
Further Quinolizidine Derivatives as Antiarrhythmic Agents- 3
 ,
,  , and
, and 
Abstract
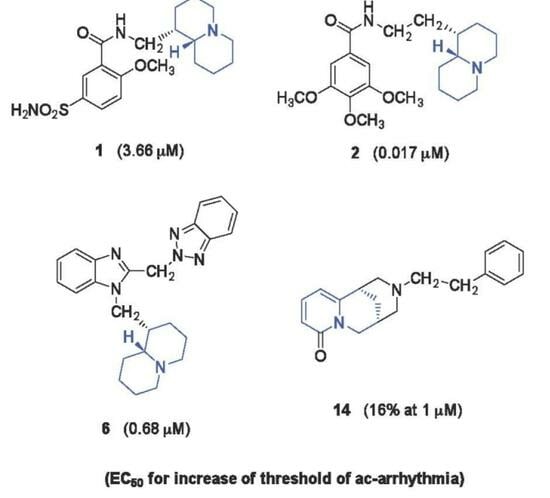
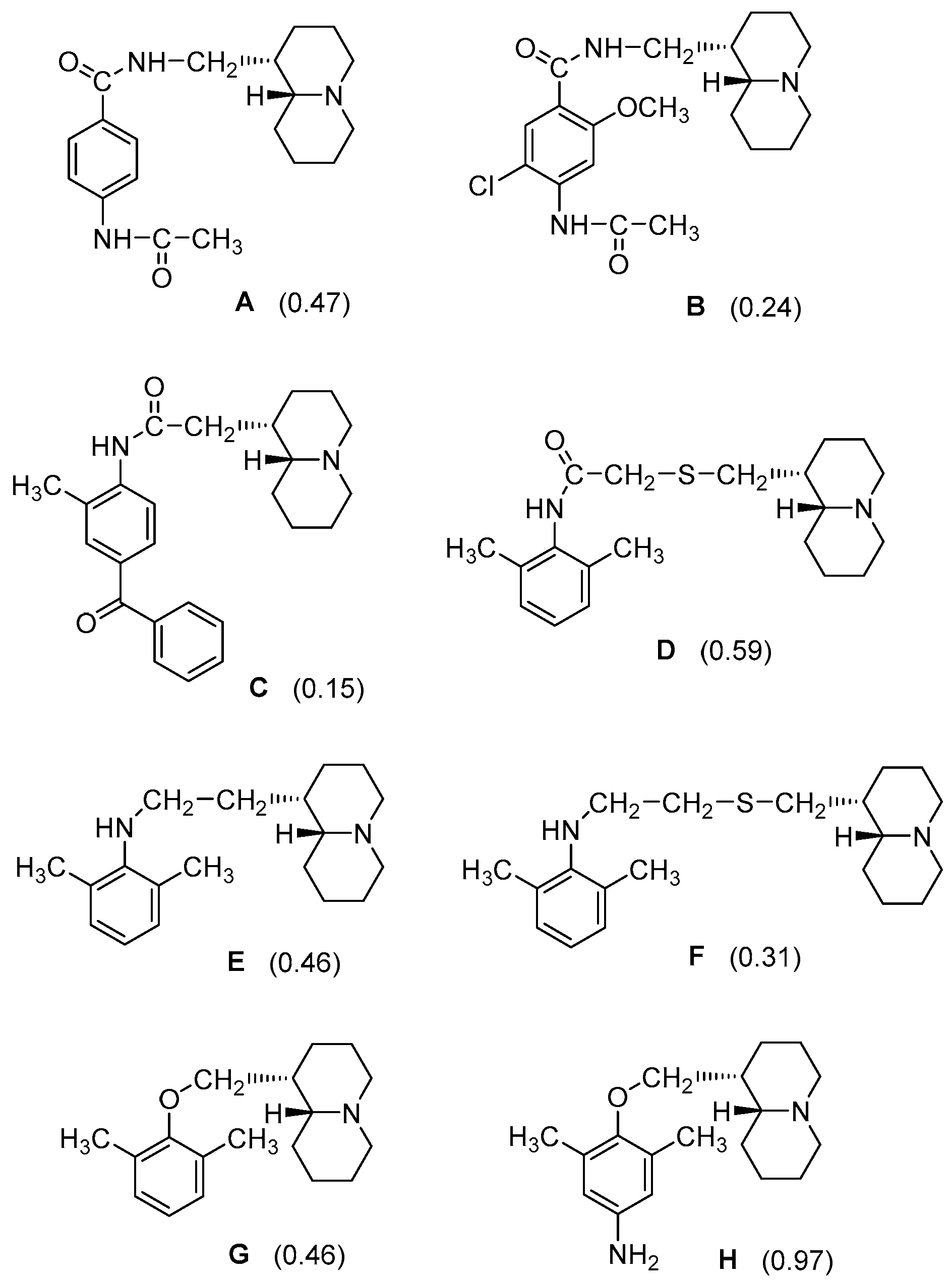
