
The Phenomenon of Self-Induced Diastereomeric Anisochrony and Its Implications in NMR Spectroscopy


Abstract
:
1. Introduction
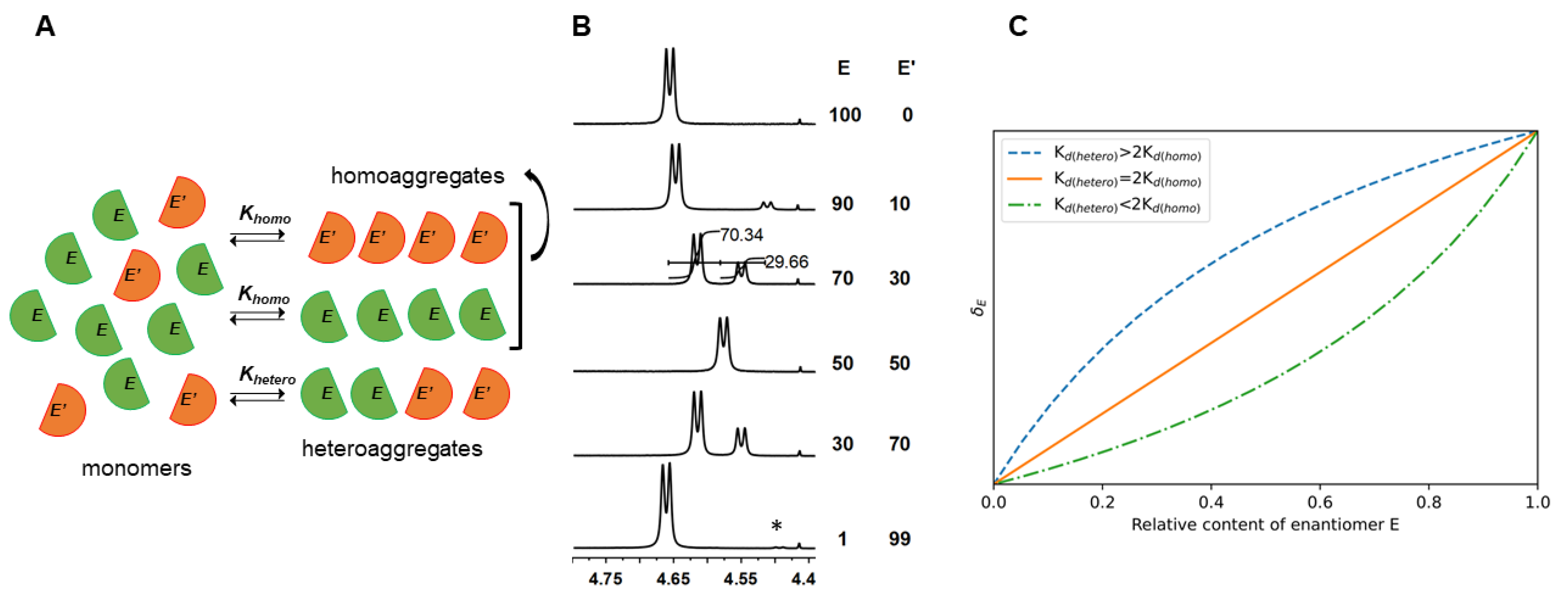
2. Theoretical Dissertation
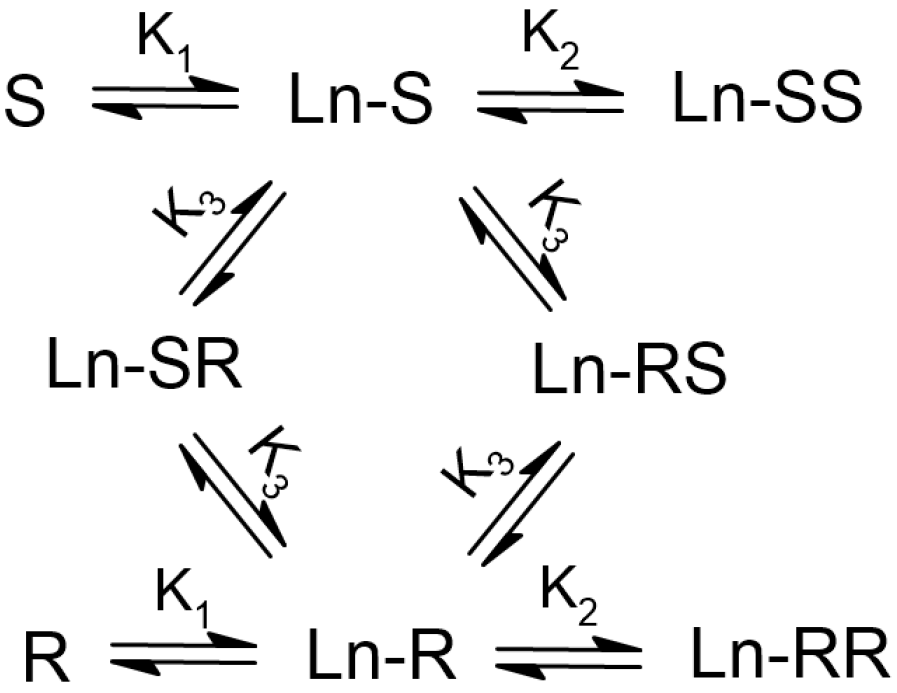
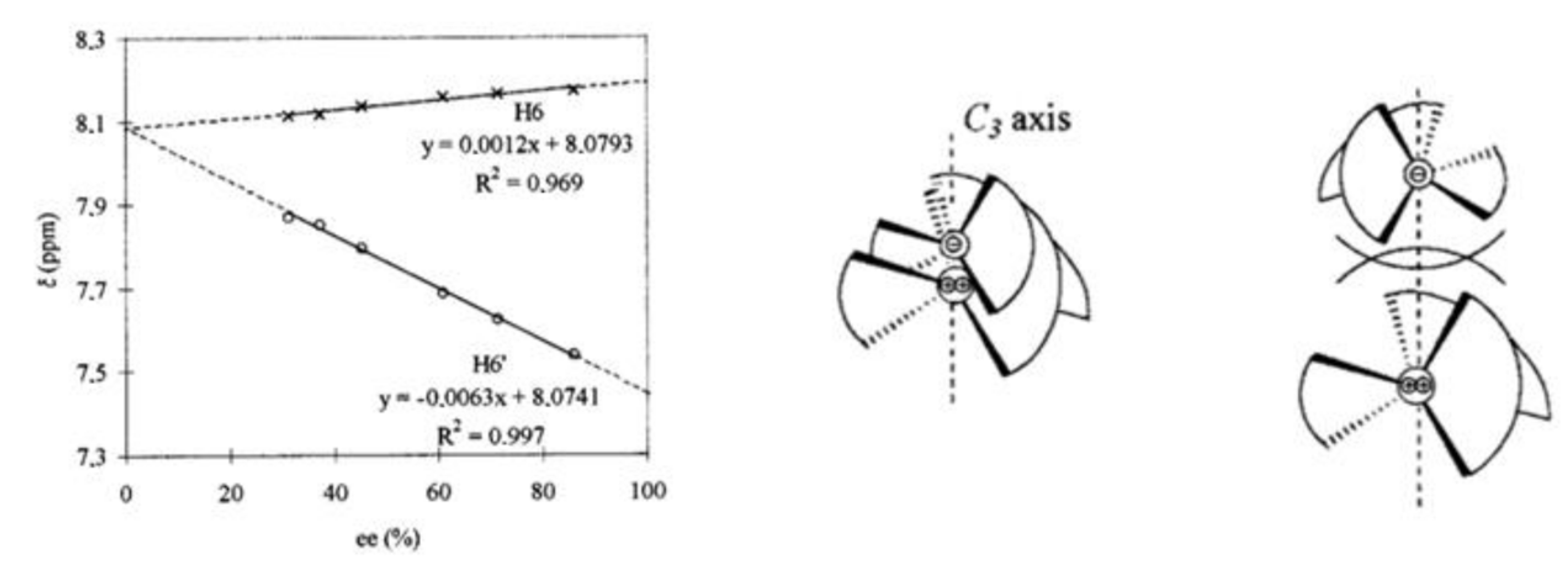
2.1. Calculation of Homodimer and Heterodimer Constants
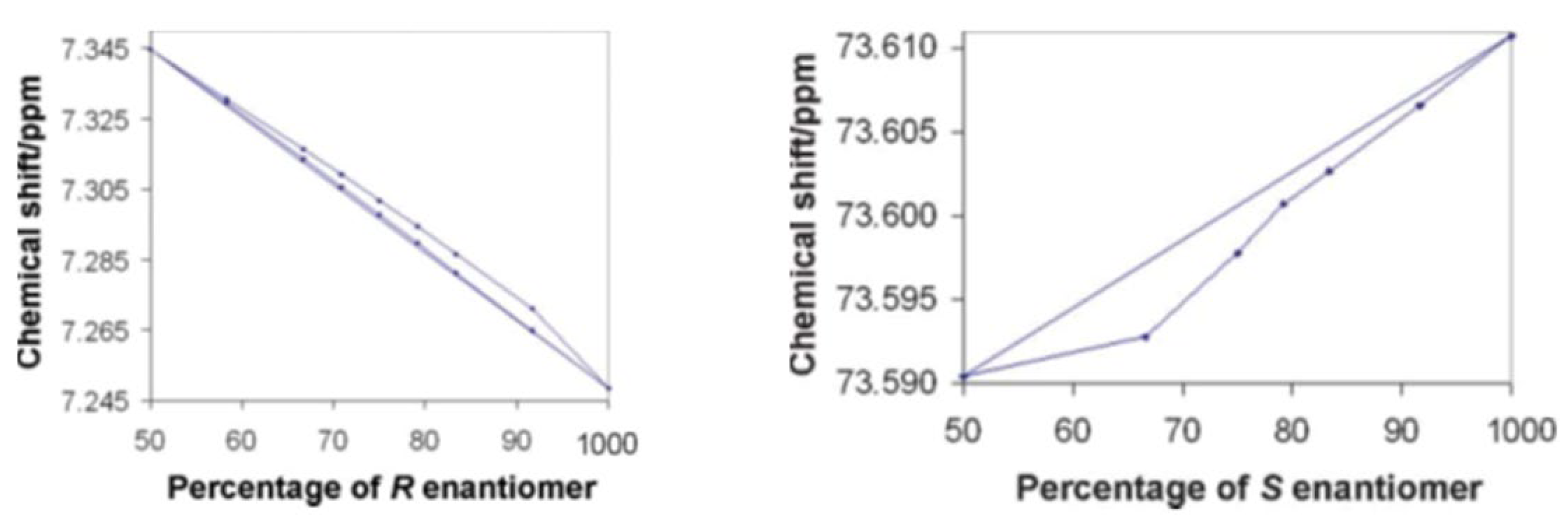
3. Classes of Chiral Compounds Involved in SIDA
3.1. P-Derivatives
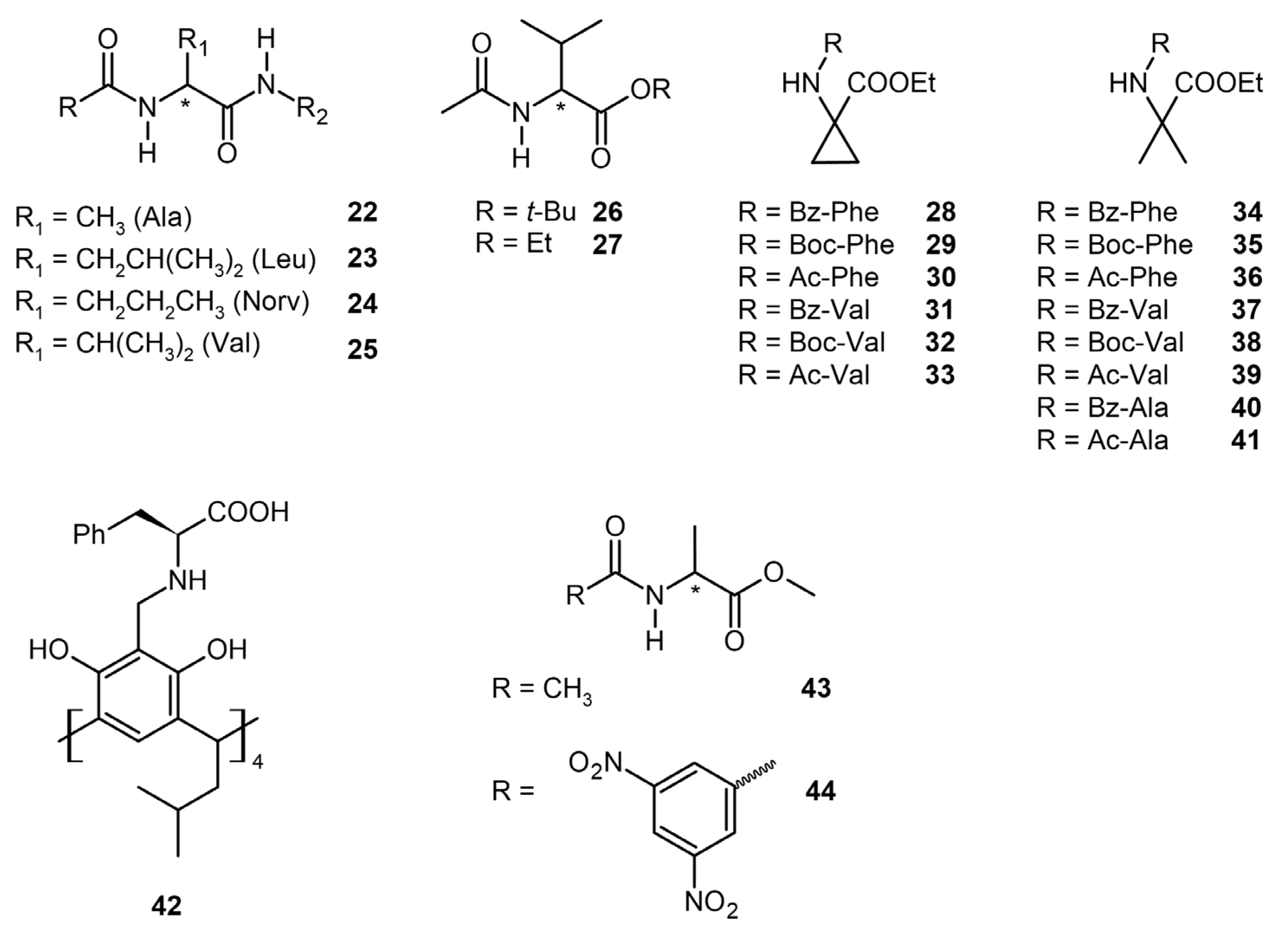
3.2. Amino Acid Derivatives
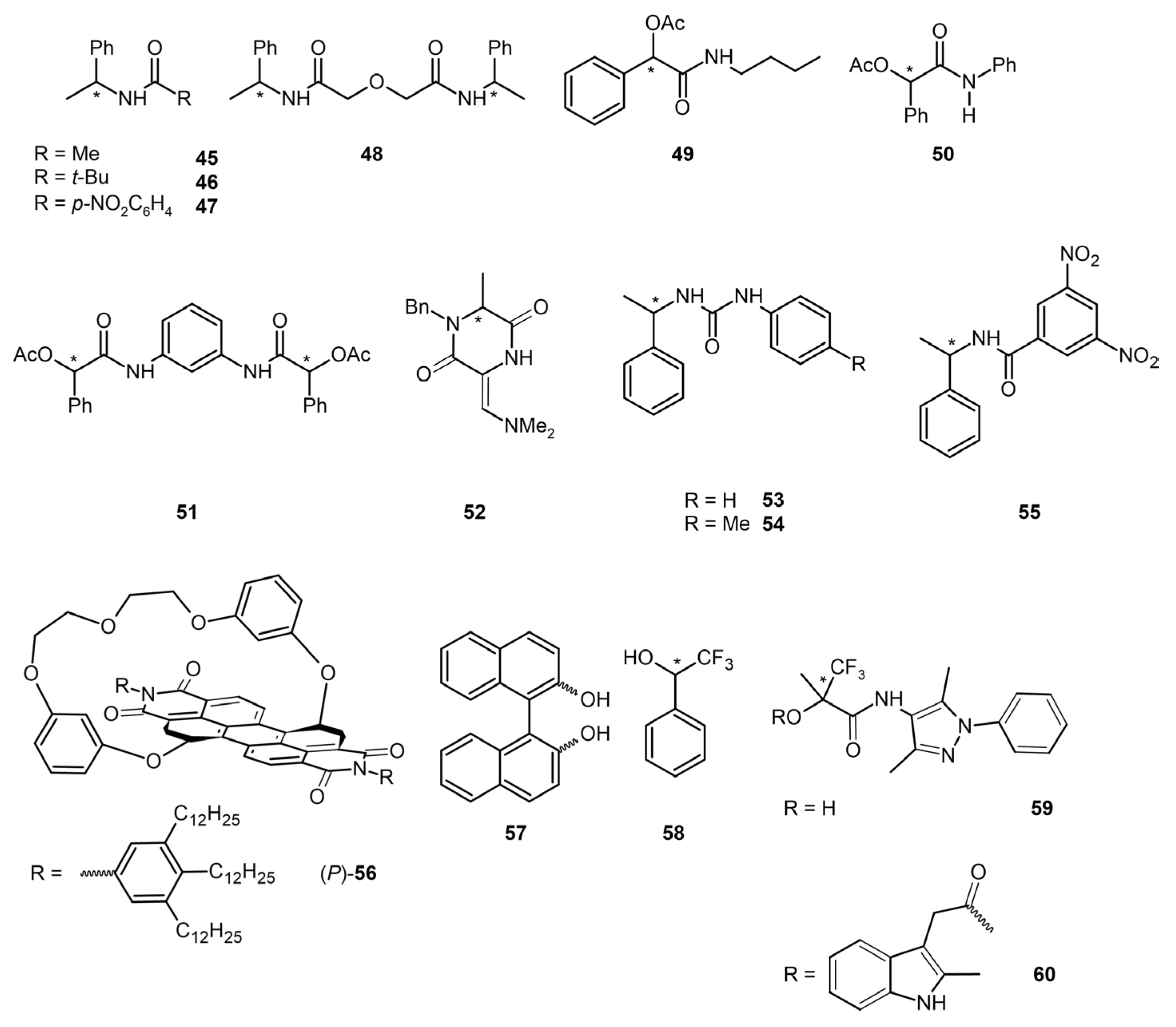
3.3. Chiral Amides, Imides, and Alcohols
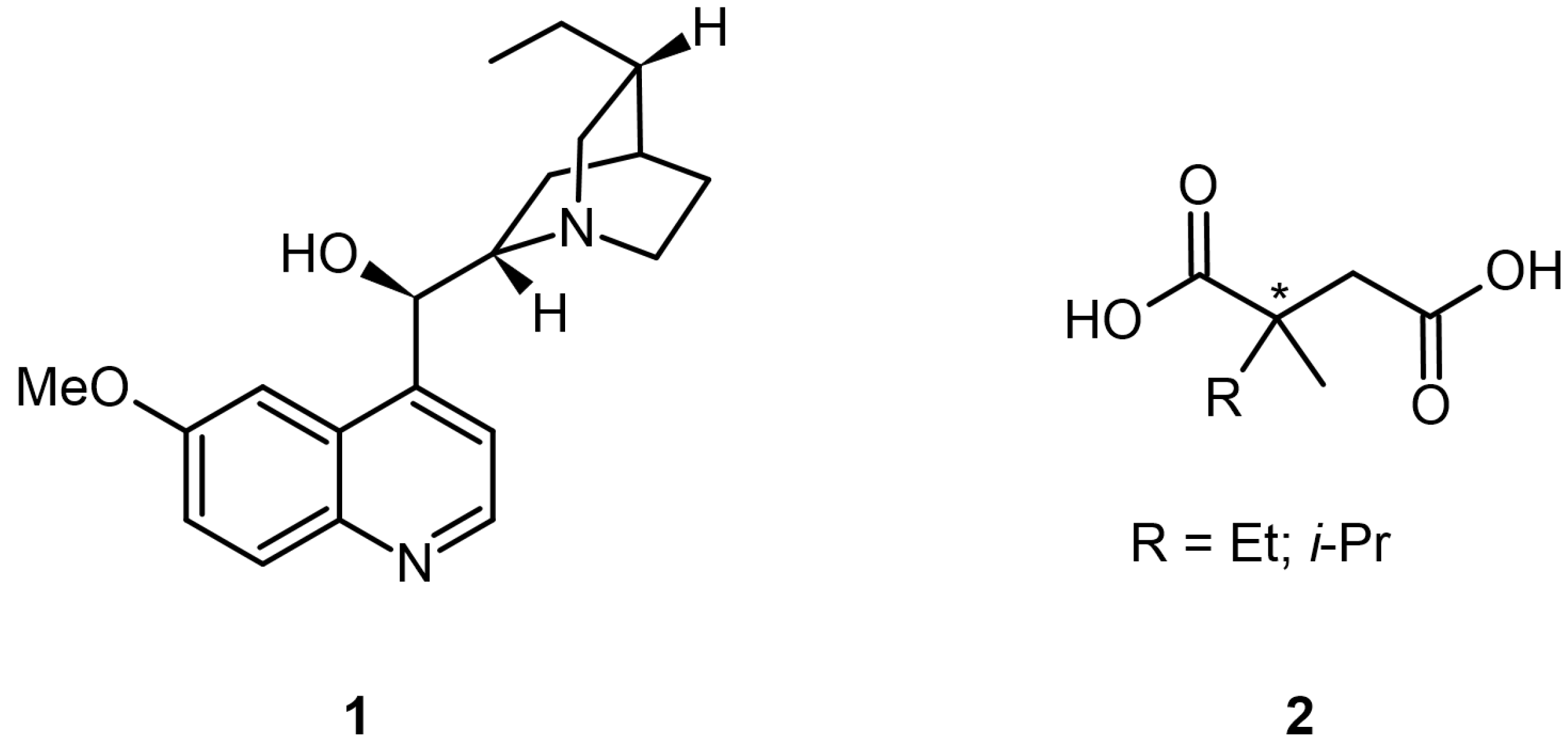
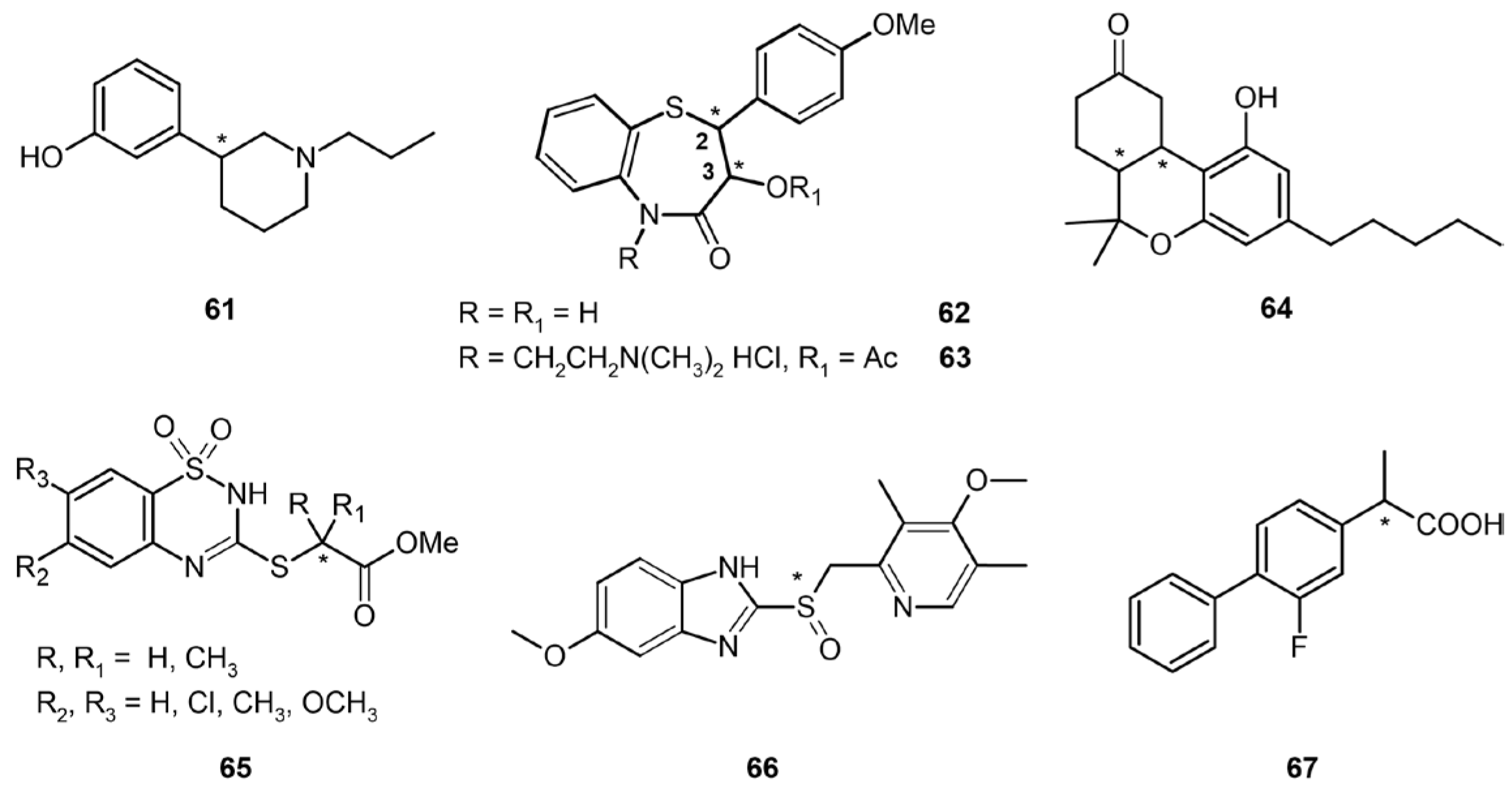
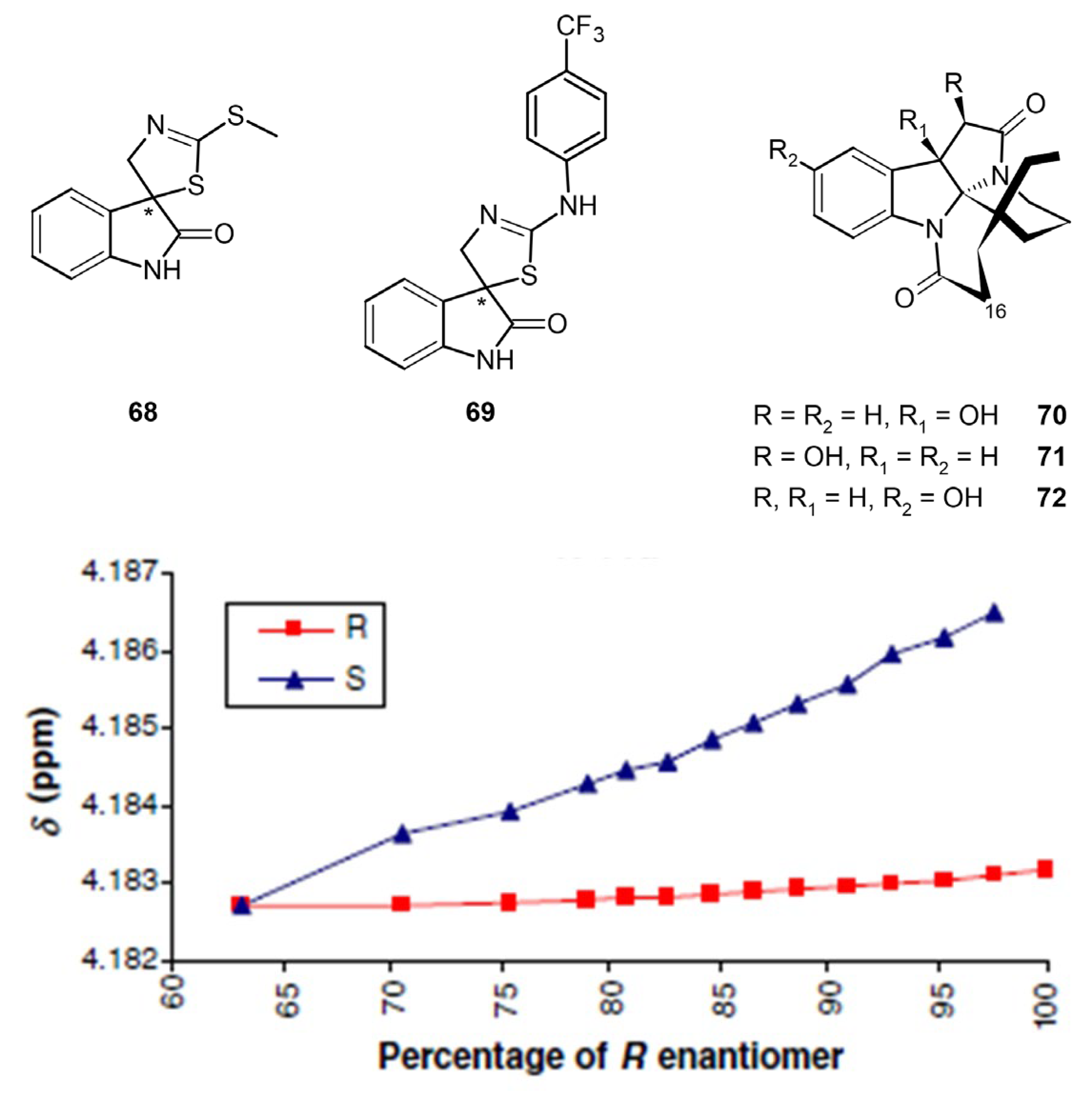
3.4. Chiral Drugs
3.5. Natural Products
3.6. Metal Complexes
4. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ouryupin, A.B.; Kadyko, M.I.; Petrovskii, P.V.; Fedin, E.I. Chiral discrimination in nonracemic mixtures of methanephosphonic acid, N,N′-bis(1-phenylethyl)diamides. Chirality 1994, 6, 1–4. [Google Scholar] [CrossRef]
- Williams, T.; Pitcher, R.G.; Bommer, P.; Gutzwiller, J.; Uskokovic, M. Diastereomeric solute-solute interactions of enantiomers in achiral solvents. Nonequivalence of the nuclear magnetic resonance spectra of racemic and optically active dihydroquinine. J. Am. Chem. Soc. 1969, 91, 1871–1872. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Mastryukova, T.A.; Fedin, E.I.; Vaisberg, M.S.; Morozov, L.L.; Petrovsky, P.V.; Shipov, A.E. An NMR study of optical isomers in solution. Tetrahedron 1976, 32, 1719–1728. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Mastryukova, T.A.; Fedin, E.I.; Vaisberg, M.S.; Morozov, L.L.; Petrovskii, P.V.; Shipov, A.E. Optical Isomers in Solution Investigated by Nuclear Magnetic Resonance. Russ. Chem. Rev. 1978, 47, 821–834. [Google Scholar] [CrossRef]
- Harger, M.J.P. Nuclear magnetic resonance non-equivalence of the enantiomers in optically active samples of phosphinic amides. J. Chem. Soc. Chem. Commun. 1976, 555–556. [Google Scholar] [CrossRef]
- Dobashi, A.; Saito, N.; Motoyama, Y.; Hara, S. Self-induced nonequivalence in the association of d- and l-amino acid derivatives. J. Am. Chem. Soc. 1986, 108, 307–308. [Google Scholar] [CrossRef]
