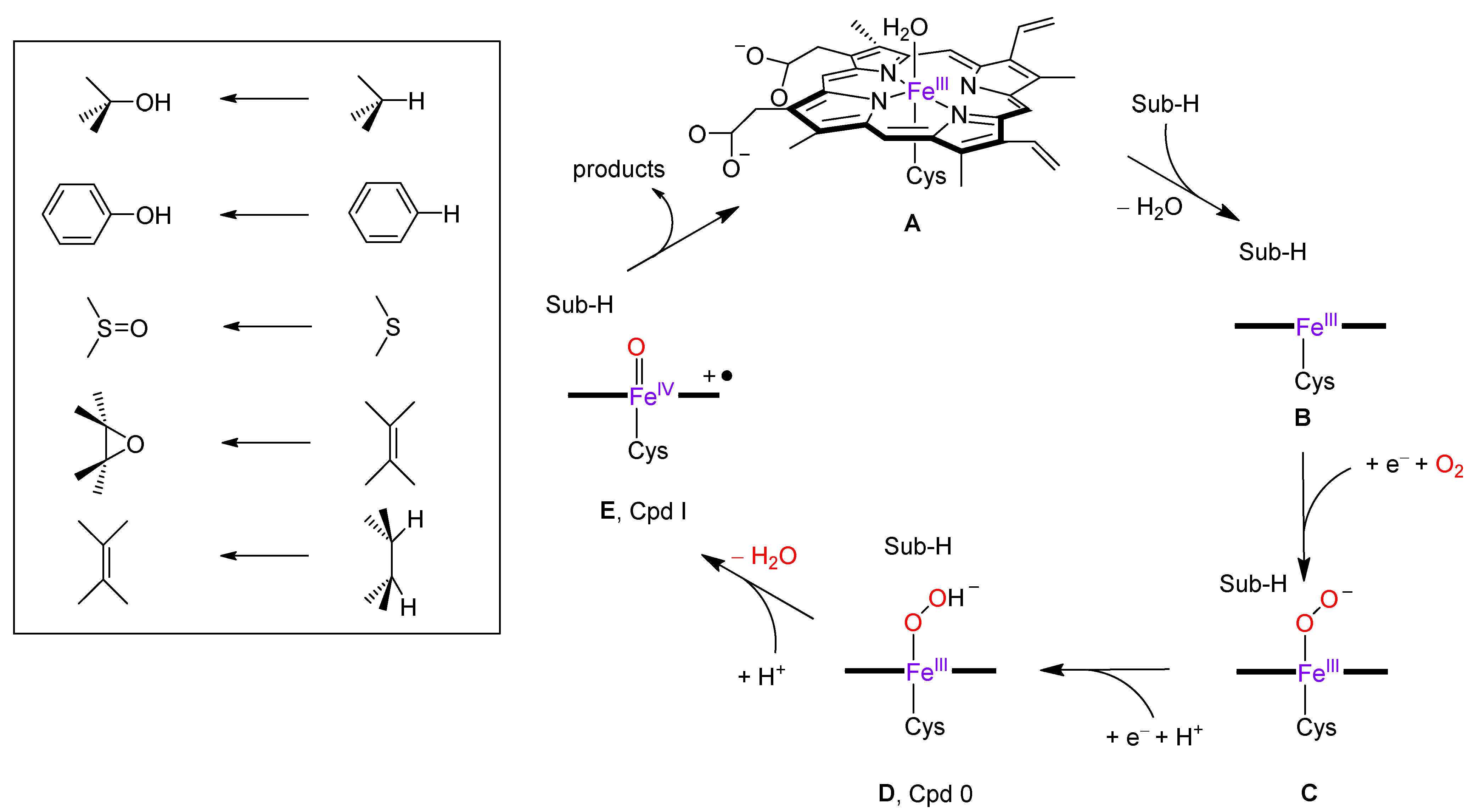
:1. Introduction
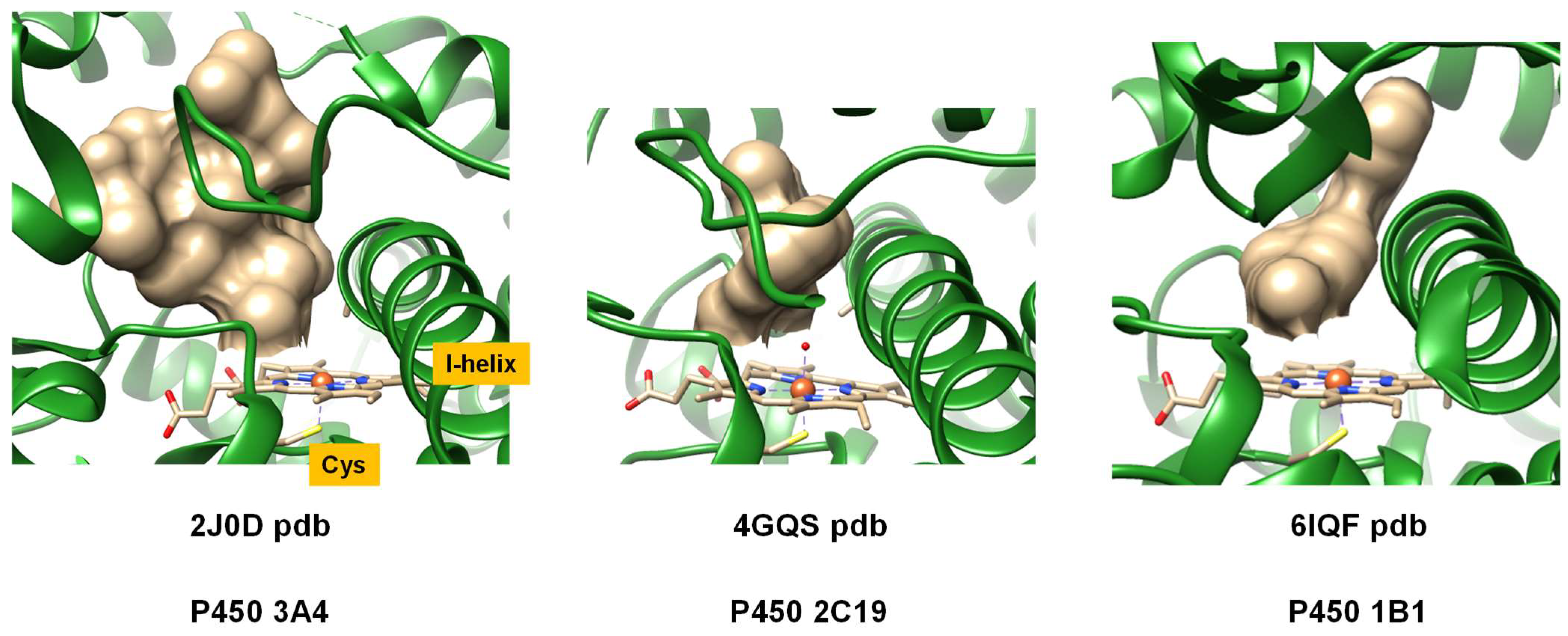
2. Results
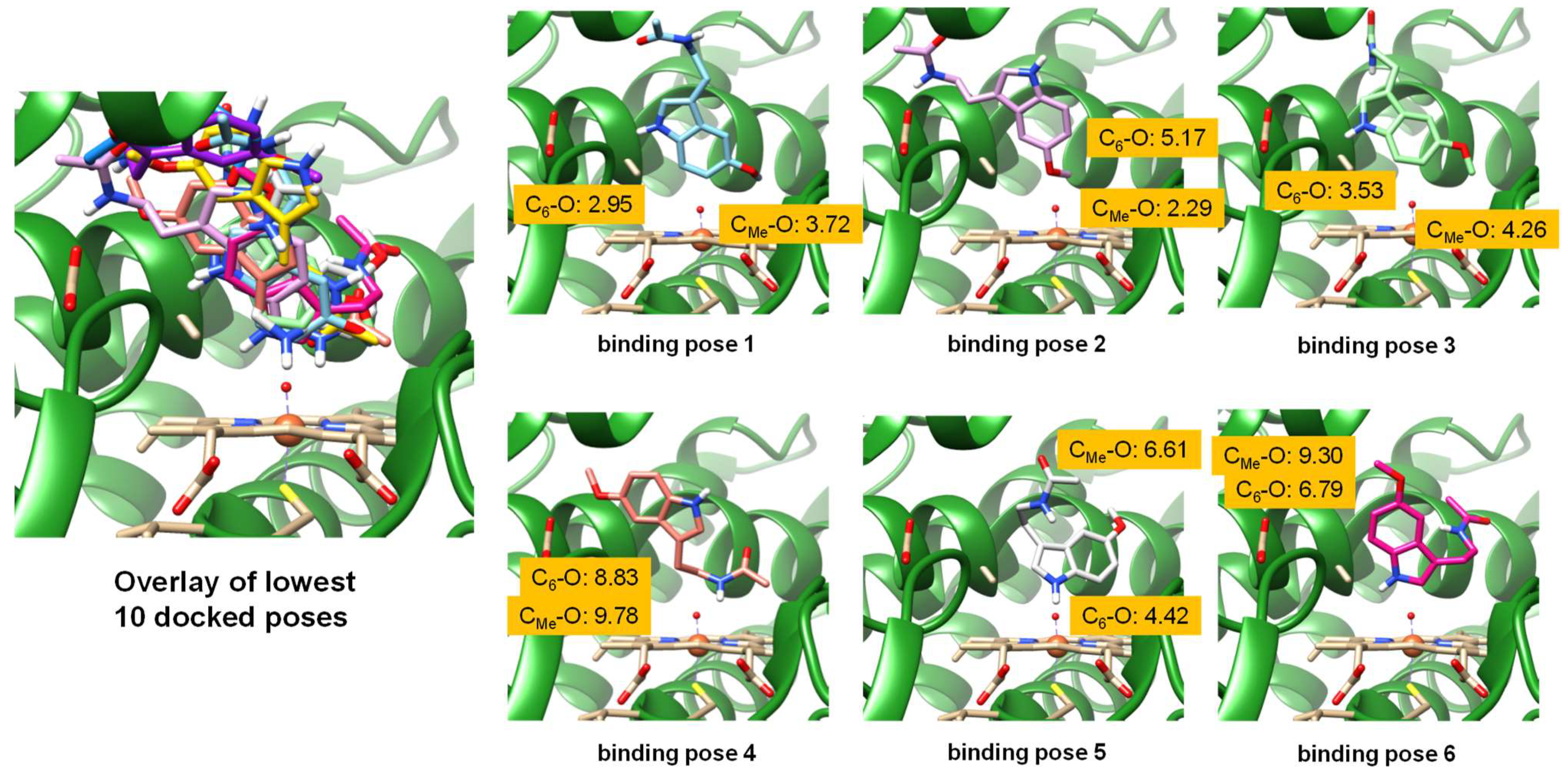
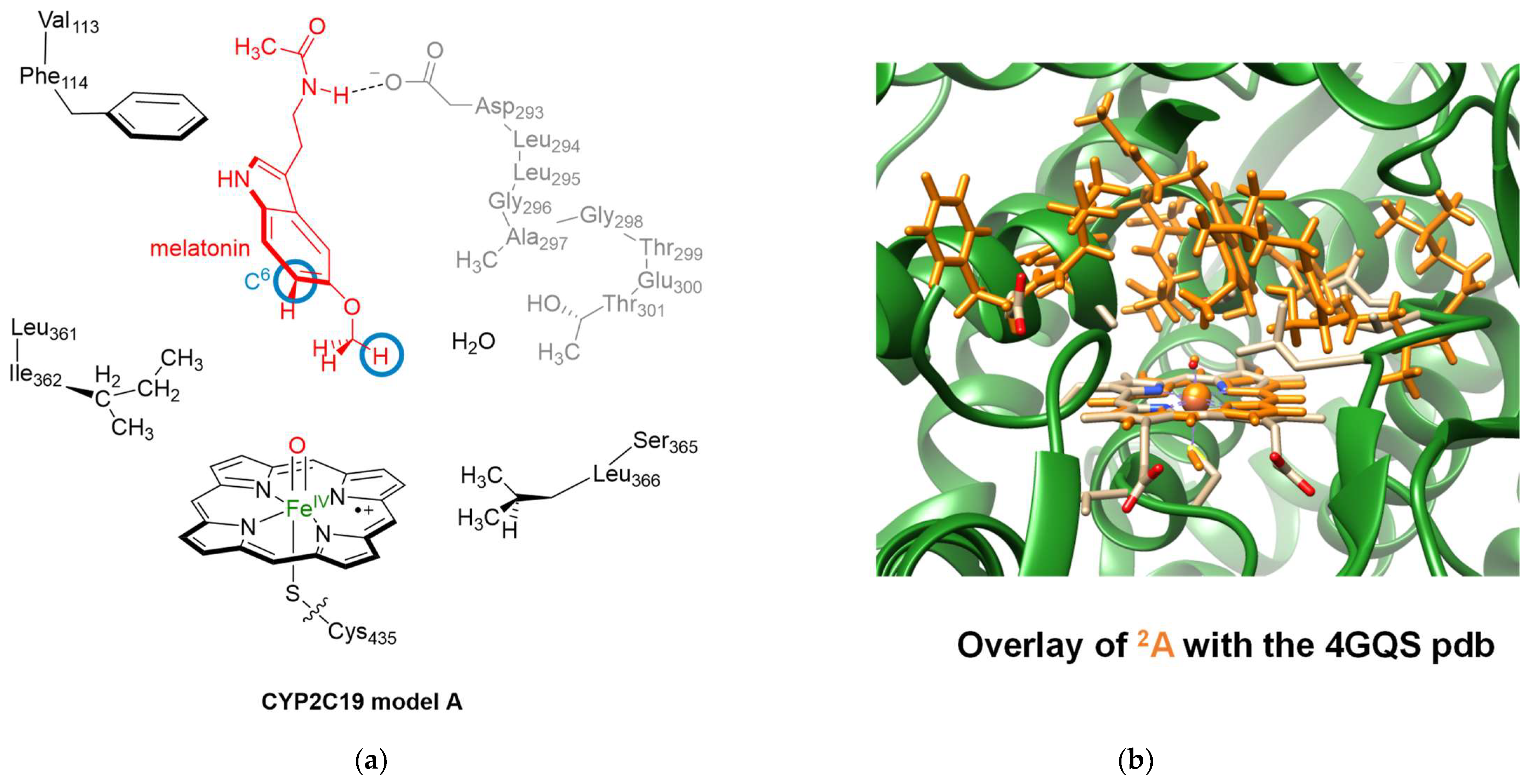
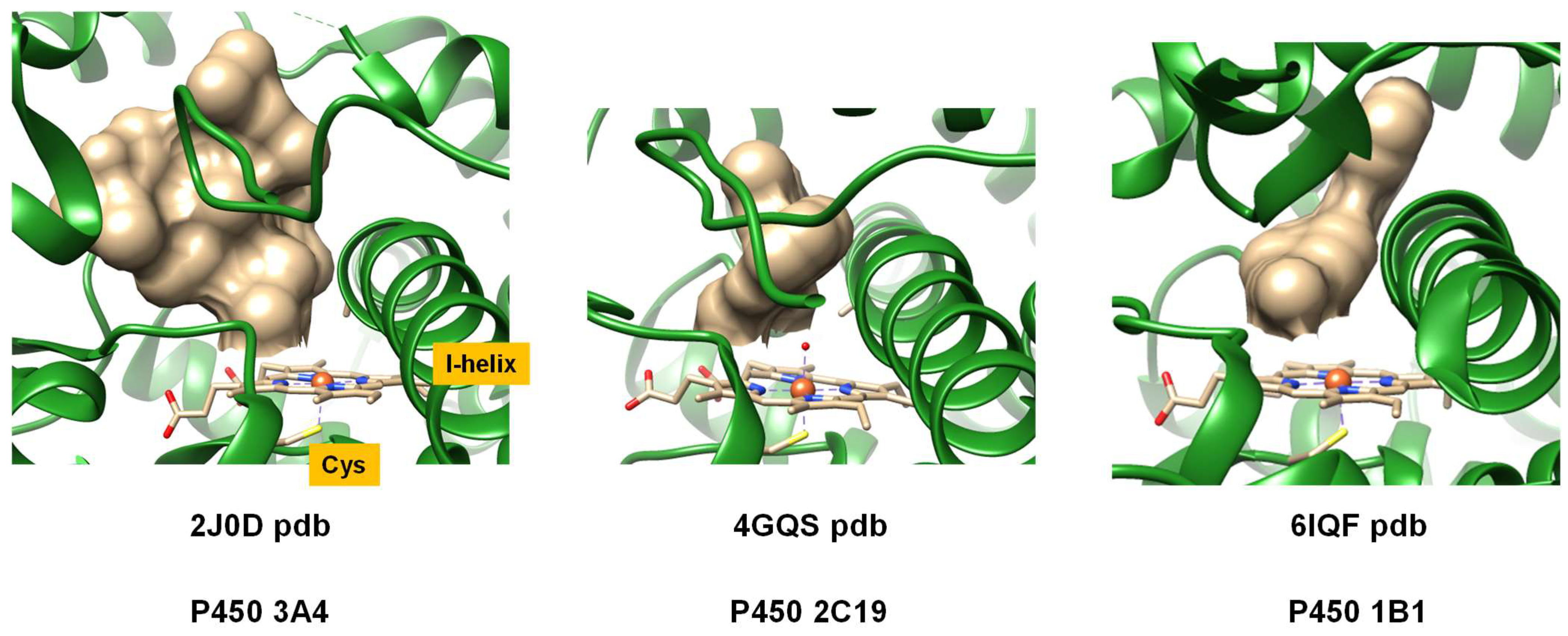
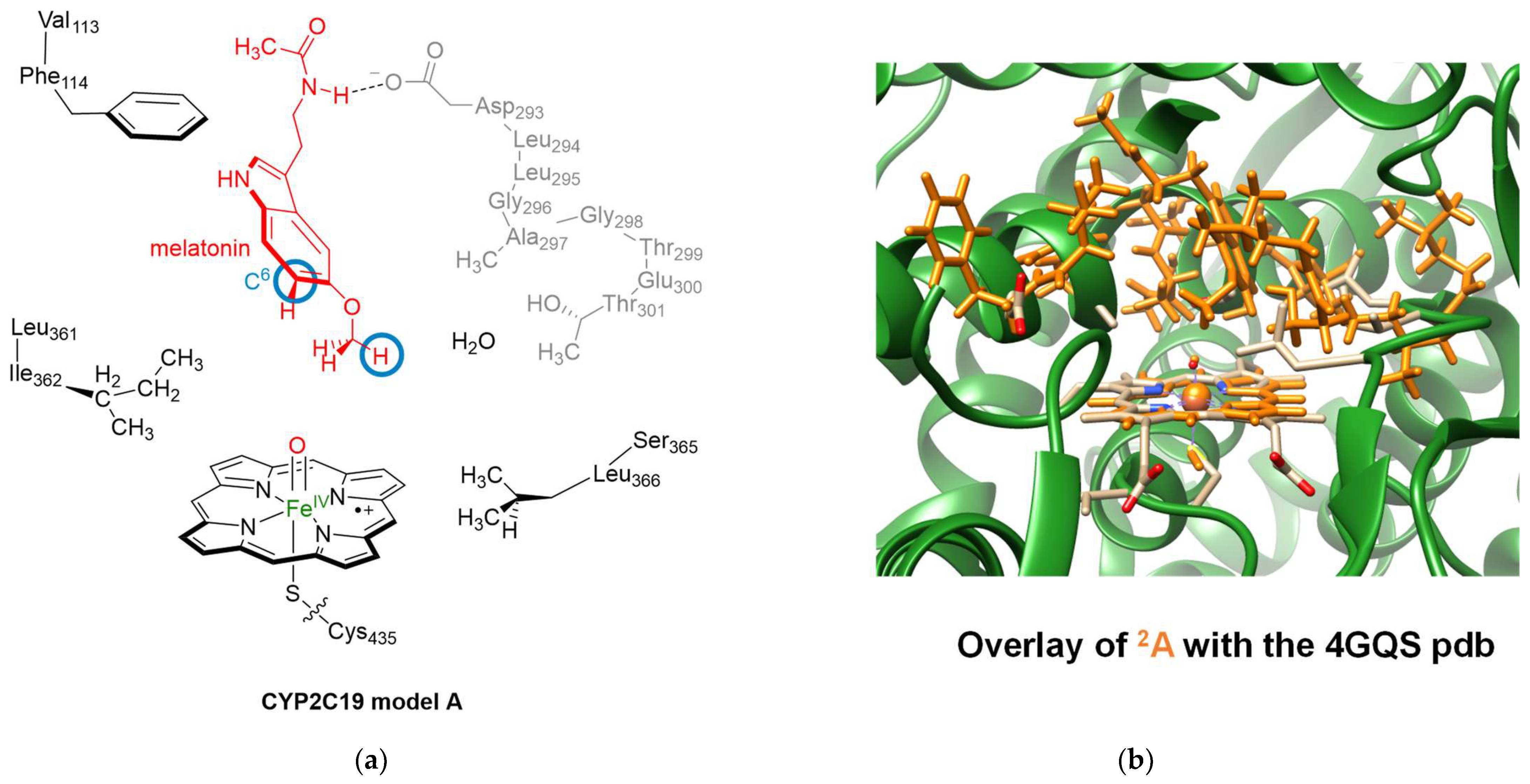
2.1. Docking Studies
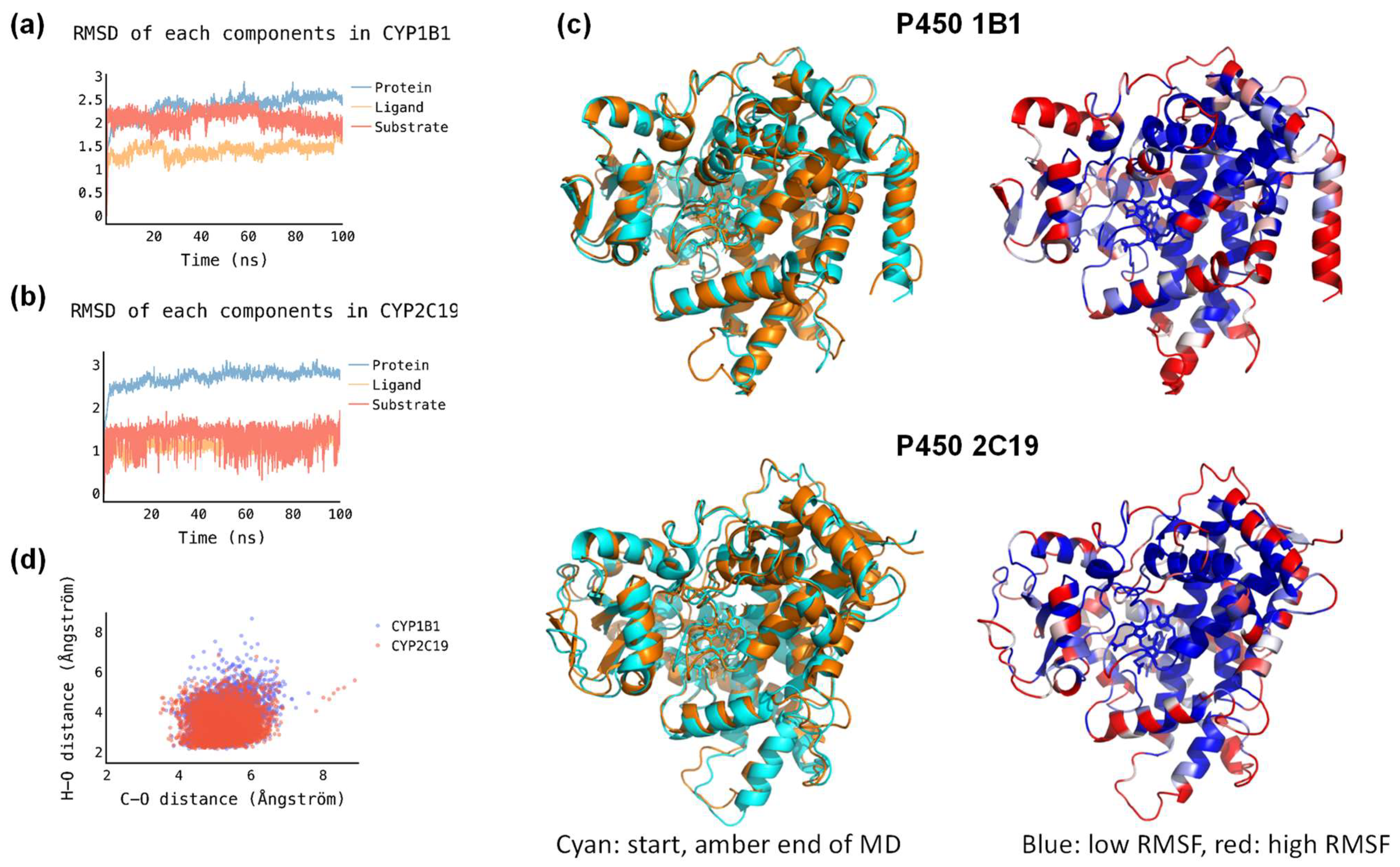
2.2. Molecular Dynamics Simulations
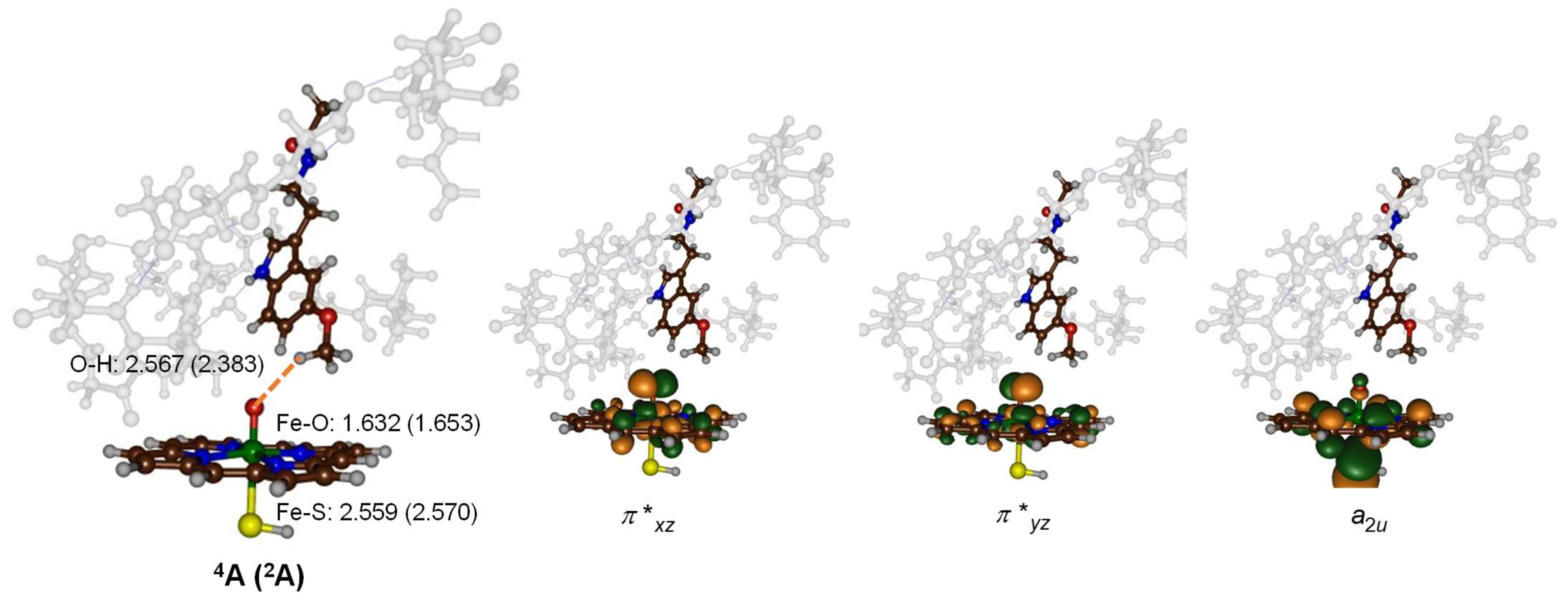
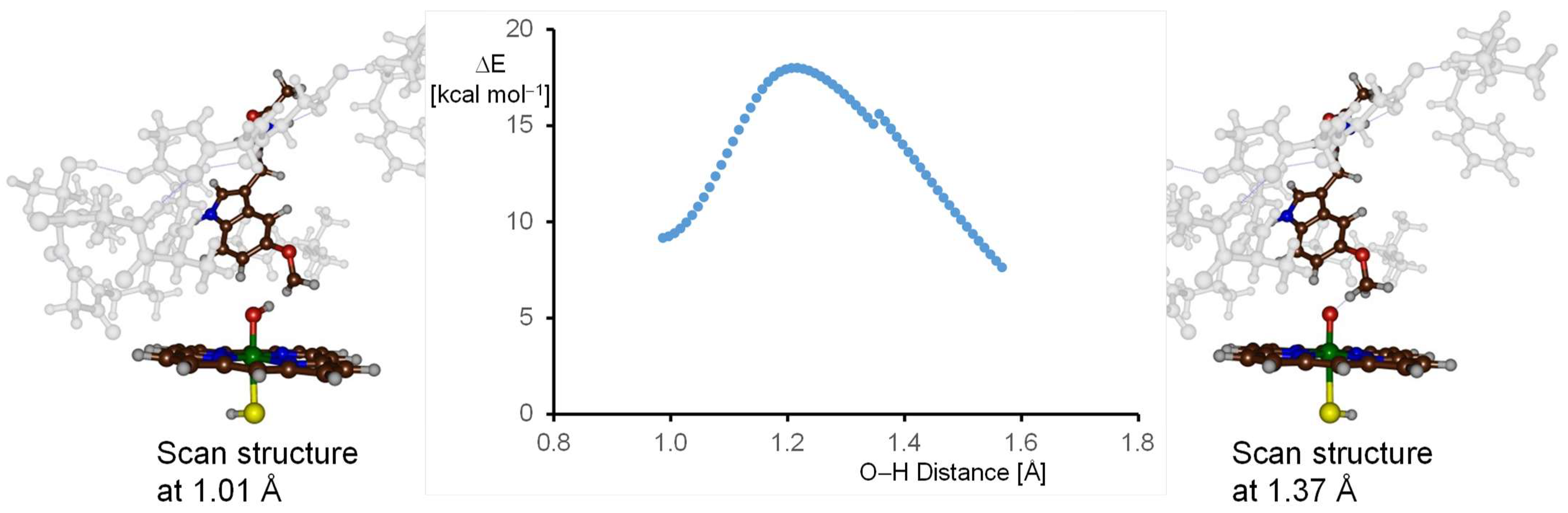
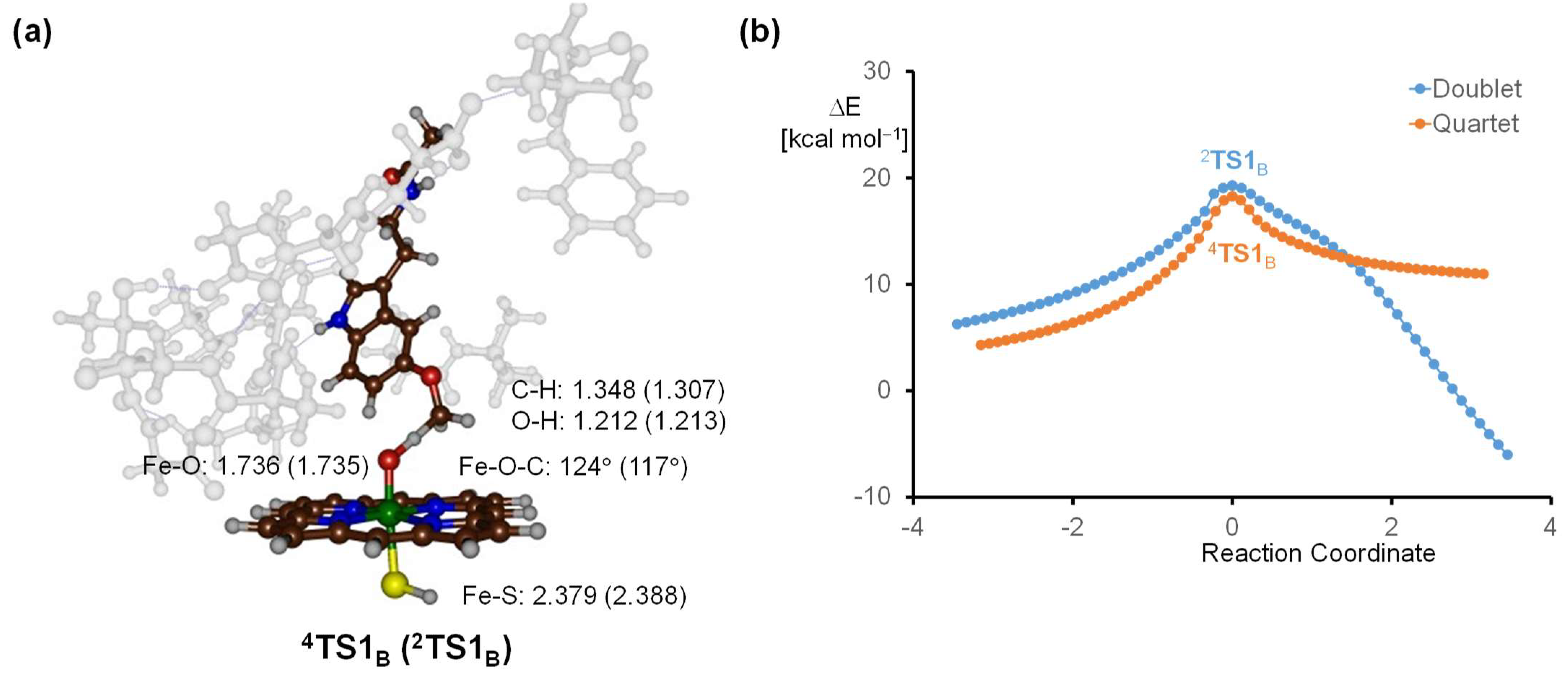
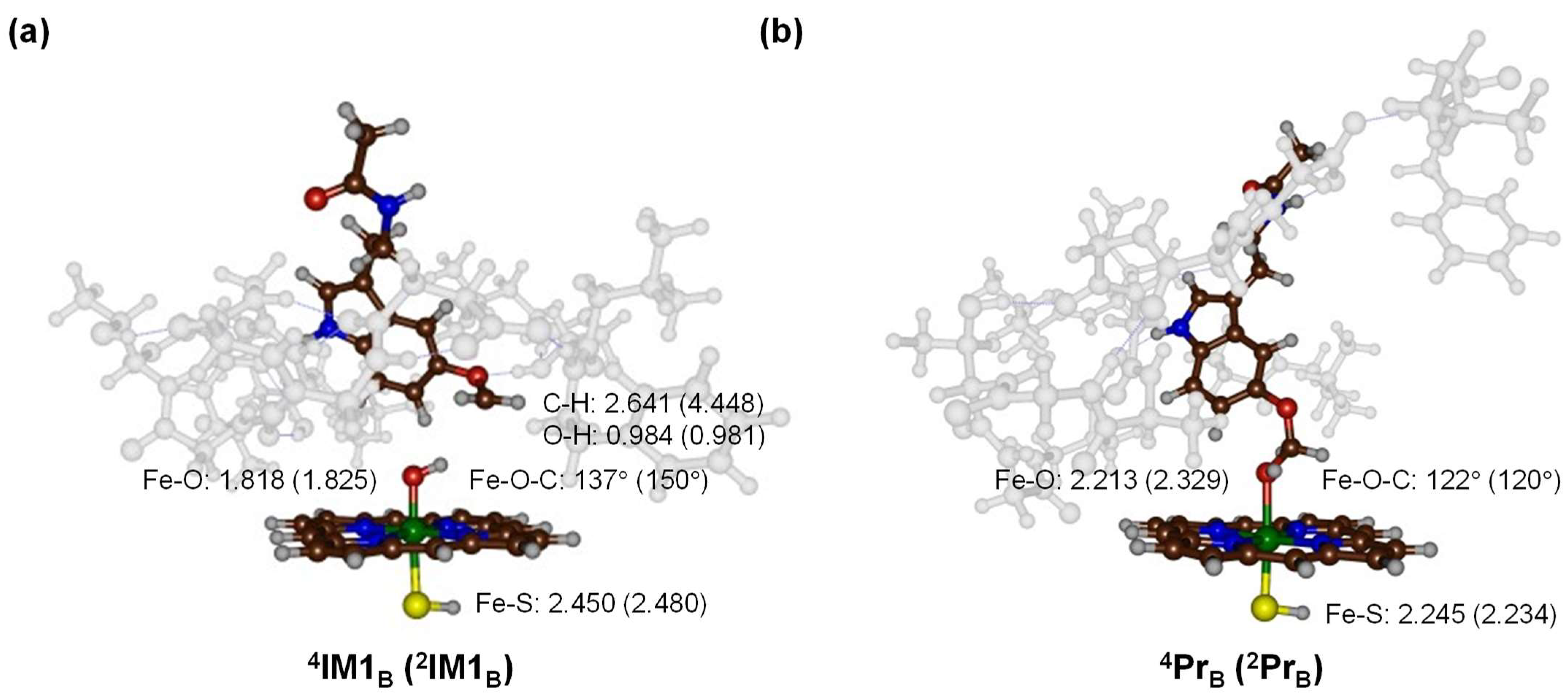
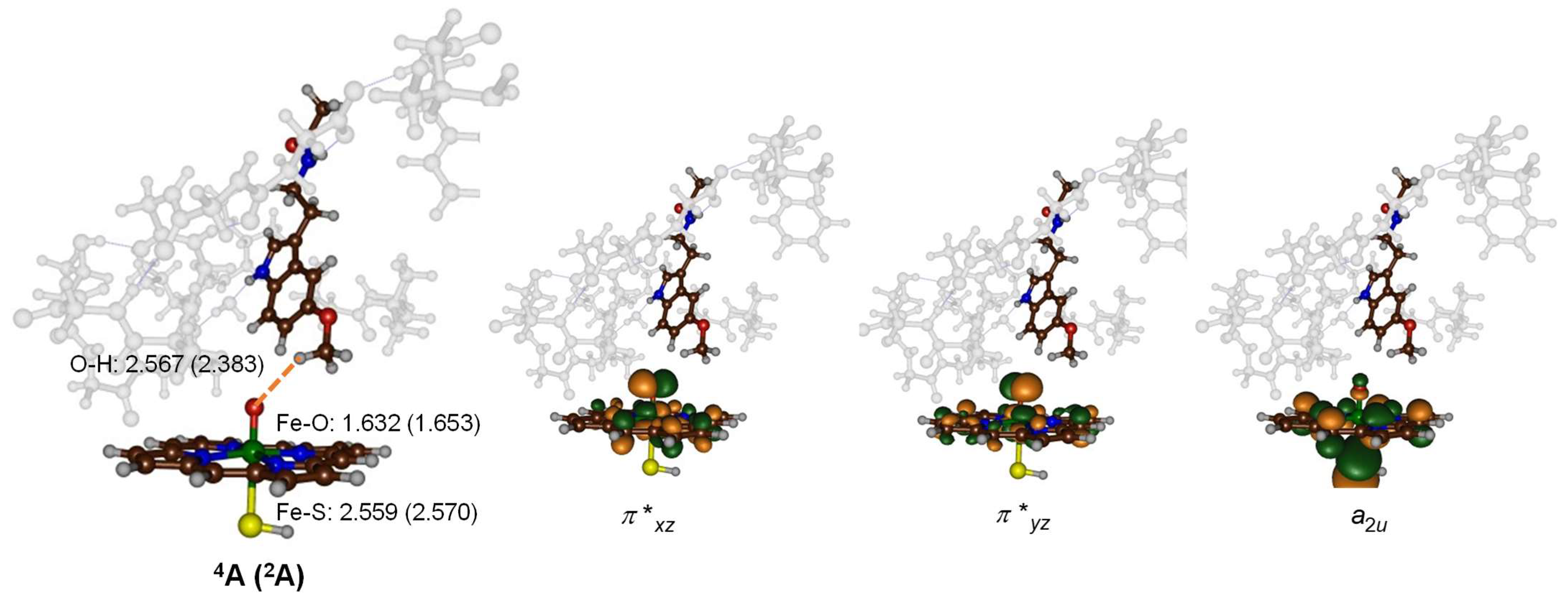
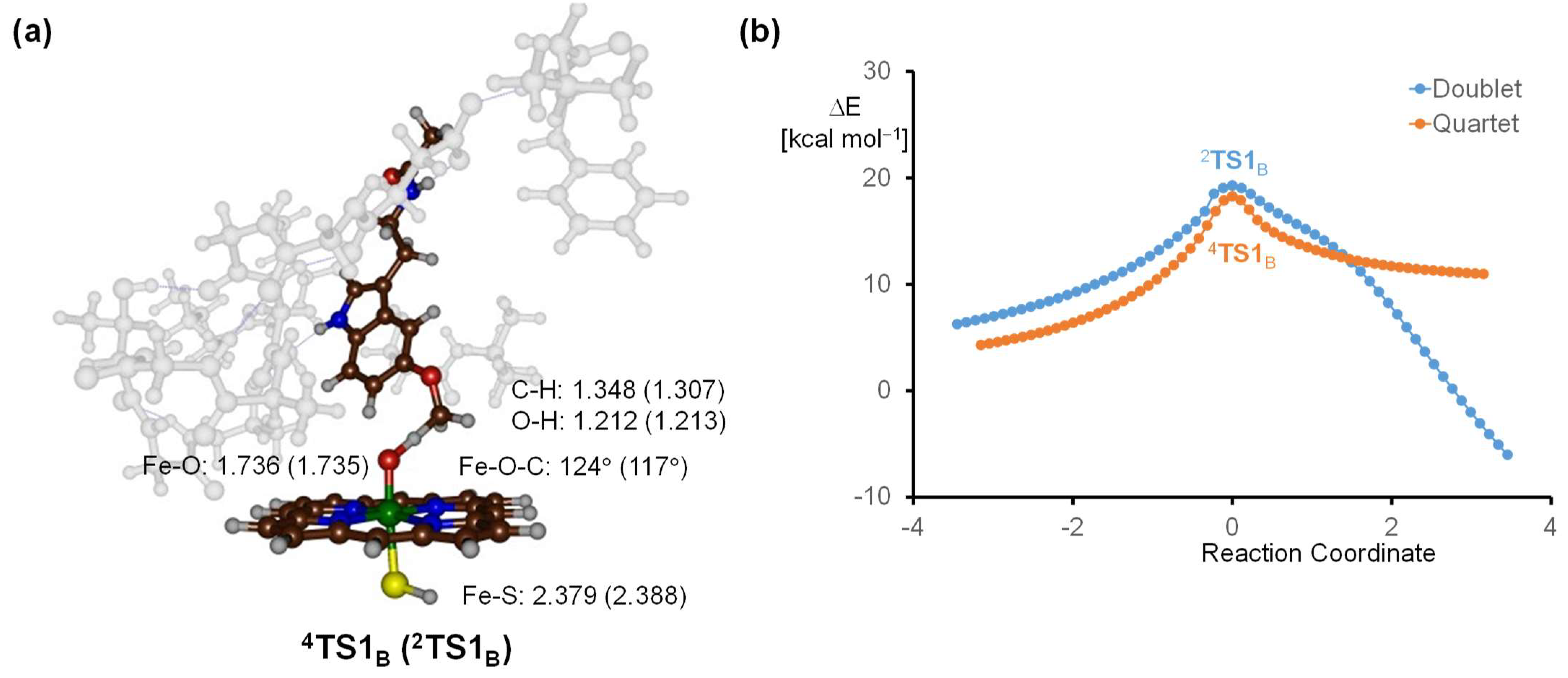
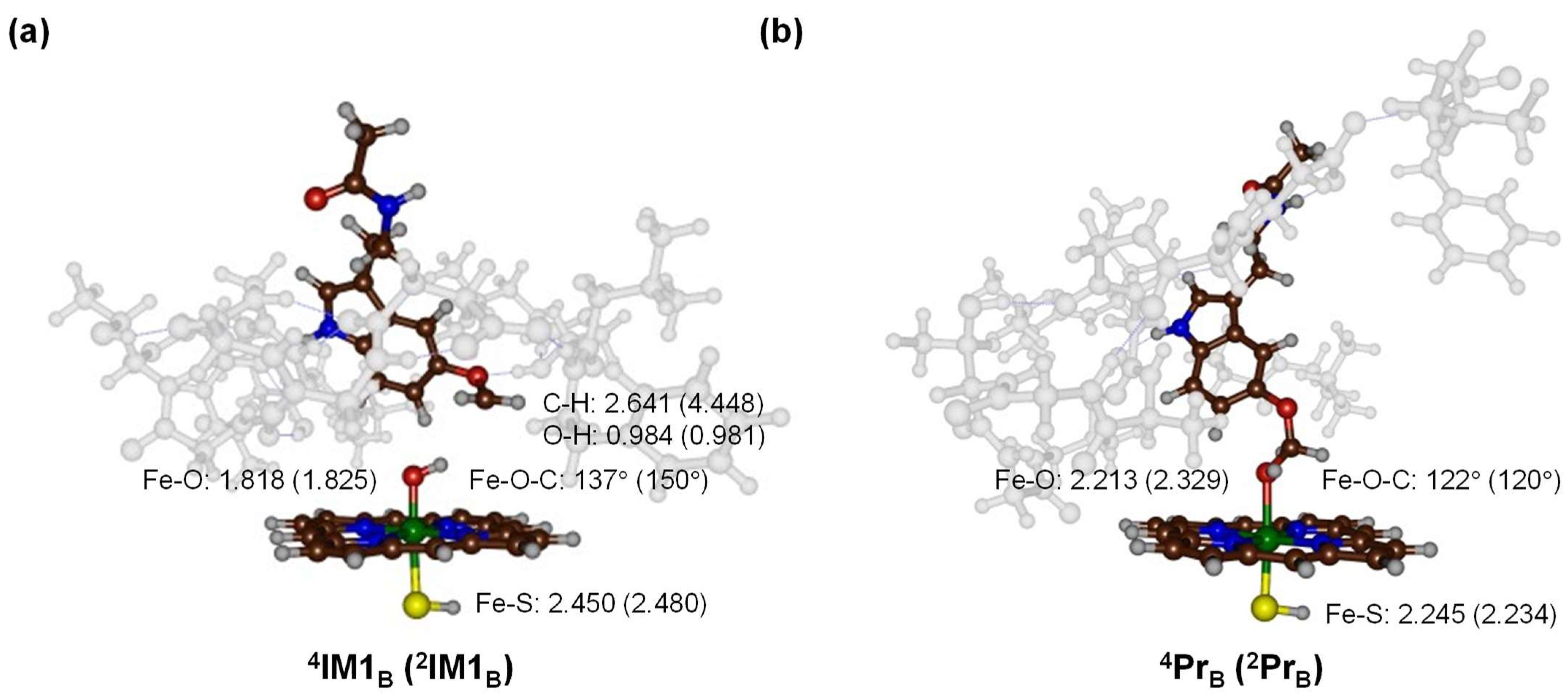
2.3. Quantum Mechanics Calculations
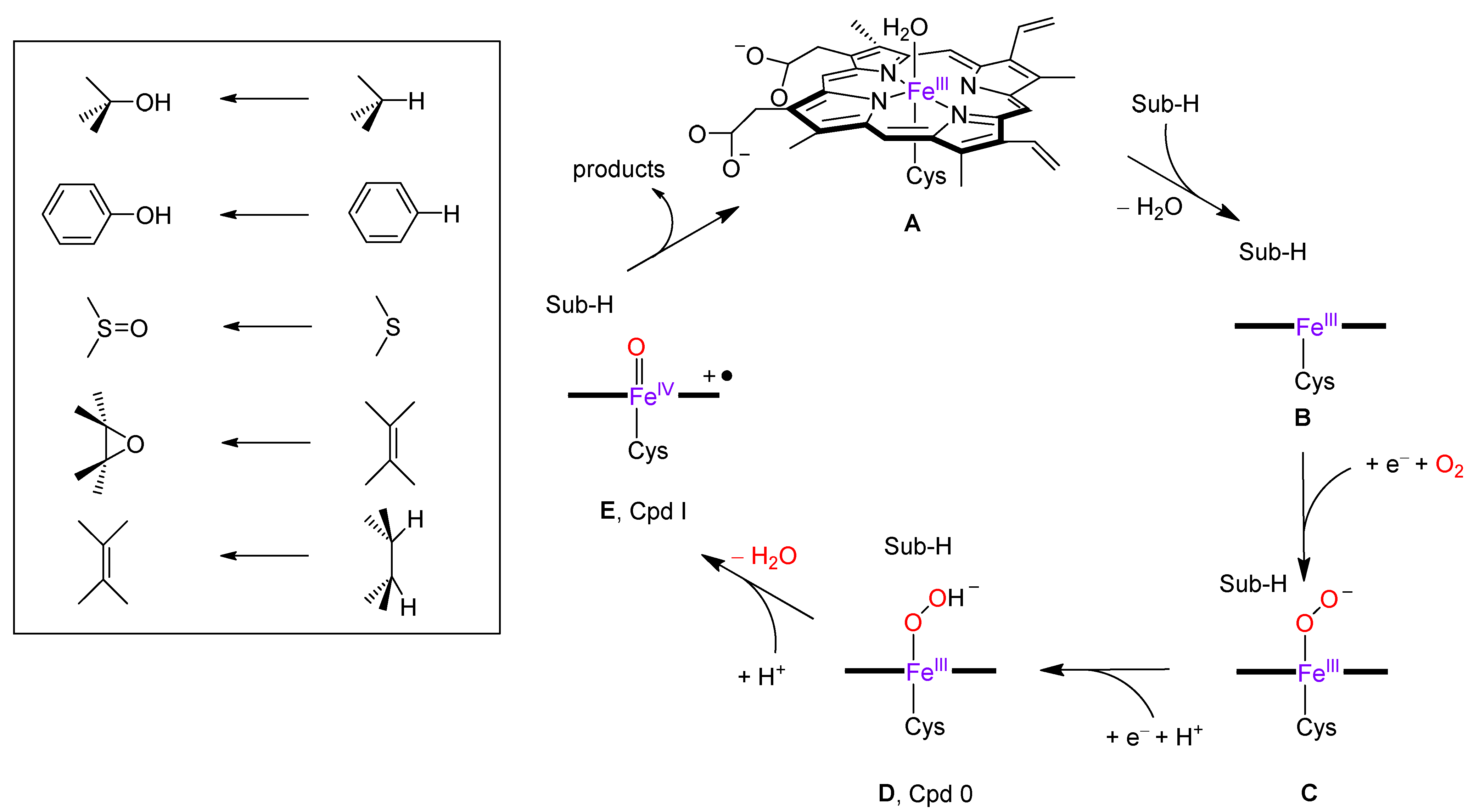
3. Discussion
4. Materials and Methods
4.1. Enzyme Set-Up and Molecular Dynamics Simulations
4.2. Cluster Model Set-Up and Quantum Chemical Calculations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Solomon, E.I.; Brunold, T.C.; Davis, M.I.; Kemsley, J.N.; Lee, S.K.; Lehnert, N.; Neese, F.; Skulan, A.J.; Yang, Y.S.; Zhou, J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100, 235–349. [Google Scholar] [CrossRef]
- Costas, M.; Mehn, M.P.; Jensen, M.P.; Que, L., Jr. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. [Google Scholar] [CrossRef]
- Abu-Omar, M.M.; Loaiza, A.; Hontzeas, N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem. Rev. 2005, 105, 2227–2252. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Kumar, D. (Eds.) Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature; Royal Society of Chemistry Publishing: Cambridge, UK, 2011. [Google Scholar]
- Martinez, S.; Hausinger, R.P. Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem. 2015, 290, 20702–20711. [Google Scholar] [CrossRef] [PubMed]
- White, M.D.; Flashman, E. Catalytic Strategies of the Non-Heme Iron Dependent Oxygenases and Their Roles in Plant Biology. Curr. Opin. Chem. Biol. 2016, 31, 126–135. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P.; Mukherjee, G.; Ali, H.S.; Sastri, C.V. Local Charge Distributions, Electric Dipole moments and Local Electric Fields Influence Reactivity Patterns and Guide Regioselectivities in α-Ketoglutarate-Dependent Nonheme Iron Dioxygenases. Acc. Chem. Res. 2022, 55, 65–74. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (t)heme-novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Grogan, G. Cytochromes P450: Exploiting Diversity and Enabling Application as Biocatalysts. Curr. Opin. Chem. Biol. 2011, 15, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Stavropoulou, E.; Pircalabioru, G.G.; Bezirtzoglou, E. The Role of Cytochromes P450 in Infection. Front. Immunol. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Dunham, N.P.; Arnold, F.H. Nature’s Machinery, Repurposed: Expanding the Repertoire of Iron-Dependent Oxygenases. ACS Catal. 2020, 10, 12239–12255. [Google Scholar] [CrossRef]
- Poulos, T.L.; Follmer, A.H. Updating the Paradigm: Redox Partner Binding and Conformational Dynamics in Cytochromes P450. Acc. Chem. Res. 2022, 55, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Wikvall, K.; Miller, W.L. Human Cytochromes P450 in Health and Disease. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120431. [Google Scholar] [CrossRef]