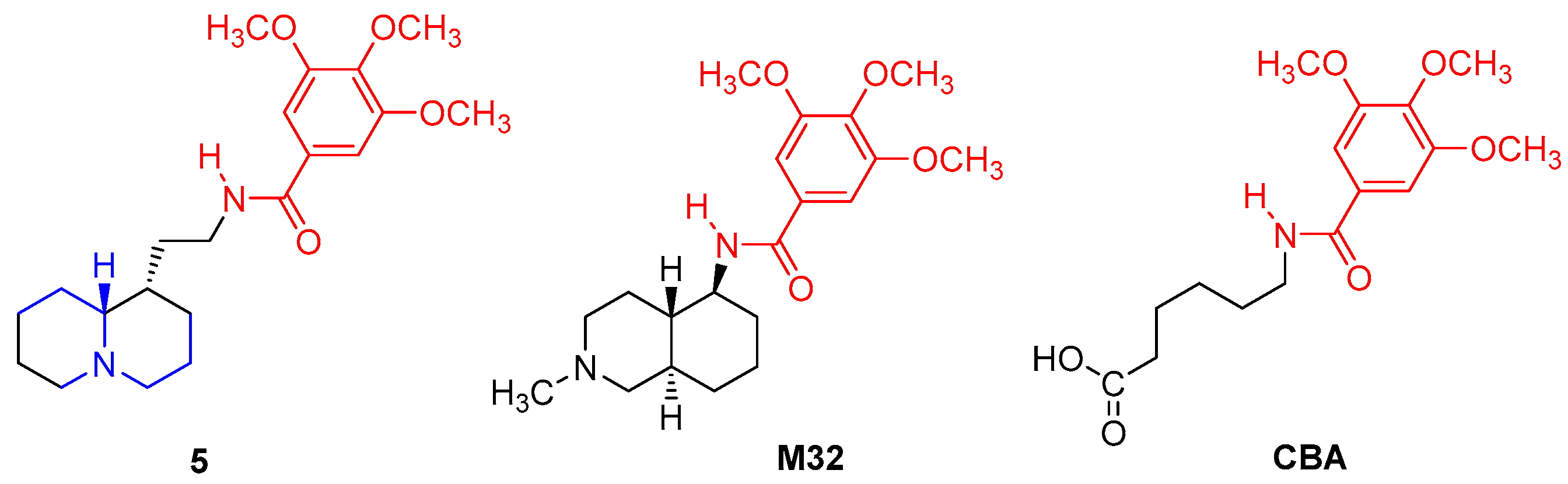
:
1. Introduction
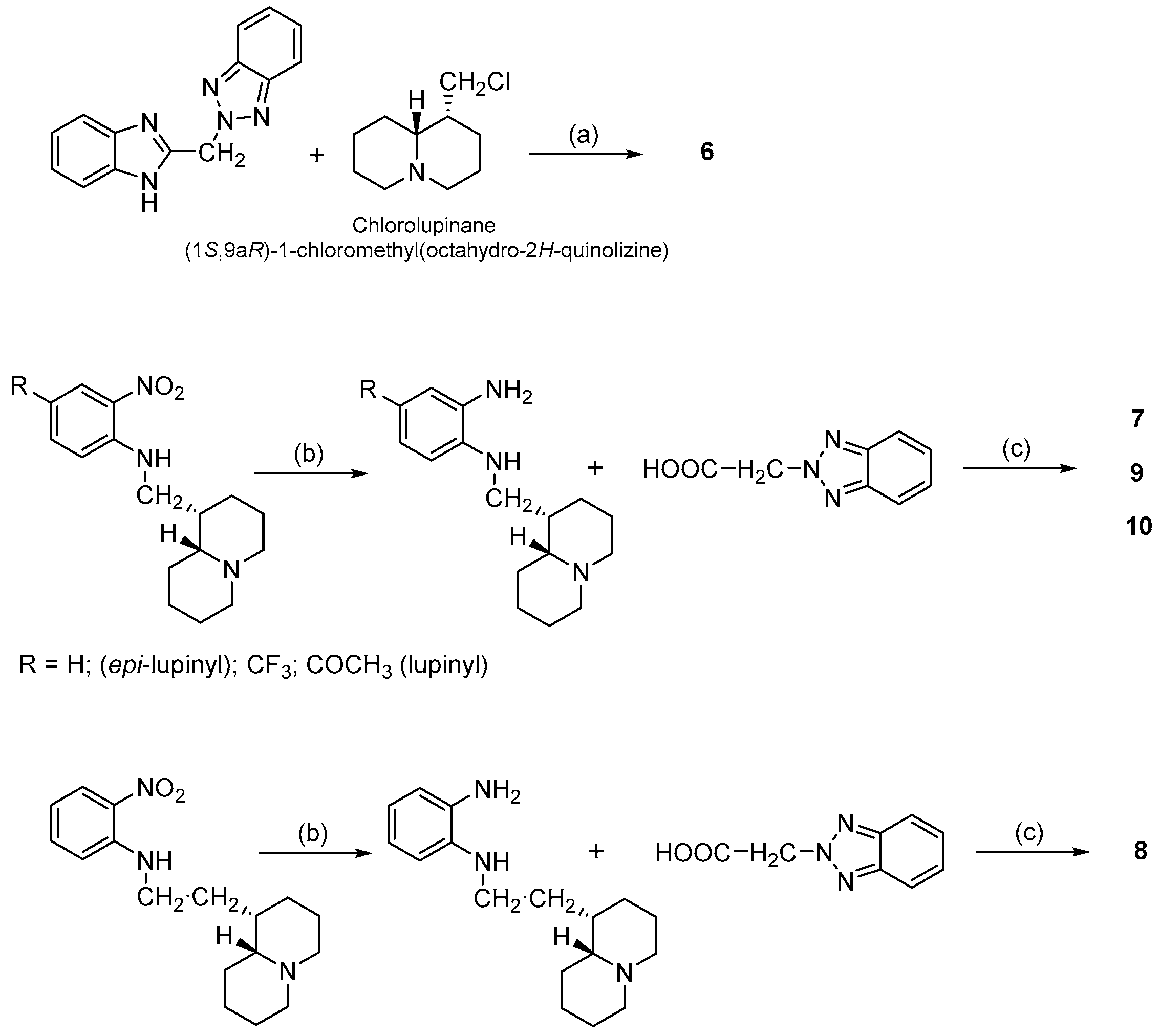
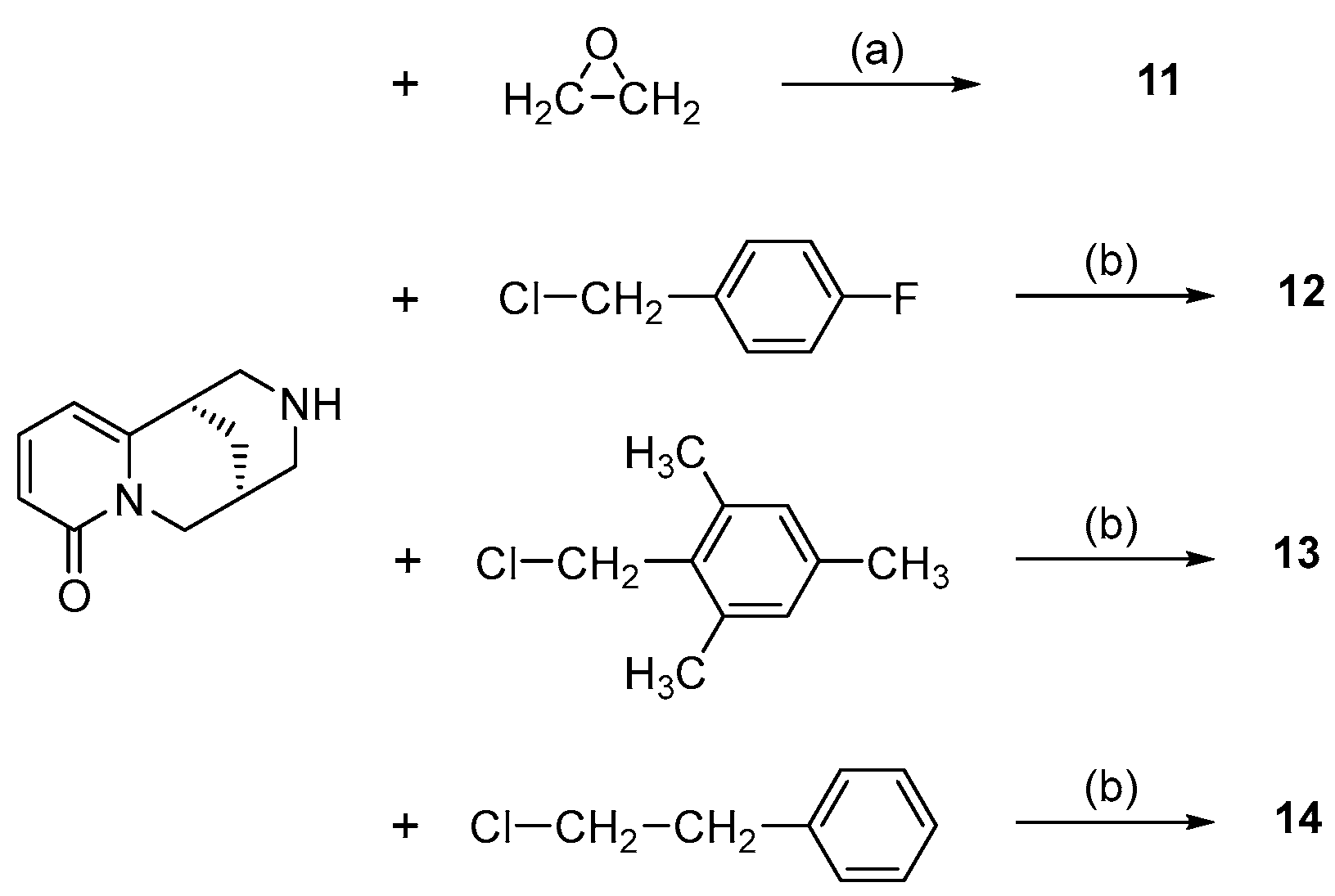
- (a)
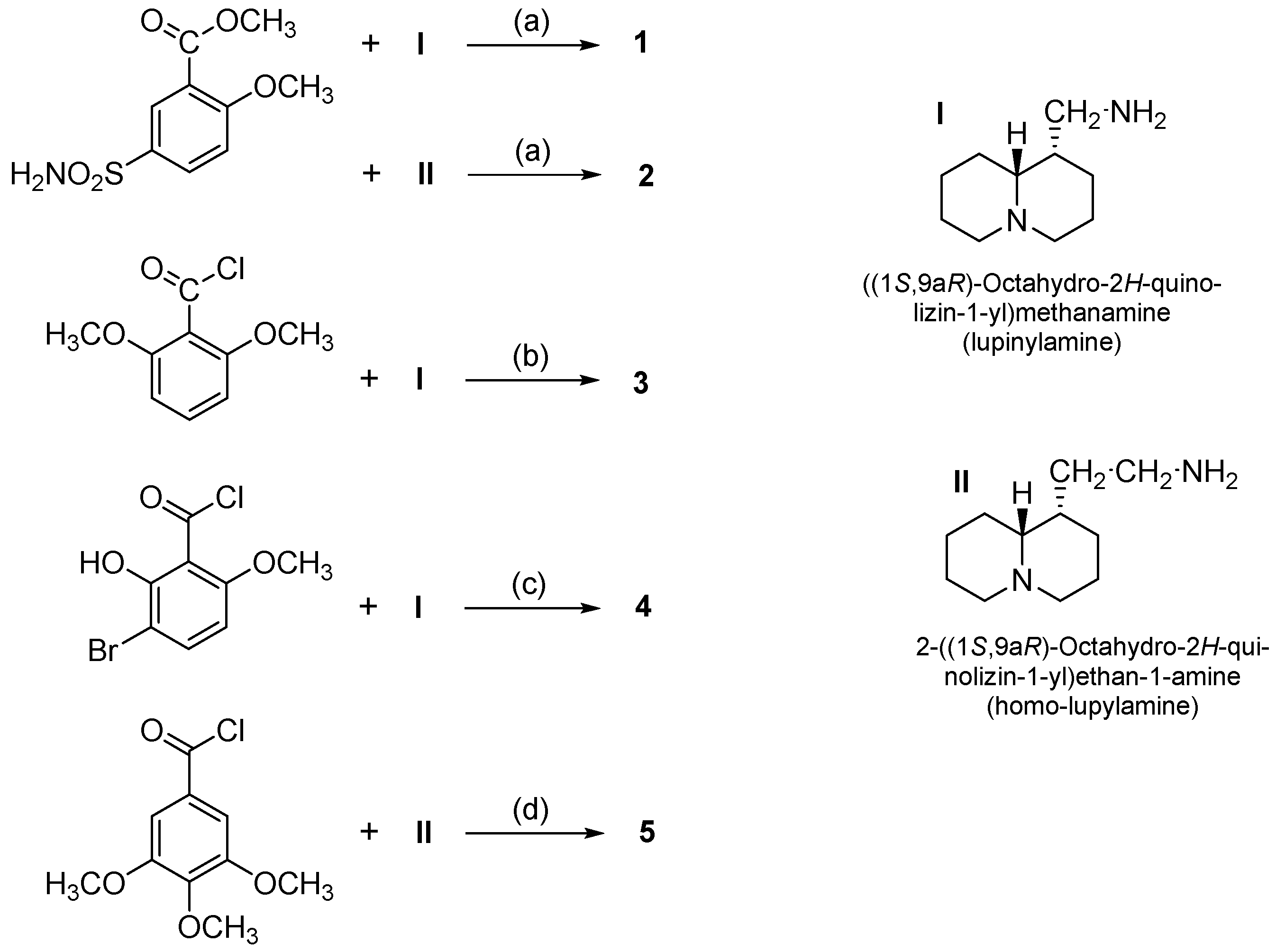
- N-(Quinolizidinyl-alkyl)-benzamides (1–5), related to the previously studied compounds A and B;
- (b)
- (c)
- N-Substituted cytisines (11–14), characterized by the presence of three of the four rings of sparteine’s molecular scaffold. The investigated compounds were selected from a large number of cytisine derivatives, previously studied by us as ligands for a neuronal nicotine receptor [29].