- Luchinat, C.; Roelens, S. Enantiomeric purity determination of 1,2-diols through NMR spectroscopy without chiral auxiliaries. J. Am. Chem. Soc. 1986, 108, 4873–4878. [Google Scholar] [CrossRef]
- Giordano, C.; Restelli, A.; Villa, M.; Annunziata, R. A case of self-induced anisochrony in the proton NMR spectra of 1,5-benzothiazepines. J. Org. Chem. 1991, 56, 2270–2272. [Google Scholar] [CrossRef]
- Szántay, C.; Demeter, Á. Chapter 14—Self-Induced Recognition of Enantiomers (SIRE) in NMR Spectroscopy. In Anthropic Awareness; Szántay, C., Ed.; Elsevier: Boston, MA, USA, 2015; pp. 401–415. [Google Scholar] [CrossRef]
- Martens, J.; Bhushan, R. Purification of Enantiomeric Mixtures in Enantioselective Synthesis: Overlooked Errors and Scientific Basis of Separation in Achiral Environment. Helv. Chim. Acta 2014, 97, 161–187. [Google Scholar] [CrossRef]
- Soloshonok, V.A. Remarkable Amplification of the Self-Disproportionation of Enantiomers on Achiral-Phase Chromatography Columns. Angew. Chem. Int. Ed. 2006, 45, 766–769. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The self-disproportionation of enantiomers (SDE) of amino acids and their derivatives. Amino Acids 2019, 51, 865–889. [Google Scholar] [CrossRef] [PubMed]
- Balzano, F.; Uccello Barretta, G.; Aiello, F. Chapter 9—Chiral Analysis by NMR Spectroscopy: Chiral Solvating Agents. In Chiral Analysis, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 367–427. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Wzorek, A.; Klika, K.D. A question of policy: Should tests for the self-disproportionation of enantiomers (SDE) be mandatory for reports involving scalemates? Tetrahedron Asymmetry 2017, 28, 1430–1434. [Google Scholar] [CrossRef]
- Horeau, A.; Guetté, J.P. Interactions diastereoisomeres d’antipodes en phase liquide. Tetrahedron 1974, 30, 1923–1931. [Google Scholar] [CrossRef]
- Kabachnik, M.I. Associate diasteromerism of organophosphorus compounds. Phosphorus Sulfur Relat. Elem. 1977, 3, 239–246. [Google Scholar] [CrossRef]
- Cung, M.T.; Marraud, M.; Neel, J. Etude experimentale de L’auto-association des molécules modèles dipeptidiques. III. Influence de la dimerisation stéréosélective sur les spectres de résonance magnétique protonique. Biopolymers 1977, 16, 715–729. [Google Scholar] [CrossRef]