- Raunio, H.; Kuusisto, M.; Juvonen, R.O.; Pentikäinen, O.T. Modeling of Interactions Between Xenobiotics and Cytochrome P450 (CYP) Enzymes. Front. Pharmacol. 2015, 6, 123. [Google Scholar] [CrossRef]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Mak, P.J.; Duggal, R.; Denisov, I.G.; Gregory, M.C.; Sligar, S.G.; Kincaid, J.R. Human Cytochrome CYP17A1: The Structural Basis for Compromised Lyase Activity with 17-Hydroxyprogesterone. J. Am. Chem. Soc. 2018, 140, 7324–7331. [Google Scholar] [CrossRef]
- Nelson, D.R. The Cytochrome P450 Homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Databank. Nucl. Acids Res. 2000, 28, 235–243. [Google Scholar] [CrossRef]
- Ekroos, M.; Sjögren, T. Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. USA 2006, 103, 13682–13687. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Yamamoto, K.; Itoh, T. Design and Synthesis of Selective CYP1B1 Inhibitor via Dearomatization of α-Naphthoflavone. Bioorg. Med. Chem. 2019, 27, 285–304. [Google Scholar] [CrossRef] [PubMed]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural Characterization of Human Cytochrome P450 2C19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef]
- Leys, D.; Mowat, C.G.; McLean, K.J.; Richmond, A.; Chapman, S.K.; Walkinshaw, M.D.; Munro, A.W. Atomic structure of Mycobacterium tuberculosis CYP121 to 1.06 A reveals novel features of cytochrome P450. J. Biol. Chem. 2003, 278, 5141–5147. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef] [PubMed]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, Characterization, and C-H bond Activation Kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef]
- Mak, P.J.; Denisov, I.G. Spectroscopic Studies of the Cytochrome P450 Reaction Mechanism. Biochim. Biophys. Acta 2018, 1866, 178–204. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef]
- Dubey, K.D.; Shaik, S. Cytochrome P450: The Wonderful Nanomachine Revealed Through Dynamic Simulations of the Catalytic Cycle. Acc. Chem. Res. 2019, 52, 389–399. [Google Scholar] [CrossRef]
- Mansuy, D. A Brief History of the Contribution of Metalloporphyrin Models to Cytochrome P450 Chemistry and Oxidation Catalysis. Comptes Rendus Chim. 2007, 10, 392–413. [Google Scholar] [CrossRef]
- Sevrioukova, I.F.; Hazzard, J.T.; Tollin, G.; Poulos, T.L. Laser Flash Induced Electron Transfer in P450cam Monooxygenase: Putidaredoxin Reductase-Putidaredoxin Interaction. Biochemistry 2001, 40, 10592–10600. [Google Scholar] [CrossRef]
- Girvan, H.M.; Heyes, D.J.; Scrutton, N.S.; Munro, A.W. Laser Photoexcitation of NAD(P)H Induces Reduction of P450BM3 Heme Domain on the Microsecond Time Scale. J. Am. Chem. Soc. 2007, 129, 6647–6653. [Google Scholar] [CrossRef]
- Davydov, R.; Im, S.; Shanmugam, M.; Gunderson, W.A.; Pearl, N.M.; Hoffman, B.M.; Waskel, L. Role of the Proximal Cysteine Hydrogen Bonding Interaction in Cytochrome P450 2B4 Studied by Cryoreduction, Electron Paramagnetic Resonance, and Electron-Nuclear Double Resonance Spectroscopy. Biochemistry 2016, 55, 869–883. [Google Scholar] [CrossRef]
- Suzuki, H.; Inabe, K.; Shirakawa, Y.; Umezawa, N.; Kato, N.; Higuchi, T. Role of Thiolate Ligand in Spin State and Redox Switching in the Cytochrome P450 Catalytic Cycle. Inorg. Chem. 2017, 56, 4245–4248. [Google Scholar] [CrossRef]