2. Results and Discussion
3. Materials and Methods
3.1. Chemistry
3.2. “In Vitro” Activity
3.2.1. Heart Preparation
3.2.2. Aorta Preparation
3.3. Statistical Analysis
4. Conclusions
- (1)
- The definition of a more complete pharmacological profile and of the mechanisms of action of the relevant compounds;
- (2)
- The synthesis of novel compounds bearing the arylcarbamido group and synthesis of novel cytisine derivatives to explore the possibility of achieving dual-acting (antiarrhythmic and positive inotropic) agents.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cely-Veloza, W.; Kato, M.J.; Coy-Barrera, E. Quinolizidine-Type Alkaloids: Chemodiversity, Occurrence, and Bioactivity. ACS Omega 2023, 8, 27862–27893. [Google Scholar] [CrossRef] [PubMed]
- Sparatore, A.; Sparatore, F. Preparation and pharmacological activities of homolupinanoyl anilides. Farmaco 1995, 50, 153–166. [Google Scholar] [PubMed]
- Iusco, G.; Boido, V.; Sparatore, F. Synthesis and preliminary pharmacological investigation of N-lupinyl-2-methoxybenzamides. Farmaco 1996, 51, 159–174. [Google Scholar] [PubMed]
- Vazzana, I.; Budriesi, R.; Terranova, E.; Joan, P.; Ugenti, M.P.; Tasso, B.; Chiarini, A.; Sparatore, F. Novel quinolizidinyl derivatives as antiarrhythmic agents. J. Med. Chem. 2007, 50, 334–343. [Google Scholar] [CrossRef]
- Tasso, B.; Budriesi, R.; Vazzana, I.; Joan, P.; Micucci, M.; Novelli, F.; Tonelli, M.; Sparatore, A.; Chiarini, A.; Sparatore, F. Novel quinolizidinyl derivatives as antiarrhythmic agents: 2. Further investigation. J. Med. Chem. 2010, 53, 4668–4677. [Google Scholar] [CrossRef]
- Vaughan Williams, E.M. Classification of antidysrhythmic drugs. Pharmacol. Ther. B 1975, 1, 115–138. [Google Scholar]
- Vaughan Williams, E.M. Classifying antiarrhythmic actions: By facts or speculation. J. Clin. Pharmacol. 1992, 32, 964–977. [Google Scholar] [CrossRef]
- Lei, M.; Wu, L.; Terrar, D.A.; Huang, C.L.-H. Modernized classification of cardiac antiarrhythmic drugs. Circulation 2018, 138, 1879–1896. [Google Scholar] [CrossRef]
- Geng, M.; Lin, A.; Nguyen, T.P. Revisiting antiarrhythmic drug therapy for atrial fibrillation: Reviewing lessons learned and redefining therapeutic paradigms. Front. Pharmacol. 2020, 11, 581837. [Google Scholar] [CrossRef]
- McCawley, E.L. Cardioactive alkaloids. In The Alkaloids: Chemistry and Physiology; Manske, R.H.F., Ed.; Academic Press: New York, NY, USA, 1955; Volume 5, pp. 93–97. [Google Scholar]
- Philipsborn, G.V.; Wilhelm, E.; Homburger, H. Untersuchungen zur Wirkung von Spartein am isolierten Vorhofmyokard von Maerschweinchen. Naunyn-Scmiedeberg’ Arch. Pharmacol. 1973, 277, 281–290. [Google Scholar] [CrossRef]
- Raschaek, M. Wirking von Spartein und Spartein derivate auf Herz und Kreislauf. Azneim. Forsch. 1974, 24, 753–759. [Google Scholar]
- Engelmann, K.; Radke, W.; Petter, A. Die Bedentung hydrophober Gruppen fur die antiarrhythmische eigenschaft alkylierte Sparteine. Arzneim. Forsch. 1974, 24, 759–764. [Google Scholar]
- Zetler, G.; Strubelt, O. Antifibrillatory, cardiovascular and toxic effects of sparteine, butylsparteine and pentylsparteine. Arznreim. Forsch. 1980, 30, 1497–1502. [Google Scholar]
- Gawall, V.S.; Simeonov, S.; Drescher, M.; Knott, T.; Scheel, O.; Kudolo, J.; Kahlig, H.; Hochenegg, U.; Roller, A.; Todt, H.; et al. C2-Modified sparteine derivatives are a new class of potentially long-acting sodium channel blockers. ChemMedChem Comm. 2017, 12, 1819–1822. [Google Scholar] [CrossRef] [PubMed]
- Ruenitz, P.C.; Mokler, C.M. Analogs of sparteine. 5. Antiarrhythmic activity of selected N,N’-disubstituted bispidines. J. Med. Chem. 1977, 20, 1668–1671. [Google Scholar] [CrossRef]
- Ruenitz, P.C.; Mokler, C.M. Anthiarrhythmic activity of some N-alkylbispidinebenzamides. J. Med. Chem. 1979, 22, 1142–1147. [Google Scholar] [CrossRef]