- Avignon, M.; Huong, P.V.; Lascombe, J.; Marraud, M.; Neel, J. Etude, par spectroscopie infra-rouge, de la conformation de quelques composés peptidiques modèles. Biopolymers 1969, 8, 69–89. [Google Scholar] [CrossRef]
- Cung, M.T.; Marraud, M.; Neel, J.; Aubry, A. Experimental study on aggregation of model dipeptide molecules. V. Stereoselective association of leucine dipeptides. Biopolymers 1978, 17, 1149–1173. [Google Scholar] [CrossRef]
- Nakao, Y.; Sugeta, H.; Kyogoku, Y. Intermolecular Hydrogen Bonding of Enantiomers of Pantolactone Studied by Infrared and 1H-NMR Spectroscopy. Bull. Chem. Soc. Jpn. 1985, 58, 1767–1771. [Google Scholar] [CrossRef]
- Sugeta, H. Spectrophotometric Determination of Formation Constants and Estimation of Molar Absorption Spectra of Individual Components in Chemical Equilibria. Infrared Study of Intermolecular Hydrogen Bonding of 2-Aminopyridine. Bull. Chem. Soc. Jpn. 1981, 54, 3706–3710. [Google Scholar] [CrossRef]
- Fedin, E.I.; Davankov, V.A. NMR investigations of the enantiomeric excess effects in solutions with weak intermolecular association. Chirality 1995, 7, 326–330. [Google Scholar] [CrossRef]
- Gut, D.; Rudi, A.; Kopilov, J.; Goldberg, I.; Kol, M. Pairing of Propellers: Dimerization of Octahedral Ruthenium(II) and Osmium(II) Complexes of Eilatin via π−π Stacking Featuring Heterochiral Recognition. J. Am. Chem. Soc. 2002, 124, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Szakács, Z.; Sánta, Z.; Lomoschitz, A.; Szántay, C. Self-induced recognition of enantiomers (SIRE) and its application in chiral NMR analysis. TrAC Trends Anal. Chem. 2018, 109, 180–197. [Google Scholar] [CrossRef]
- Ouryupin, A.B.; Kadyko, M.I.; Petrovskii, P.V.; Fedin, E.I.; Okruszek, A.; Kinas, R.; Stec, W.J. Enantiomeric 2-anilino-2-oxo-1,3,2-oxazaphosphorinanes: Synthesis and NMR-investigation of their non-racemic mixtures. Tetrahedron Asymmetry 1995, 6, 1813–1824. [Google Scholar] [CrossRef]
- Han, J.; Fustero, S.; Moriwaki, H.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D. The Self-Disproportionation of Enantiomers (SDE): Fluorine as an SDE-Phoric Substituent. In Organofluorine Chemistry; Szabó, K., Selander, N., Eds.; Wiley: Weinheim, Germany, 2021; pp. 281–306. [Google Scholar]
