
The Triterpenoids from Munronia pinnata and Their Anti-Proliferative Effects

 ,
,
Abstract
:1. Introduction
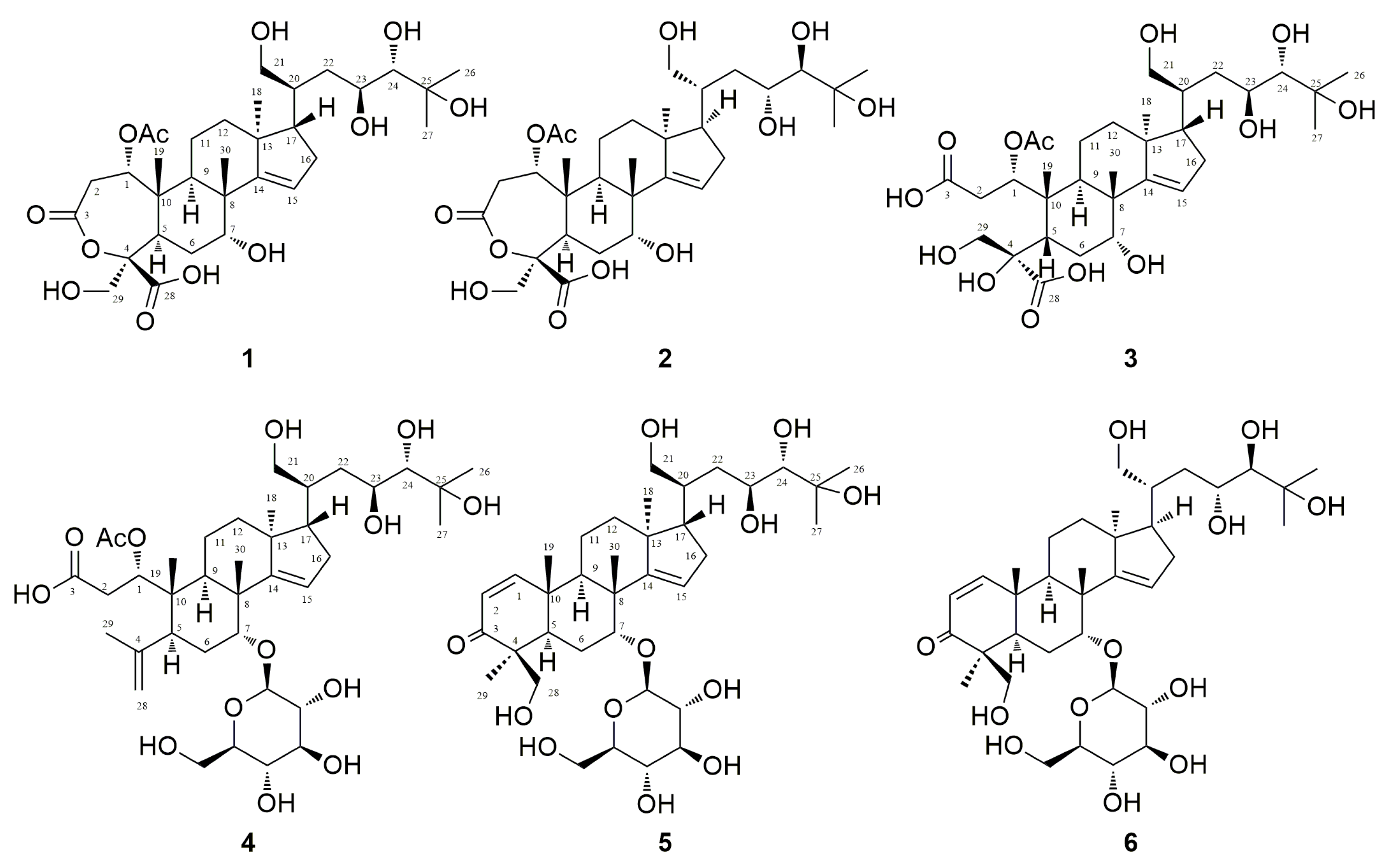
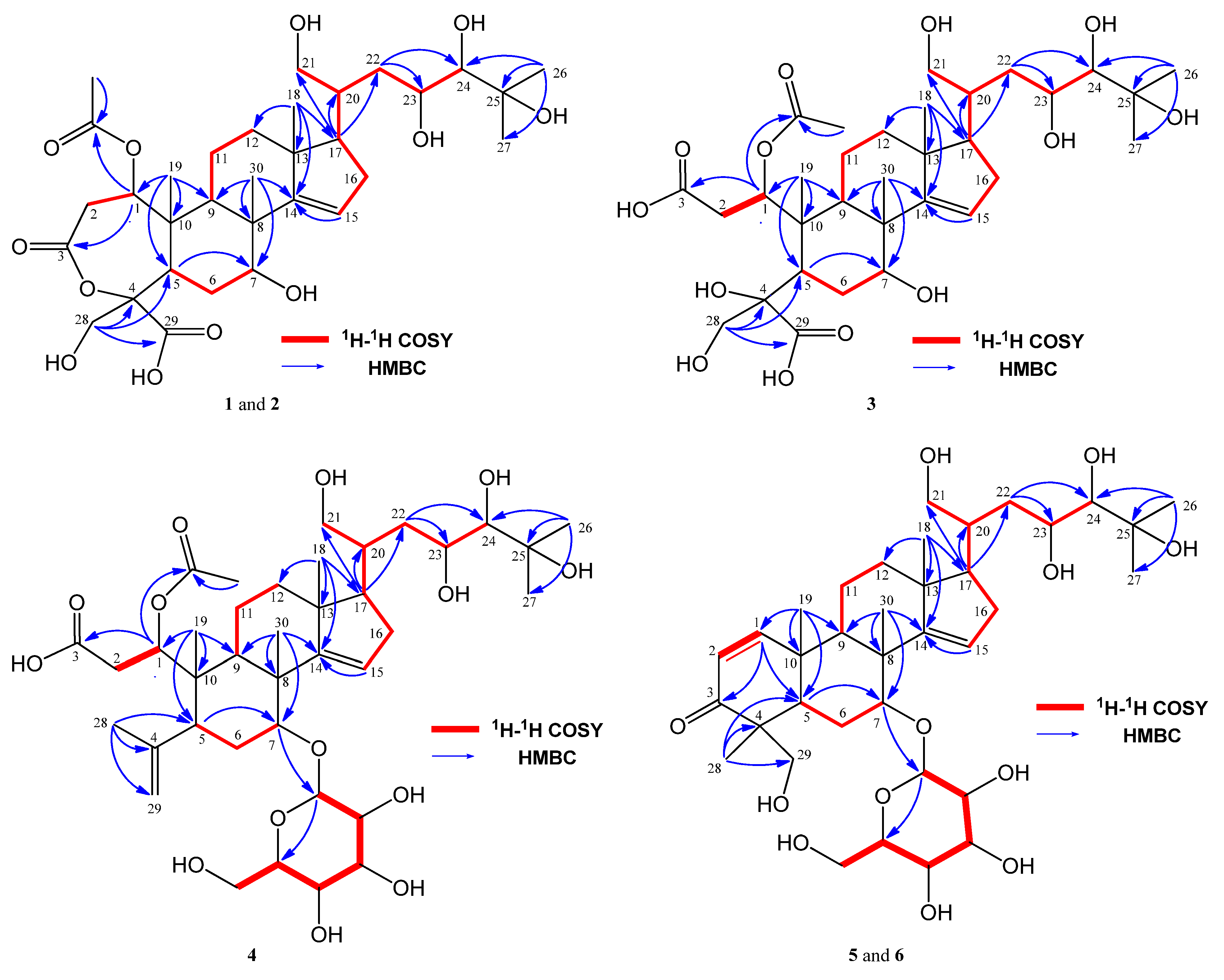
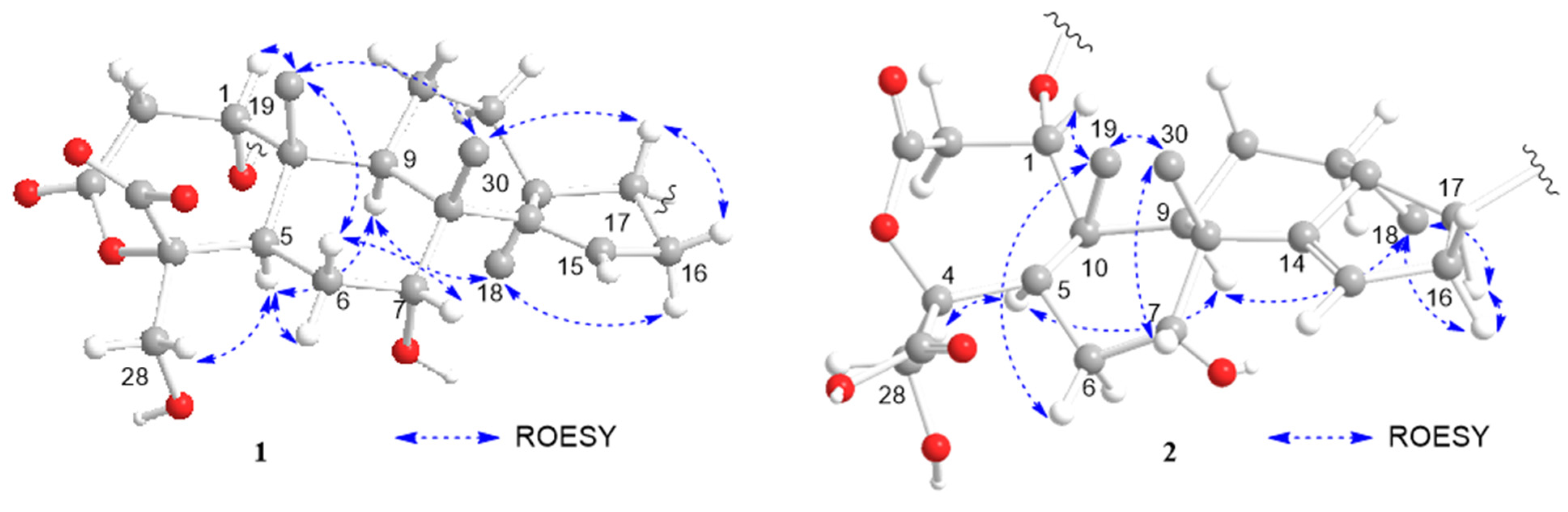
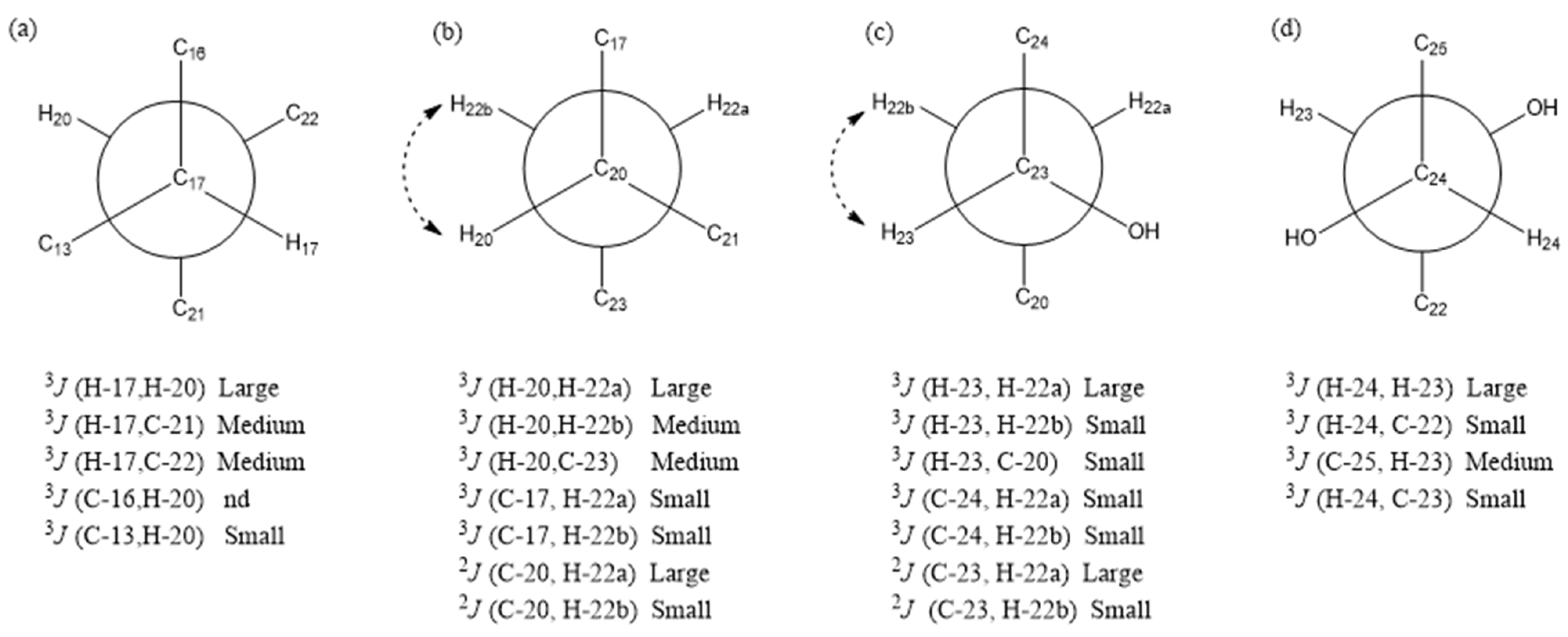
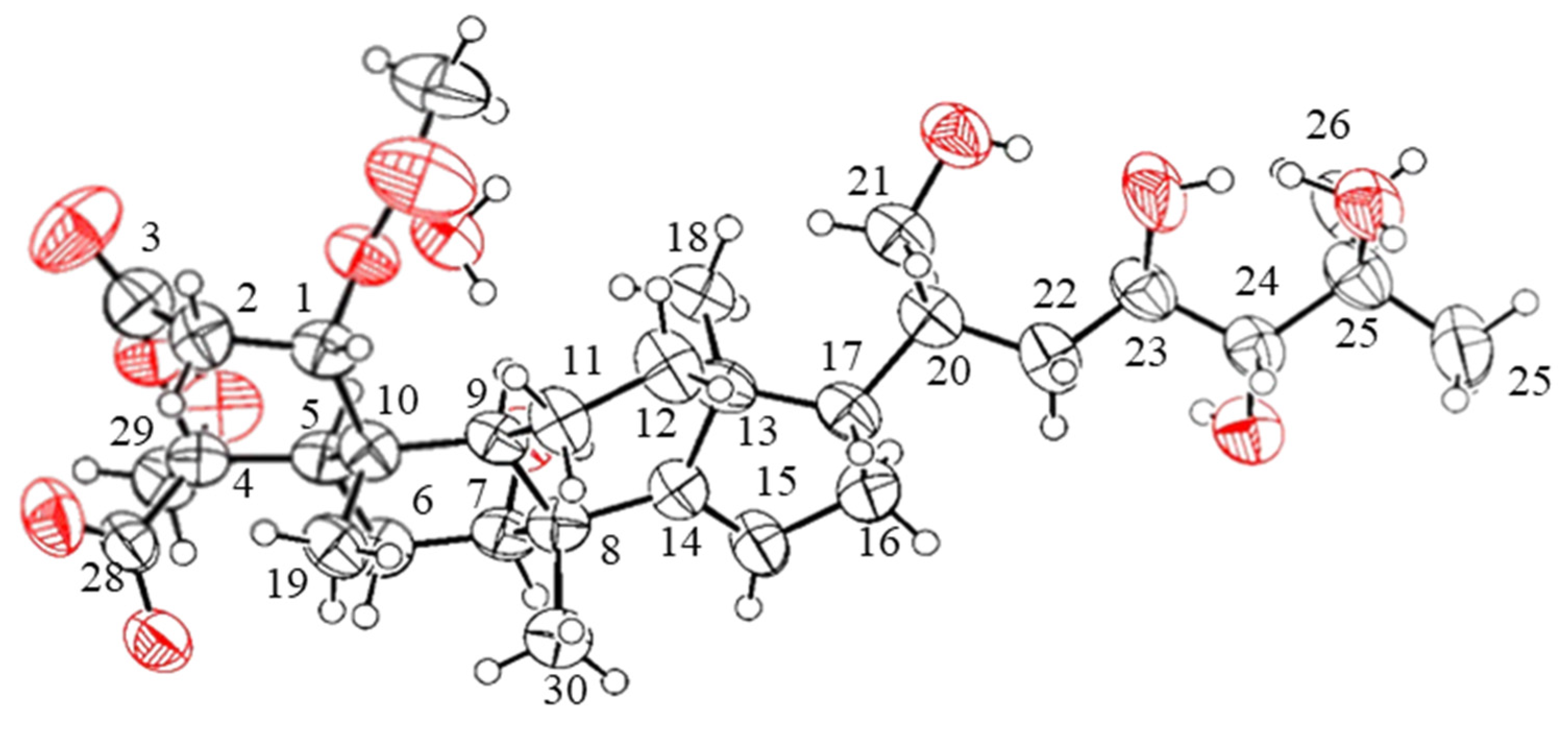
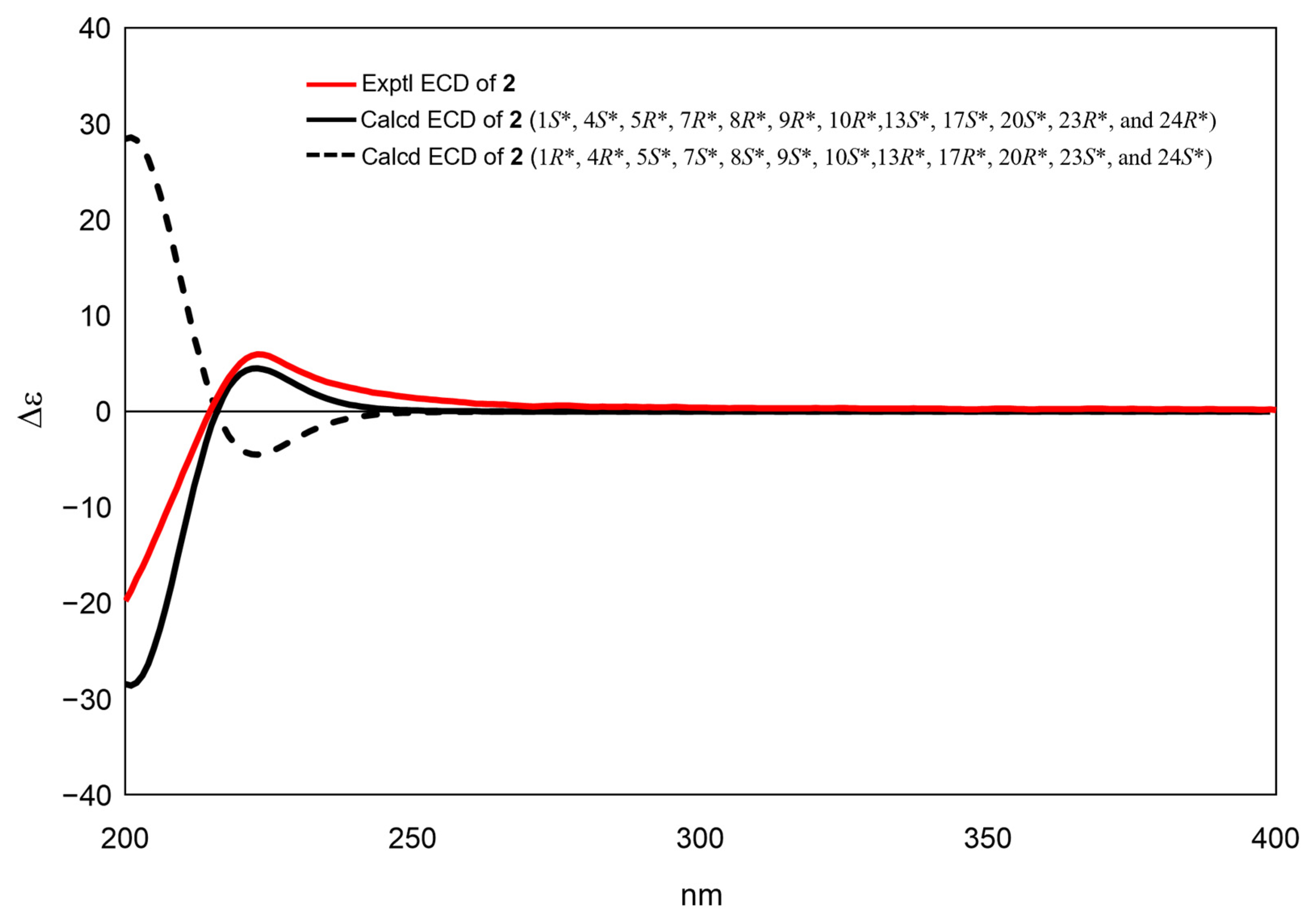
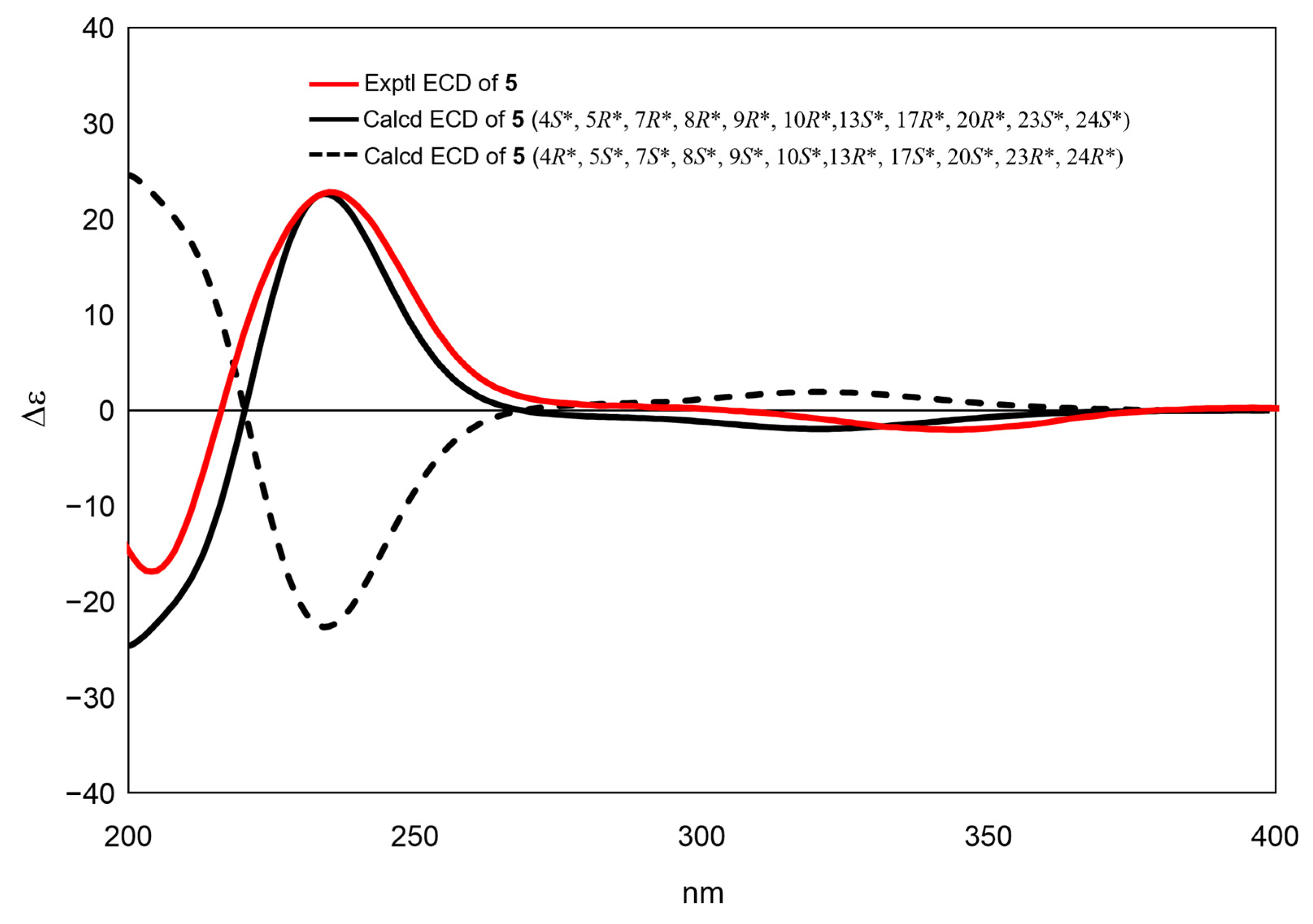
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Protocols
3.2. Plant Material
3.3. Extraction and Isolation
3.3.1. Munropene A (Compound 1)
3.3.2. Munropene B (Compound 2)
3.3.3. Munropene C (Compound 3)
3.3.4. Munropene D (Compound 4)