- Hiraoka, M.; Sunami, A.; Tajima, K. Bisaramil, a new class I antiarrhythmic agent. Cardiovasc. Drug Rev. 1993, 11, 516–524. [Google Scholar] [CrossRef]
- Schoen, U.; Antel, J.; Bruckner, R.; Messinger, J.; Franke, R.; Gruska, A. Synthesis, pharmacological characterization, and quantitative structure-activity relationship analyses of 3,7,9,9-tetraalkylbispidines: Derivatives with specific bradycardic activity. J. Med. Chem. 1998, 41, 318–331. [Google Scholar] [CrossRef]
- Takanaka, C.; Sarma, J.S.; Singh, B.N. Electrophysiological effects of ambasilide (LU 47110), a novel class II antiarrhythmic agent, on the properties of isolated rabbit and canine cardiac muscle. J. Cardiovasc. Pharmacol. 1992, 19, 290–298. [Google Scholar] [CrossRef]
- Pugsley, M.; Walker, M.J.A.; Garrison, G.B.; Howard, P.G.; Lazzara, R.; Patterson, E.; Penz, W.P.; Scherlag, B.J.; Berlin, K.D. The cardiovascular and antiarrhythmic properties of a series of novel sparteine analogs. Proc. West. Pharmacol. Soc. 1992, 35, 87–91. [Google Scholar]
- Pugsley, M.K.; Saint, D.A.; Hayes, E.; Berlin, K.D.; Walker, M.J. The cardiac electrophysiological effects of sparteine and its analog BRD-1-28 in the rat. Eur. Pharmacol. 1995, 294, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Tomassoli, J.; Gundish, D. Bispidine as a priviliged scaffold. Curr. Top. Med. Chem. 2016, 16, 1314–1342. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, G.; Liu, J.; Ouyang, L. Quinolizidine alkaloids derivatives from Sophora alopecuroides Linn: Bioactivities, structure-activity relationship and preliminary molecular mechanisms. Eur. J. Med. Chem. 2020, 188, 111972. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.-H.; Guo, H.; Xie, B. Structural modifications of Matrine-type alkaloids. Mini Rev. Med. Chem. 2016, 18, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Sparatore, F. Benzotriazolylalkyl-benzimidazoles and their dialkylaminoalkyl derivatives. Boll. Chim. Far. 1965, 104, 427–431. [Google Scholar]
- Paglietti, G.; Boido, V.; Sparatore, F. Dialkylaminoalkylbenzimidazoles of pharmacological interest. Farm. Ed. Sci. 1975, 30, 505–511. [Google Scholar]
- Tonelli, M.; Paglietti, G.; Boido, V.; Sparatore, F.; Marongiu, F.; Marongiu, E.; La Colla, P.; Loddo, R. Antiviral activity of benzimidazole derivatives: I. Antiviral activity of 1-substituted-2-[(benzotriazol-1/2-yl)methyl]benzimidazoles. Chem. Biodivers. 2008, 5, 2386–2401. [Google Scholar] [CrossRef]
- Canu Boido, C.; Sparatore, F. Synthesis and preliminary pharmacological evaluation of some cytisine derivative. Farmaco 1999, 54, 438–451. [Google Scholar] [CrossRef]
- Ciofalo, E.; Levitt, B.; Roberts, J. Some aspects of the antiarrhythmic activity of reserpine. Brit. J. Pharmacol. Chemother. 1966, 28, 44–50. [Google Scholar] [CrossRef]
- Lawson, J.W. Antiarrhythmic activity of some isoquinoline derivatives determined by a rapid screening procedure in the mouse. J. Pharmacol. Expl. Therap. 1968, 160, 22–31. [Google Scholar]
- Mathison, J.W.; Gueldner, R.C.; Lawson, J.W.; Fawler, S.J.; Peters, E.R. The stereochemistry of 5-substituted decahydroisoquinolines and their antiarrhythmic activity. J. Med. Chem. 1968, 11, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Mathison, S.W.; Pennington, R.J. Synthesis and antiarrhythmic properties of some 5-benzamido-2-methyl-trans-decahydroisoquinolines. J. Med. Chem. 1980, 23, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Garzia, A. Pharmaceutical omega-(trimethoxybenzamido)fatty acids. DE 2034192 (1971). Chem. Abstr. 1971, 75, 55230c. [Google Scholar]
- Boido, V.; Boido, A.; Boido Canu, C.; Sparatore, F. Quinolizidinylalkylamines with antihypertensive activity. Farm. Ed. Sci. 1979, 34, 2–16. [Google Scholar]
- Boido, V.; Sparatore, F. Derivatives of natural aminoalcohols and diamines of pharmacological interest: 5. Novel derivatives of lupinine and aminolupinane: Preliminary observations on their pharmacological activity. Ann. Chim. 1969, 59, 526–538. [Google Scholar]