- Klika, K.D.; Budovská, M.; Kutschy, P. Enantiodifferentiation of phytoalexin spirobrassinin derivatives using the chiral solvating agent (R)-(+)-1,1′-bi-2-naphthol in conjunction with molecular modeling. Tetrahedron Asymmetry 2010, 21, 647–658. [Google Scholar] [CrossRef]
- Nieminen, V.; Murzin, D.Y.; Klika, K.D. NMR and molecular modeling of the dimeric self-association of the enantiomers of 1,1′-bi-2-naphthol and 1-phenyl-2,2,2-trifluoroethanol in the solution state and their relevance to enantiomer self-disproportionation on achiral-phase chromatography (ESDAC). Org. Biomol. Chem. 2009, 7, 537–542. [Google Scholar] [CrossRef]
- Horman, I.; Dreux, B. Estimation of Dimerisation Constants from Complexatin-Induced Displacements of 1H-NMR Chemical Shifts: Dimerisation of Caffeine. Helv. Chim. Acta 1984, 67, 754–764. [Google Scholar] [CrossRef]
- Chen, J.S.; Shirts, R.B. Iterative determination of the NMR monomer shift and dimerization constant in a self-associating system. J. Phy. Chem. 1985, 89, 1643–1646. [Google Scholar] [CrossRef]
- Jenn-shing, C.; Rosenberger, F. Accurate nmr data evaluation for monomer shift, dimer shift and dimerization constant in a self-associating system. Tetrahedron Lett. 1990, 31, 3975–3978. [Google Scholar] [CrossRef]
- Brynn Hibbert, D.; Thordarson, P. The death of the Job plot, transparency, open science and online tools, uncertainty estimation methods and other developments in supramolecular chemistry data analysis. Chem. Commun. 2016, 52, 12792–12805. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Mastryukova, T.A.; Fedin, É.I.; Shipov, É.A.; Vaisberg, M.S.; Petrovskii, P.V.; Morozov, L.L. Study of diastereomeric anisochronism of some O-ethyl methyldithiophosphonates that contain a second chiral center in the thiol group. Bull. Acad. Sci. USSR Div. Chem. Sci. 1975, 24, 537–543. [Google Scholar] [CrossRef]
- Kabachnik, M.I.; Fedin, É.I.; Morozov, L.L.; Vaisberg, M.S.; Petrovskii, P.V.; Shipov, A.É.; Mastryukova, T.A. Diastereomeric anisochronism in rapid interaggregate exchange. Bull. Acad. Sci. USSR Div. Chem. Sci. 1976, 25, 58–67. [Google Scholar] [CrossRef]