3.3.5. Munropene E (Compound 5)
3.3.6. Munropene F (Compound 6)
3.4. Acid Hydrolysis and Sugar Analysis of Munropenes D–F (Compounds 4–6)
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Guoqiang, W. Compilation of China Medicinal Herbal; People’s Medical Publishing House: Beijing, China, 1975; Volume 2, p. 1081. [Google Scholar]
- Guangxi Institute of Botany. Flora of Guangxi: Volume 3 Specermatophyta; Guangxi Science &Technology Publishing House: Nanning, China, 2011; p. 333. [Google Scholar]
- Huang, M.-H.; Zhang, Y.-M.; Li, J.; Shao, F.; Yang, M.; Zhang, P.-Z. Research progress on chemical constituents of plants from Munronia Wight and their biological activities. Chin. Tradit. Herb. Drugs 2017, 48, 1240–1249. [Google Scholar]
- Su, L.; Huang, L.-Z.; Cai, Y. Preliminary study on the analgesic, anti-inflammation and anti-acute liver injury effects of the water extract from Zhuang medicine Munronia delavayi Franch. J. Youjiang Med. Univ. Natl. 2021, 43, 741–745. [Google Scholar] [CrossRef]
- Cai, X.-H.; Du, Z.-Z.; Luo, X.-D. Tirucallane Triterpenoid Saponins from Munronia delavayi Franch. Helv. Chim. Acta 2007, 90, 1980–1986. [Google Scholar] [CrossRef]