- O’Connor, S.E.; Brown, R.A. The pharmacology of sulpiride—A dopamine receptor antagonist. Gen. Pharmacol. 1982, 13, 185–193. [Google Scholar] [CrossRef]
- Silvestre, J.S.; Prous, J. Comparative evaluation of hERG potassium channel blockade by antipsychotics. Methods Find Exp. Clin. Pharmacol. 2007, 29, 457–465. [Google Scholar] [CrossRef]
- Iusco, G.; Boido, V.; Sparatore, F.; Colombo, G.; Saba, P.L.; Rossetti, Z.; Vaccari, A. New benzamide-derived 5-HT3 receptor antagonists which prevent the effects of ethanol on extracellular dopamine and fail to reduce voluntary alcohol intake in rats. Farmaco 1997, 52, 141–146. [Google Scholar] [CrossRef]
- De Paulis, T.; Kumar, Y.; Johansson, L.; Raemsby, S.; Florvell, L.; Hall, H.; Aengeby-Muller, K.; Ogren, S.O. Potential neuroleptic agents. 3. Chemistry and antidopaminergic properties of 6-methoxysalicylamides. J. Med. Chem. 1985, 28, 1263–1269. [Google Scholar] [CrossRef]
- De Paulis, T.; Kumar, Y.; Johansson, L.; Raemsby, S.; Hall, H.; Saellemark, M.; Aengeby-Muller, K.; Ogren, S.O. Potential neuroleptic agents. 4. Chemistry, behavioral pharmacology and inhibition of [3H]spiperone binding of 3,5-disubstituted N-[(1-ethyl-2-pyrrolidinyl)methyl]-6-methoxysalicylamides. J. Med. Chem. 1986, 29, 61–69. [Google Scholar] [CrossRef]
- Khisamatdinova, R.Y.; Yarmukhamedov, N.N.; Gabdrakhmanova, S.F.; Karachurina, L.T.; Sapozhnikova, T.A.; Baibulatova, N.Z.; Baschenko, N.Z.; Zarudi, F.S. Synthesis and antiarrhythmic activity of N-(2-hydroxyethyl)cytisine hydrochloride and 3-(2-hydroxyethyl)-1,5-dinitro-3-azabicyclo-[3.3.1]non-3-ene hydrochloride. Pharm. Chem. J. 2004, 38, 311–313. [Google Scholar] [CrossRef]
- Shishkin, D.V.; Shaimuratova, A.R.; Lobov, A.N.; Baibulatova, N.Z.; Spirikhin, L.; Yunusov, M.S.; Makara, N.S.; Baschenko, N.Z.; Dokichev, V.A. Synthesis and biological activity of N-(2-hydroxyethyl)cytisine derivatives. Chem. Nat. Comp. 2007, 43, 190–196. [Google Scholar] [CrossRef]
- Tsipisheva, J.P.; Kovolskaya, A.V.; Khalilova, I.U.; Bakhtina, Y.Y.; Khisamutdinova, R.; Gabdrakhmanova, S.F.; Lobov, A.N.; Zarudi, F.S.; Yunusov, S.Y. New 12-N-β-Hydroxyethylcytisine derivatives with potential antiarrhythmic activity. Chem Nat. Comp. 2014, 56, 333–338. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, H.; Qiao, G.; Sui, X.; Lu, Y.; Yang, B. Inotropic effects and mechanisms of Matrine, a main alkaloid from Sophora flavescens Ait. Biol. Pharm. Bull. 2008, 31, 2057–2067. [Google Scholar] [CrossRef]
- Borchard, U.; Bosken, R.; Greeff, K. Characterization of antiarrhythmic drugs by alternating current induced arrhythmias in isolated heart tissue. Arch. Int. Pharmacodyn. 1982, 256, 253–268. [Google Scholar]
- Bhatt, L.K.; Naudakumar, K.; Bodhankar, L.S. Experimental animal models to induce cardiac arrhythmia. Indian J. Pharmacol. 2005, 37, 348–357. [Google Scholar]
- Roselli, M.; Carrocci, A.; Budriesi, R.; Micucci, M.; Toma, M.; Di Cesare Mannelli, L.; Lovece, A.; Catalano, A.; Cavalluzzi, M.M.; Bruno, C.; et al. Synthesis, antiarrhythmic activity, and toxicological evaluation of mexiletine analogues. Eur. J. Med. Chem. 2016, 121, 300–307. [Google Scholar] [CrossRef]
- Zeka, K.; Marrazzo, P.; Micucci, M.; Ruparelia, K.C.; Arroo, R.R.J.; Macchiarelli, G.; Nottola, S.A.; Continenza, M.A.; Chiarini, A.; Angeloni, C.; et al. Activity af antioxidants from Crocus sativus L. petals. Preventive effects towards cardiovascular system. Antioxidants 2020, 9, 1102. [Google Scholar] [CrossRef]
- Tallarida, R.J.; Murray, R.B. Manual of Pharmacologic Calculations with Computer Programs, 2nd ed.; Springer: New York, NY, USA, 1987. [Google Scholar]
- GraphPad Prism 4.03; Gaphpad Software Inc.: San Diego, CA, USA, 2005.
- GraphPad Prism 3.02; Gaphpad Software Inc.: San Diego, CA, USA, 2002.