- Harger, M.J.P. Proton magnetic resonance non-equivalence of the enantiomers of alkylphenylphosphinic amides. J. Chem. Soc. Perkin Trans. 2 1977, 1882–1887. [Google Scholar] [CrossRef]
- Harger, M.J.P. Chemical shift non-equivalence of enantiomers in the proton magnetic resonance spectra of partly resolved phosphinothioic acids. J. Chem. Soc. Perkin Trans. 2 1978, 326–331. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Żurawiński, R.; Kiełbasiński, P.; Wieczorek, M.W.; Błaszczyk, J.; Majzner, W.R. Total Synthesis of Racemic and Optically Active Sarkomycin. Synthesis 1997, 1997, 356–365. [Google Scholar] [CrossRef]
- Dašková, V.; Padín, D.; Feringa, B.L. Turning Enantiomeric Relationships into Diastereomeric Ones: Self-Resolving α-Ureidophosphonates and Their Organocatalytic Enantioselective Synthesis. J. Am. Chem. Soc. 2022, 144, 23603–23613. [Google Scholar] [CrossRef]
- Cung Manh, T.; Marraud, M.; Neel, J. Estimation of the optical purity of chiral peptide derivatives by proton NMR spectroscopy. C. R. Hebd. Seances Acad. Sci. Ser. C 1975, 281, 691–694. [Google Scholar]
- Cung, M.T.; Marraud, M.; Neel, J. Étude expérimentale de l’auto-association des molécules modèles dipeptidiques. II. Association stéréosélective des molécules énantiomères. Biopolymers 1976, 15, 2081–2095. [Google Scholar] [CrossRef]
- Ghosh, S.K. Chiral recognition in dipeptides containing 1-aminocyclopropane carboxylic acid or α-aminoisobutyric acid: NMR studies in solution. J. Peptide Res. 1999, 53, 261–274. [Google Scholar] [CrossRef]
- Kuberski, B.; Szumna, A. A self-assembled chiral capsule with polar interior. Chem. Commun. 2009, 1959–1961. [Google Scholar] [CrossRef]
- Storch, G.; Haas, M.; Trapp, O. Attracting Enantiomers: Chiral Analytes That Are Simultaneously Shift Reagents Allow Rapid Screening of Enantiomeric Ratios by NMR Spectroscopy. Chem. Eur. J. 2017, 23, 5414–5418. [Google Scholar] [CrossRef]
- Jursic, B.S.; Goldberg, S.I. Enantiomer discrimination arising from solute-solute interactions in partially resolved chloroform solutions of chiral carboxamides. J. Org. Chem. 1992, 57, 7172–7174. [Google Scholar] [CrossRef]