- Katariya, M.M.; Snee, M.; Tunnicliffe, R.B.; Kavanagh, M.E.; Boshoff, H.I.M.; Amadi, C.N.; Levy, C.W.; Munro, A.W.; Abell, C.; Leys, D.; et al. Structure Based Discovery of Inhibitors of CYP125 and CYP142 from Mycobacterium tuberculosis. Chem. Eur. J. 2023, 29, e202203868. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Hirao, H.; de Visser, S.P.; Zheng, J.; Wang, D.; Thiel, W.; Shaik, S. New Features in the Catalytic Cycle of Cytochrome P450 During the Formation of Compound I from Compound 0. J. Phys. Chem. B 2005, 109, 19946–19951. [Google Scholar] [CrossRef] [PubMed]
- Davydov, R.; Gilep, A.A.; Strushkevich, N.V.; Usanov, S.A.; Hoffman, B.M. Compound I Is the Reactive Intermediate in the First Monooxygenation Step During Conversion of Chloresterol to Pregnenolone by Cytochrome P450scc: EPR/ENDOR/Cryoreduction/Annealing Studies. J. Am. Chem. Soc. 2012, 134, 17149–17156. [Google Scholar] [CrossRef]
- Mittra, K.; Green, M.T. Reduction Potentials of P450 Compounds I and II: Insight into the Thermodynamics of C-H Bond Activation. J. Am. Chem. Soc. 2019, 141, 5504–5510. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Nemec, J.; Schuetz, E.G.; Schuetz, J.D.; Gonzalez, F.J.; Korzekwa, K.R. O-Demethylation of Epipodophyllotoxins Is Catalyzed by Human Cytochrome P450 3A4. Mol. Pharmacol. 1994, 45, 352–358. [Google Scholar] [PubMed]
- Hagel, J.; Facchini, P. Biochemistry and Occurrence of O-Demethylation in Plant Metabolism. Front. Physiol. 2010, 1, 14. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. [Google Scholar] [CrossRef]
- Butler, C.F.; Peet, C.; Mason, A.E.; Voice, M.W.; Leys, D.; Munro, A.W. Key Mutations Alter the Cytochrome P450 BM3 Conformational Landscape and Remove Inherent Substrate Bias. J. Biol. Chem. 2013, 288, 25387–25399. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, J.D.; Dong, L.-B.; Zhang, X.; Renata, H.; Shen, B. Cytochrome P450-Catalyzed Hydroxylation Initiating Ether Formation in Platensimycin Biosynthesis. J. Am. Chem. Soc. 2018, 140, 12349–12353. [Google Scholar] [CrossRef] [PubMed]
- Mallinson, S.J.B.; Machovina, M.M.; Silveira, R.L.; Garcia-Borràs, M.; Gallup, N.; Johnson, C.W.; Allen, M.D.; Skaf, M.S.; Crowley, M.F.; Neidle, E.L.; et al. A Promiscuous Cytochrome P450 Aromatic O-Demethylase for Lignin Bioconversion. Nat. Commun. 2018, 9, 2487. [Google Scholar] [CrossRef] [PubMed]
- Pickl, M.; Kurakin, S.; Cantú Reinhard, F.G.; Schmid, P.; Pöcheim, A.; Winkler, C.K.; Kroutil, W.; de Visser, S.P.; Faber, K. Mechanistic Studies of Fatty Acid Activation by CYP152 Peroxygenases Reveal Unexpected Desaturase Activity. ACS Catal. 2019, 9, 565–577. [Google Scholar] [CrossRef]
- Morita, I.; Mori, T.; Abe, I. Enzymatic Formation of Indolactam Scaffold by C-N Bond-Forming Cytochrome P450 Oxidases in Teleocidin Biosynthesis. Chem. Eur. J. 2021, 27, 2963–2972. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P.; Fekry, M.I. Methylene Oxidation of Alkyl Sulfates by Cytochrome P450BM-3 and a Role for Conformational Selection in Substrate Recognition. ACS Catal. 2020, 10, 5008–5022. [Google Scholar] [CrossRef]