- Li, X.-L.; He, Q.-X.; Zhang, F.-L.; Zhao, Y.-L.; Liu, K.-C.; Jiang, S.-P. Chemical constituents from Munronia sinica and their bioactivities. Nat. Prod. Bioprospecting 2012, 2, 76–80. [Google Scholar] [CrossRef]
- Qi, S.-H.; Wu, D.-G.; Ma, Y.-B.; Luo, X.-D. The chemical constituents of Munronia Henryi. J. Asian Nat. Prod. Res. 2003, 5, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.-H.; Wu, D.-G.; Chen, L.; Ma, Y.-B.; Luo, X.-D. Insect Antifeedants from Munronia henryi: Structure of Munroniamide. J. Agric. Food Chem. 2003, 51, 6949–6952. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, J.-X.; Huang, T.; Mao, X.-Y.; Gu, W.; He, H.-P.; Di, Y.-T.; Li, S.-L.; Chen, D.-Z.; Zhang, Y.; et al. Bioactive Limonoid Constituents of Munronia henryi. J. Nat. Prod. 2015, 78, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-R.; Tanaka, N.; Tsuji, D.; Lu, F.-L.; Yan, X.-J.; Itoh, K.; Li, D.-P.; Kashiwada, Y. Limonoids from the aerial parts of Munronia pinnata. Tetrahedron 2019, 75, 130779. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical Determination of Acyclic Structures Based on Carbon−Proton Spin-Coupling Constants. A Method of Configuration Analysis for Natural Products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Otting, G.; Wüthrich, K. Heteronuclear filters in two-dimensional [ 1 