- Saeed, M.; Bhandohal, J.S.; Visco, F.; Pekler, G.; Mushiyev, S. Gastrocardiac syndrome A forgotten entity. Am. J. Emerg. Med. 2018, 36, 1525e5–1525e7. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Max% Increase in Threshold of Ac-Arrhythmia after Pretreatment with Compound a (M ± SEM) | EC50 b (μM) | 95% Conf Lim (μM) |
|---|---|---|---|
| Amiodarone | 10 ± 0.5 c | ||
| Lidocaine | 34 ± 2.6 | ||
| Procainamide | 11 ± 0.4 | ||
| Quinidine | 69 ± 0.4 | 10.26 | 8.44–12.46 |
| N-(Quinolizidinyl-alkyl)-benzamides-related compounds | |||
| 1 | 59 ± 1.7 | 3.66 | 2.10–6.38 |
| 2 | 9 ± 0.6 d | ||
| 3 | 17 ± 0.4 e | ||
| 4 | 92 ± 1.3 | 10.67 | 7.56–14.91 |
| 5 | 104 ± 3.4 | 0.017 | 0.0068–0.046 |
| 1-(Quinolizidinyl)alkyl-2-(benzotriazol-2-yl)methyl benzimidazoles-related compounds | |||
| 6 | 75 ± 1.8 d | 0.68 | 0.47–0.98 |
| 7 | 37 ± 3.4 f | ||
| 8 | 39 ± 1.7 d | ||
| 9 | 16 ± 0.9 | ||
| 10 | 20 ± 0.4 | ||
| N-Substituted cytisines-related compounds | |||
| 11 | 26 ± 0.3 c | ||
| 12 | 4 ± 0.2 | ||
| 13 | 27 ± 1.3 c | ||
| 14 | 16 ± 0.7 g | ||
| Heart | Aorta | ||||||
|---|---|---|---|---|---|---|---|
| Left Atria | Right Atria | ||||||
| Negative Inotropy | Negative Chronotropy | Vasorelaxant | |||||
| Compd | IA a (M ± SEM) | EC50 b (μM) | 95% conf lim (μM) | IA c (M ± SEM) | EC50 b (μM) | 95% conf lim (μM) | IA d (M ± SEM) |
| Amiodarone | 30 ± 2.6 e | 72 ± 4.5 e | 14.95 | 11.07–20.16 | 3 ± 0.1 g | ||
| Lidocaine | 88 ± 3.0 | 0.017 | 0.012–0.024 | 29 ± 0.9 #,j | 14 ± 0.9 | ||
| Procainamide | 92 ± 1.4 f | 0.014 | 0.011–0.017 | 9 ± 0.6 #,e | 3 ± 0.2 | ||
| Quinidine | 71 ± 3.6 g | 3.38 | 2.69–4.25 | 86 ± 0.5 | 25.31 | 14.45–44.32 | 30 ± 1.6 g |
| N-(Quinolizidinyl-alkyl)-benzamides-related compondds | |||||||
| 1 | 92 ± 1.4 h | 0.037 | 0.027–0.051 | 24 ± 1.3 k | 5 ± 0.2 | ||
| 2 | 93 ± 1.4 i | 0.0091 | 0.002–0.021 | 2 ± 0.1 h | 3 ± 0.2 | ||
| 3 | 75 ± 2.3 i | 0.011 | 0.0079–0.014 | 25 ± 1.6# | 2 ± 0.1 | ||
| 4 | 98 ± 1.3 | 0.021 | 0.016–0.027 | 46 ± 2.2 | 36 ± 1.3 | ||
| 5 | 85 ± 2.2 | 0.050 | 0.035–0.071 | 25 ± 0.9 h | 22 ± 1.6 | ||
| 1-(Quinolizidinyl)alkyl-2-(benzotriazol-2-yl)methyl benzimidazoles-related compounds | |||||||
| 6 | 91 ± 2.4 h | 0.046 | 0.035–0.061 | 67 ± 0.7 | 11.15 | 9.05–13.74 | 25 ± 1.7 g |
| 7 | 93 ± 2.7 j | 0.083 | 0.064–0.11 | 69 ± 1.3 h | 0.49 | 0.43–0.65 | 16 ± 1.1 |
| 8 | 92 ± 1.3 | 0.022 | 0.015–0.031 | 83 ± 2.4 | 0.019 | 0.014–0.026 | 19 ± 1.2 |
| 9 | 87 ± 1.1 j | 0.056 | 0.042–0.076 | 44 ± 1.5 h | 24 ± 1.6 | ||
| 10 | 94 ± 3.4 | 0.021 | 0.014–0.032 | 22 ± 1.2 e | 11 ± 1.0 | ||
| N-Substituted cytisines-related compounds | |||||||
| 11 | 86 ± 2.2 h | 0.14 | 0.095–0.20 | 35 ± 1.4 k | 0.3 ± 00.1 | ||
| 12 | 92 ± 1.8 h | 0.018 | 0.013–0.026 | 26 ± 1.9 | 25 ± 1.4 | ||
| 13 | 87 ± 1.4 h | 0.044 | 0.028–0.068 | 20 ± 0.3 h | 32 ± 2.2 | ||
| 14 | 71 ± 0.7 i | 0.016 | 0.0081–0.023 | 47 ± 1.1 e | 20 ± 1.6 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tasso, B.; Mattioli, L.B.; Tonelli, M.; Boido, V.; Chiarini, A.; Sparatore, F.; Budriesi, R. Further Quinolizidine Derivatives as Antiarrhythmic Agents- 3. Molecules 2023, 28, 6916. https://doi.org/10.3390/molecules28196916
Tasso B, Mattioli LB, Tonelli M, Boido V, Chiarini A, Sparatore F, Budriesi R. Further Quinolizidine Derivatives as Antiarrhythmic Agents- 3. Molecules. 2023; 28(19):6916. https://doi.org/10.3390/molecules28196916
Chicago/Turabian StyleTasso, Bruno, Laura Beatrice Mattioli, Michele Tonelli, Vito Boido, Alberto Chiarini, Fabio Sparatore, and Roberta Budriesi. 2023. "Further Quinolizidine Derivatives as Antiarrhythmic Agents- 3" Molecules 28, no. 19: 6916. https://doi.org/10.3390/molecules28196916
APA StyleTasso, B., Mattioli, L. B., Tonelli, M., Boido, V., Chiarini, A., Sparatore, F., & Budriesi, R. (2023). Further Quinolizidine Derivatives as Antiarrhythmic Agents- 3. Molecules, 28(19), 6916. https://doi.org/10.3390/molecules28196916