- Wang, J.; Huang, Q.; Peng, W.; Wu, P.; Yu, D.; Chen, B.; Wang, B.; Reetz, M.T. P450-BM3-Catalyzed Sulfoxidation versus Hydroxylation: A Common or Two Different Catalytically Active Species? J. Am. Chem. Soc. 2020, 142, 2068–2073. [Google Scholar] [CrossRef]
- Podgorski, M.N.; Coleman, T.; Churchman, L.R.; Bruning, J.B.; De Voss, J.J.; Bell, S.G. Investigating the Active Oxidants Involved in Cytochrome P450 Catalyzed Sulfoxidation Reactions. Chem. Eur. J. 2022, 28, e202202428. [Google Scholar] [CrossRef]
- Coleman, T.; Kirk, A.M.; Lee, J.H.Z.; Doherty, D.Z.; Bruning, J.B.; Krenske, E.H.; De Voss, J.J.; Bell, S.G. Different Geometric Requirements for Cytochrome P450-Catalyzed Aliphatic Versus Aromatic Hydroxylation Results in Chemoselective Oxidation. ACS Catal. 2022, 12, 1258–1267. [Google Scholar] [CrossRef]
- Espinoza, R.V.; Maskeri, M.A.; Turlik, A.; Nangia, A.; Khatri, Y.; Montgomery, J.; Houk, K.N.; Sherman, D.H. Epoxidation and Late-Stage C–H Functionalization by P450 TamI Are Mediated by Variant Heme-Iron Oxidizing Species. ACS Catal. 2022, 12, 3731–3742. [Google Scholar] [CrossRef]
- McLean, K.J.; Warman, A.J.; Seward, H.E.; Marshall, K.R.; Girvan, H.M.; Cheeseman, M.J.; Waterman, M.R.; Munro, A.W. Biophysical Characterization of the Sterol Demethylase P450 from Myobacterium tuberculosis, Its Cognate Ferrodoxin, and Their Interactions. Biochemistry 2006, 45, 8427–8443. [Google Scholar] [CrossRef] [PubMed]
- Matthews, S.; Belcher, J.D.; Tee, K.L.; Girvan, H.M.; McLean, K.J.; Rigby, S.E.J.; Levy, C.W.; Leys, D.; Parker, D.A.; Blankley, R.T.; et al. Catalytic Determinants of Alkene Production by the Cytochrome P450 Peroxygenase OleTJE. J. Biol. Chem. 2017, 292, 5128–5143. [Google Scholar] [CrossRef] [PubMed]
- Taxak, N.; Kalra, S.; Bharatam, P.V. Mechanism-Based Inactivation of Cytochromes by Furan Epoxide: Unraveling the Molecular Mechanism. Inorg. Chem. 2013, 52, 13496–13508. [Google Scholar] [CrossRef] [PubMed]
- Slessor, K.E.; Stok, J.E.; Chow, S.; De Voss, J.J. Significance of Protein-Substrate Hydrogen Bonding for the Selectivity of P450-Catalyzed Oxidations. Chem. Eur. J. 2019, 25, 4149–4155. [Google Scholar] [CrossRef]
- Quan, Z.; Awakawa, T.; Wang, D.; Hu, Y.; Abe, I. Multidomain P450 Epoxidase and a Terpene Cyclase from the Ascochlorin Biosynthetic Pathway in Fusarium sp. Org. Lett. 2019, 21, 2330–2334. [Google Scholar] [CrossRef]
- Jiang, G.; Zhang, Y.; Powell, M.M.; Hylton, S.M.; Hiller, N.W.; Loria, R.; Ding, Y. A Promiscuous Cytochrome P450 Hydroxylates Aliphatic and Aromatic C-H Bonds of Aromatic 2,5-Diketopiperazines. ChemBioChem 2019, 20, 1068–1077. [Google Scholar] [CrossRef]
- Alkhalaf, L.M.; Barry, S.M.; Rea, D.; Gallo, A.; Griffiths, D.; Lewandowski, J.R.; Fulop, V.; Challis, G.L. Binding of Distinct Substrate Conformations Enables Hydroxylation of Remote Sites in Thaxtomin D by Cytochrome P450 TxtC. J. Am. Chem. Soc. 2019, 141, 216–222. [Google Scholar] [CrossRef]
- Schiavini, P.; Cheong, K.J.; Moiterssier, N.; Auclair, K. Active Site Crowding of Cytochrome P450 3A4 As a Strategy to Alter Its Selectivity. ChemBioChem 2017, 18, 248–252. [Google Scholar] [CrossRef]
- Kawakami, N.; Shoji, O.; Watanabe, Y. Direct Hydroxylation of Primary Carbons in Small Alkanes by Wild-Type Cytochrome P450BM3 Containing Perfluorocarboxylic Acids as Decoy Molecules. Chem. Sci. 2013, 4, 2344–2348. [Google Scholar] [CrossRef]