- Wagger, J.; Grdadolnik, S.G.; Grošelj, U.; Meden, A.; Stanovnik, B.; Svete, J. Chiral solvating properties of (S)-1-benzyl-6-methylpiperazine-2,5-dione. Tetrahedron Asymmetry 2007, 18, 464–475. [Google Scholar] [CrossRef]
- Huang, S.-H.; Bai, Z.-W.; Feng, J.-W. Chiral self-discrimination of the enantiomers of α-phenylethylamine derivatives in proton NMR. Magn. Reson. Chem. 2009, 47, 423–427. [Google Scholar] [CrossRef]
- Safont-Sempere, M.M.; Osswald, P.; Radacki, K.; Würthner, F. Chiral Self-Recognition and Self-Discrimination of Strapped Perylene Bisimides by π-Stacking Dimerization. Chem. Eur. J. 2010, 16, 7380–7384. [Google Scholar] [CrossRef]
- Safont-Sempere, M.M.; Osswald, P.; Stolte, M.; Grüne, M.; Renz, M.; Kaupp, M.; Radacki, K.; Braunschweig, H.; Würthner, F. Impact of Molecular Flexibility on Binding Strength and Self-Sorting of Chiral π-Surfaces. J. Am. Chem. Soc. 2011, 133, 9580–9591. [Google Scholar] [CrossRef]
- Baumann, A.; Wzorek, A.; Soloshonok, V.A.; Klika, K.D.; Miller, A.K. Potentially Mistaking Enantiomers for Different Compounds Due to the Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon. Symmetry 2020, 12, 1106–1121. [Google Scholar] [CrossRef]
- Arnold, W.; Daly, J.J.; Imhof, R.; Kyburz, E. An efficient resolution of 3-PPP and assignment of the absolute configuration. Tetrahedron Lett. 1983, 24, 343–346. [Google Scholar] [CrossRef]
- Sonstrom, R.E.; Neill, J.L.; Mikhonin, A.V.; Doetzer, R.; Pate, B.H. Chiral analysis of pantolactone with molecular rotational resonance spectroscopy. Chirality 2022, 34, 114–125. [Google Scholar] [CrossRef]
- Hui, R.A.H.F.; Salamone, S.; Williams, T.H. Self-induced nonequivalence in the 1H-NMR spectra of the (+)- and (−)-isomers of a cannabinoid ketone intermediate. Pharmacol. Biochem. Behav. 1991, 40, 491–496. [Google Scholar] [CrossRef]
- Srinivasan, K. Chapter 42—Polyphenols in Vision and Eye Health. In Handbook of Nutrition, Diet and the Eye; Preedy, V.R., Ed.; Academic Press: San Diego, CA, USA, 2014; pp. 413–421. [Google Scholar]
- Tait, A.; Colorni, E.; Di Bella, M. Stereospecific synthesis of 2-[(2H-1,2,4-benzothiadiazine-1,1-dioxide-3-yl)thio]propanoic acids: Enantiomeric excess evaluation by 1H NMR. Tetrahedron Asymmetry 1997, 8, 2199–2207. [Google Scholar] [CrossRef]