- Gregory, M.C.; Denisov, I.; Grinkova, Y.V.; Khatri, Y.; Sligar, S.G. Kinetic Solvent Isotope Effect in Human P450 CYP17A1-Mediated Androgen Formation: Evidence for a Reactive Peroxoanion Intermediate. J. Am. Chem. Soc. 2013, 135, 16245–16247. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shafer, B.M.; Demars, M.D.; Stern, H.A.; Fasan, R. Controlled Oxidation of Remote sp3 C-H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J. Am. Chem. Soc. 2012, 134, 18695–18704. [Google Scholar] [CrossRef]
- Cantú Reinhard, F.G.; Sainna, M.A.; Upadhyay, P.; Balan, G.A.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A Systematic Account on Aromatic Hydroxylation by a Cytochrome P450 Model Compound I: A Low-Pressure Mass Spectrometry and Computational Study. Chem. Eur. J. 2016, 22, 18608–18619. [Google Scholar] [CrossRef]
- Zhao, Y.; Marschall, E.; Treisman, M.; McKay, A.; Padva, L.; Crüsemann, M.; Nelson, D.R.; Steer, D.L.; Schittenhelm, R.B.; Tailhades, J.; et al. Cytochrome P450Blt Enables Versatile Peptide Cyclisation to Generate Histidine- and Tyrosine-Containing Crosslinked Tripeptide Building Blocks. Angew. Chem. Int. Ed. 2022, 61, e202204957. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug Metabolism by Cytochrome P450 Enzymes: What Distinguishes the Pathways Leading to Substrate Hydroxylation Over Desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef]
- Escribano, B.; Colín-González, A.; Santamaria, A.; Túnez, I. The Role of Melatonin in Multiple Sclerosis, Huntington’s Disease and Cerebral Ischemia. CNS Neurol. Disord. Drug Targets 2014, 13, 1096–1119. [Google Scholar] [CrossRef]
- Slominski, A.T.; Semak, I.; Fischer, T.W.; Kim, T.-K.; Kleszczyński, K.; Hardeland, R.; Reiter, R.J. Metabolism of Melatonin in the Skin: Why Is It Important? Exp. Dermatol. 2017, 26, 563–568. [Google Scholar] [CrossRef]
- Hardeland, R. Taxon- and Site-Specific Melatonin Catabolism. Molecules 2017, 22, 2015. [Google Scholar] [CrossRef]
- Salehi, B.; Sharopov, F.; Fokou, P.V.T.; Kobylinska, A.; de Jonge, L.; Tadio, K.; Sharifi-Rad, J.; Posmyk, M.M.; Martorell, M.; Martins, N.; et al. Melatonin in Medicinal and Food Plants: Occurrence, Bioavailability, and Health Potential for Humans. Cells 2019, 8, 681. [Google Scholar] [CrossRef]
- Ma, X.; Idle, J.; Krausz, K.; Gonzalez, F. Metabolism of Melatonin by Human Cytochromes P450. Drug Metab. Dispos. 2005, 33, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Schyman, P.; Usharani, D.; Wang, Y.; Shaik, S. Brain Chemistry: How Does P450 Catalyze the O-Demethylation Reaction of 5-Methoxytryptamine to Yield Serotonin? J. Phys. Chem. B 2010, 114, 7078–7089. [Google Scholar] [CrossRef] [PubMed]
- Mokkawes, T.; Lim, Z.Q.; de Visser, S.P. Mechanism of Melatonin Metabolism by CYP1A1. What Determines the Bifurcation Pathways of Hydroxylation Versus Deformylation? J. Phys. Chem. B 2022, 126, 9591–9606. [Google Scholar] [CrossRef] [PubMed]
- Mokkawes, T.; de Visser, S.P. Melatonin Activation by Cytochrome P450 Isozymes. How Does CYP1A2 Compare to CYP1A1? Int. J. Mol. Sci. 2023, 24, 3651. [Google Scholar] [CrossRef]