- Hentschel, P.; Holtin, K.; Steinhauser, L.; Albert, K. Monitoring the On-line Titration of Enantiomeric Omeprazole Employing Continuous-flow Capillary Microcoil 1H NMR Spectroscopy. Chirality 2012, 24, 1074–1076. [Google Scholar] [CrossRef]
- Kwiatkowska, M.; Wzorek, A.; Kolbus, A.; Urbaniak, M.; Han, J.; Soloshonok, V.A.; Klika, K.D. Flurbiprofen: A Study of the Behavior of the Scalemate by Chromatography, Sublimation, and NMR. Symmetry 2021, 13, 543–560. [Google Scholar] [CrossRef]
- Klika, K.D.; Budovská, M.; Kutschy, P. NMR spectral enantioresolution of spirobrassinin and 1-methoxyspirobrassinin enantiomers using (S)-(−)-ethyl lactate and modeling of spirobrassinin self-association for rationalization of its self-induced diastereomeric anisochromism (SIDA) and enantiomer self-disproportionation on achiral-phase chromatography (ESDAC) phenomena. J. Fluor. Chem. 2010, 131, 467–476. [Google Scholar] [CrossRef]
- Budovská, M.; Tischlerová, V.; Mojžiš, J.; Harvanová, M.; Kozlov, O.; Gondová, T.; Tomášková, N. 2′-Aminoanalogues of the cruciferous phytoalexins spirobrassinin, 1-methoxyspirobrassinin and 1-methoxyspirobrassinol methyl ether: Synthesis and anticancer properties. Tetrahedron 2017, 73, 6356–6371. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, Q.; Zhu, J. Total Syntheses of (−)-Mersicarpine, (−)-Scholarisine G, (+)-Melodinine E, (−)-Leuconoxine, (−)-Leuconolam, (−)-Leuconodine A, (+)-Leuconodine F, and (−)-Leuconodine C: Self-Induced Diastereomeric Anisochronism (SIDA) Phenomenon for Scholarisine G and Leuconodines A and C. J. Am. Chem. Soc. 2015, 137, 6712–6724. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, M.; Hans, M.; Jauch, J. Enantioselective Synthesis of Myrtucommulone A. Eur. J. Org. Chem. 2013, 2013, 4078–4084. [Google Scholar] [CrossRef]
- Ajisaka, K.; Kainosho, M. Diastereomeric interaction of partially resolved amines facilitated by lanthanide chelates. Evidence for dynamic equilibrium between seven-coordinate and eight-coordinate alkylamine-lanthanide chelate adducts. J. Am. Chem. Soc. 1975, 97, 1761–1765. [Google Scholar] [CrossRef]
- Reuben, J. Aqueous lanthanide shift reagents. 8. Chiral interactions and stereochemical assignments of chemically and isotopically chiral ligands. J. Am. Chem. Soc. 1980, 102, 2232–2237. [Google Scholar] [CrossRef]