- De Visser, S.P. Second-Coordination Sphere Effects on Selectivity and Specificity of Heme and Nonheme Iron Enzymes. Chem. Eur. J. 2020, 26, 5308–5327. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ghafoor, S.; Mansha, A.; de Visser, S.P. Selective Hydrogen Atom Abstraction from Dihydroflavonol by a Nonheme Iron Center Is the Key Step in the Enzymatic Flavonol Synthesis and Avoids Byproducts. J. Am. Chem. Soc. 2019, 141, 20278–20292. [Google Scholar] [CrossRef]
- Louka, S.; Barry, S.M.; Heyes, D.J.; Mubarak, M.Q.E.; Ali, H.S.; Alkhalaf, L.M.; Munro, A.W.; Scrutton, N.S.; Challis, G.L.; de Visser, S.P. The Catalytic Mechanism of Aromatic Nitration by Cytochrome P450 TxtE: Involvement of a Ferric-PeroxyNitrite Intermediate. J. Am. Chem. Soc. 2020, 142, 15764–15779. [Google Scholar] [CrossRef]
- Himo, F.; de Visser, S.P. Status Report on the Quantum Chemical Cluster Approach for Modeling Enzyme Reactions. Commun. Chem. 2022, 5, 29. [Google Scholar] [CrossRef]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-Selective Formation of an Iron(IV)-Oxo Species at the More Electron-Rich Iron Atom of Heteroleptic μ-Nitrido Diiron Phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Cohen, S.; Kaneti, J.; Shaik, S. The Experimentally Elusive Oxidant of Cytochrome P450: A Theoretical “Trapping” Defining More Closely the “Real” Species. ChemBioChem 2001, 2, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Ogliaro, F.; de Visser, S.P.; Shaik, S. The “Push” Effect of the Thiolate Ligand in Cytochrome P450: A Theoretical Gauging. J. Inorg. Biochem. 2002, 91, 554–567. [Google Scholar] [CrossRef]
- Hirao, H.; Kumar, D.; Thiel, W.; Shaik, S. Two States and Two More in the Mechanisms of Hydroxylation and Epoxidation by Cytochrome P450. J. Am. Chem. Soc. 2005, 127, 13007–13018. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 Enzymes: Their Structure, Reactivity, and Selectivity—Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. [Google Scholar] [CrossRef]
- Green, M.T. Evidence for Sulfur-Based Radicals in Thiolate Compound I Intermediates. J. Am. Chem. Soc. 1999, 121, 7939–7940. [Google Scholar] [CrossRef]
- Schöneboom, J.C.; Lin, H.; Reuter, N.; Thiel, W.; Cohen, S.; Ogliaro, F.; Shaik, S. The Elusive Oxidant Species of Cytochrome P450 Enzymes: Characterization by Combined Quantum Mechanical/Molecular Mechanical (QM/MM) Calculations. J. Am. Chem. Soc. 2002, 124, 8142–8151. [Google Scholar] [CrossRef]
- De Visser, S.P.; Shaik, S.; Sharma, P.K.; Kumar, D.; Thiel, W. Active Species of Horseradish Peroxidase (HRP) and Cytochrome P450: Two Electronic Chameleons. J. Am. Chem. Soc. 2003, 125, 15779–15788. [Google Scholar] [CrossRef]
- Bathelt, C.M.; Zurek, J.; Mulholland, A.J.; Harvey, J.N. Electronic Structure of Compound I in Human Isoforms of Cytochrome P450 from QM/MM Modeling. J. Am. Chem. Soc. 2005, 127, 12900–12908. [Google Scholar] [CrossRef]
- Radoń, M.; Broclawik, E.; Pierloot, K. DFT and Ab Initio Study of Iron-Oxo Porphyrins: May They Have a Low-Lying Iron(V)-Oxo Electromer? J. Chem. Theory Comput. 2011, 7, 898–908. [Google Scholar] [CrossRef]
- Lonsdale, R.; Oláh, J.; Mulholland, A.J.; Harvey, J.N. Does Compound I Vary Significantly Between Isoforms of Cytochrome P450? J. Am. Chem. Soc. 2011, 133, 15464–15474. [Google Scholar] [CrossRef]