- Maury, O.; Lacour, J.; Le Bozec, H. Diastereoselective Homochiral Self-Assembly Between Anions and Cation in Solution. Eur. J. Inorg. Chem. 2001, 2001, 201–204. [Google Scholar] [CrossRef]
- Bergman, S.D.; Kol, M. π-Stacking Induced NMR Spectrum Splitting in Enantiomerically Enriched Ru(II) Complexes: Evaluation of Enantiomeric Excess. Inorg. Chem. 2005, 44, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Morita, F.; Nogami, J.; Araujo Dias, A.J.; Kinoshita, S.; Nagashima, Y.; Tanaka, K. Regiodivergent Synthesis and π-Stacking-Induced Chiral Self-Recognition of Hexabenzocoronene-Based [6]Helicenes. Eur. J. Org. Chem. 2022, 2022, e202200690–e202200697. [Google Scholar] [CrossRef]
- Fedin, E.I. NMR of Chiral Molecules. In Proceedings of the 20th Congress AMPERE, Tallinn, Estonia, 21–26 August 1978. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Definition | δ | Definition |
|---|---|---|---|
| E/E′ | enantiomer/opposite enantiomer | δm | monomer chemical shift |
| EE/E′E′ | homodimers | δhomo | homodimer chemical shift |
| EE′ | heterodimer | δhetero | heterodimer chemical shift |
| D/D′ | diastereomer pairs | δD/δD′ | diastereomers chemical shift |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, F.; Uccello Barretta, G.; Balzano, F.; Spiaggia, F. The Phenomenon of Self-Induced Diastereomeric Anisochrony and Its Implications in NMR Spectroscopy. Molecules 2023, 28, 6854. https://doi.org/10.3390/molecules28196854
Aiello F, Uccello Barretta G, Balzano F, Spiaggia F. The Phenomenon of Self-Induced Diastereomeric Anisochrony and Its Implications in NMR Spectroscopy. Molecules. 2023; 28(19):6854. https://doi.org/10.3390/molecules28196854
Chicago/Turabian StyleAiello, Federica, Gloria Uccello Barretta, Federica Balzano, and Fabio Spiaggia. 2023. "The Phenomenon of Self-Induced Diastereomeric Anisochrony and Its Implications in NMR Spectroscopy" Molecules 28, no. 19: 6854. https://doi.org/10.3390/molecules28196854
APA StyleAiello, F., Uccello Barretta, G., Balzano, F., & Spiaggia, F. (2023). The Phenomenon of Self-Induced Diastereomeric Anisochrony and Its Implications in NMR Spectroscopy. Molecules, 28(19), 6854. https://doi.org/10.3390/molecules28196854