- Hirao, H.; Cheong, Z.H.; Wang, X. Pivotal Role of Water in Terminating Enzymatic Function: A Density Functional Theory Study of the Mechanism-Based Inactivation of Cytochromes P450. J. Phys. Chem. B 2012, 116, 7787–7794. [Google Scholar] [CrossRef]
- Hirao, H.; Chuanprasit, P.; Cheong, Y.Y.; Wang, X. How Is a Metabolic Intermediate Formed in the Mechanism-Based Inactivation of Cytochrome P450 by Using 1,1-Dimethylhydrazine: Hydrogen Abstraction or Nitrogen Oxidation? Chem. Eur. J. 2013, 19, 7361–7369. [Google Scholar] [CrossRef] [PubMed]
- Kepp, K.P. Heme Isomers Substantially Affect Heme’s Electronic Structure and Function. Phys. Chem. Chem. Phys. 2017, 19, 22355–22362. [Google Scholar] [CrossRef] [PubMed]
- Phung, Q.M.; Pierloot, K. Low-Lying Electromeric States in Chloro-Ligated Iron(IV)-Oxo Porphyrin As a Model for Compound I, Studied with Second-Order Perturbation Theory Based on Density Matrix Renormalization Group. J. Chem. Theory Comput. 2019, 15, 3033–3043. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; DeYonker, N.J. QM-Cluster Model Study of the Guaiacol Hydrogen Atom Transfer and Oxygen Rebound with Cytochrome P450 Enzyme GcoA. J. Phys. Chem. B 2021, 125, 3296–3306. [Google Scholar] [CrossRef]
- Mokkawes, T.; de Visser, S.P. Caffeine Biodegradation by Cytochrome P450 1A2. What Determines the Product Distributions? Chem. Eur. J. 2023, 29, e202203875. [Google Scholar] [CrossRef]
- Kumar, D.; de Visser, S.P.; Shaik, S. Oxygen Economy of Cytochrome P450: What Is the Origin of the Mixed Functionality as a Dehydrogenase–Oxidase Enzyme Compared with Its Normal Function? J. Am. Chem. Soc. 2004, 126, 5072–5073. [Google Scholar] [CrossRef]
- De Visser, S.P.; Kumar, D.; Cohen, S.; Shacham, R.; Shaik, S. A Predictive Pattern of Computed Barriers for C–H Hydroxylation by Compound I of Cytochrome P450. J. Am. Chem. Soc. 2004, 126, 8362–8363. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P. A Valence Bond Modeling of Trends in Hydrogen Abstraction Barriers and Transition States of Hydroxylation Reactions Catalyzed by Cytochrome P450 Enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef]
- Li, D.; Wang, Y.; Yang, C.; Han, K. Theoretical Study of N-Dealkylation of N-Cyclopropyl-N-Methylaniline Catalyzed by Cytochrome P450: Insight into the Origin of the Regioselectivity. Dalton Trans. 2009, 38, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Tahsini, L.; de Visser, S.P.; Kang, H.Y.; Kim, S.J.; Nam, W. The Effect of Porphyrin Ligands on the Regioselective Dehydrogenation Versus Epoxidation of Olefins by Oxoiron(IV) Mimics of Cytochrome P450. J. Phys. Chem. A 2009, 113, 11713–11722. [Google Scholar] [CrossRef] [PubMed]
- Rydberg, P.; Olsen, L. Do Two Different Reaction Mechanisms Contribute to the Hydroxylation of Primary Amines by Cytochrome P450? J. Chem. Theory Comput. 2011, 7, 3399–3404. [Google Scholar] [CrossRef] [PubMed]
- Taxak, N.; Dixit, V.A.; Bharatam, P.V. Density Functional Study on the Cytochrome-Mediated S-Oxidation: Identification of Crucial Reactive Intermediate on the Metabolic Path of Thiazolidinediones. J. Phys. Chem. A 2012, 116, 10441–10450. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Schuurmann, G. Model and Mechanism: N-Hydroxylation of Primary Aromatic Amines by Cytochrome P450. Angew. Chem. Int. Ed. 2013, 52, 744–748. [Google Scholar]
- Wang, X.; Shi, J.; Liu, Y. Oxidative Rearrangement Mechanism of Pentalenolactone F Catalyzed by Cytochrome P450 CYP161C2 (PntM). Inorg. Chem. 2018, 57, 8933–8941. [Google Scholar] [CrossRef]
- Chatfield, D.C.; Morozov, A.N. Proximal Pocket Controls Alkene Oxidation Selectivity of Cytochrome P450 and Chloroperoxidase Toward Small, Nonpolar Substrates. J. Phys. Chem. B 2018, 122, 7828–7838. [Google Scholar] [CrossRef]
- Oláh, J.; Mulholland, A.J.; Harvey, J.N. Understanding the Determinants of Selectivity in Drug Metabolism Through Modeling of Dextromethorphan Oxidation by Cytochrome P450. Proc. Natl. Acad. Sci. USA 2011, 108, 6050–6055. [Google Scholar] [CrossRef]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity Patterns of (Protonated) Compound II and Compound I of Cytochrome P450: Which Is the Better Oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef]
- Ali, H.S.; Henchman, R.H.; de Visser, S.P. Lignin Biodegradation by a Cytochrome P450 Enzyme: A Computational Study into Syringol Activation by GcoA. Chem. Eur. J. 2020, 26, 13093–13102. [Google Scholar] [CrossRef]
- Dias, A.H.S.; Yadav, R.; Mokkawes, T.; Kumar, A.; Skaf, M.S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Biotransformation of Bisphenol by Human Cytochrome P450 2C9 Enzymes: A Density Functional Theory Study. Inorg. Chem. 2023, 62, 2244–2256. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham III, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber Biomolecular Simulation Program. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Merz, K.M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Ali, H.S.; de Visser, S.P. Electrostatic Perturbations in the Substrate-Binding Pocket of Taurine/α-Ketoglutarate Dioxygenase Determine Its Selectivity. Chem. Eur. J. 2022, 28, e202104167. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.S.; de Visser, S.P. How Does the Nonheme Iron Enzyme NapI React Through L-Arginine Desaturation Rather Than Hydroxylation? A QM/MM Study. ACS Catal. 2023, 13, 10705–10721. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–272. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements. J. Chem. Phys. 1982, 77, 3654–3658. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokkawes, T.; De Visser, T.; Cao, Y.; De Visser, S.P. Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes. Molecules 2023, 28, 6961. https://doi.org/10.3390/molecules28196961
Mokkawes T, De Visser T, Cao Y, De Visser SP. Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes. Molecules. 2023; 28(19):6961. https://doi.org/10.3390/molecules28196961
Chicago/Turabian StyleMokkawes, Thirakorn, Tamar De Visser, Yuanxin Cao, and Sam P. De Visser. 2023. "Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes" Molecules 28, no. 19: 6961. https://doi.org/10.3390/molecules28196961
APA StyleMokkawes, T., De Visser, T., Cao, Y., & De Visser, S. P. (2023). Melatonin Activation by Human Cytochrome P450 Enzymes: A Comparison between Different Isozymes. Molecules, 28(19), 6961. https://doi.org/10.3390/molecules28196961