H, 1 H]-NMR spectroscopy: Combined use with isotope labelling for studies of macromolecular conformation and intermolecular interactions. Q. Rev. Biophys. 1990, 23, 39–96. [Google Scholar] [CrossRef] [PubMed]
- Wollborn, U.; Leibfritz, D. Measurements of heteronuclear long-range coupling constants from inverse homonuclear 2D NMR spectra. J. Magn. Reson. 1969 1992, 98, 142–146. [Google Scholar] [CrossRef]
- Rodriguez, A.D. Two-dimensional NMR spectra of asbestinin diterpenes isolated from Briareum asbestinum (Pallas). Total stereochemical assignment by two-dimensional NMR techniques. Magn. Reson. Chem. 1992, 30, 977–986. [Google Scholar] [CrossRef]
- Zhu, G.; Bax, A. Measurement of Long-Range 1H-13C Coupling Constants from Quantitative 2D Heteronuclear Multiple-Quantum Correlation Spectra. J. Magn. Reson. A 1993, 104, 353–357. [Google Scholar] [CrossRef]
- Zhu, G.; Live, D.; Bax, A. Analysis of Sugar Puckers and Glycosidic Torsion Angles in a DNA G-Tetrad Structure by Heteronuclear Three-Bond J Couplings. J. Am. Chem. Soc. 1994, 116, 8370–8371. [Google Scholar] [CrossRef]
- Yang, X.-R.; Tanaka, N.; Song, J.-R.; Lu, F.-L.; Yan, X.-J.; Li, J.-X.; Zhao, X.-Y.; Kashiwada, Y.; Li, D.-P. Rhodomollosides A and B, glycosides of methyl everninate from the aerial parts of Rhododendron molle. J. Asian Nat. Prod. Res. 2022, 24, 1025–1032. [Google Scholar] [CrossRef]
- Murphy, W.S. The octant rule: Its place in organic stereochemistry. J. Chem. Educ. 1975, 52, 774. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 4.66 (1H, d, 7.2) | 71.3 | 4.78 (1H, d, 7.5) | 73.3 | 6.21 (1H, d, 7.5) | 78.8 | 5.55 (1H, dd, 11.1, 1.6) | 79.1 |
| 2 | 3.31 (1H, d,16.0)/ 2.77 (1H, m) | 35.5 | 3.84 (1H, m)/ 2.93 (1H, m) | 37.1 | 2.41 (1H, m)/ 2.99 (1H, m) | 36.2 | 2.41 (1H, dd, 15.1, 11.0)/ 3.08 (1H, m) | 36.3 |
| 3 | - | 170.5 | - | 175.8 | - | 175.5 | - | 175.5 |
| 4 | - | 87.4 | - | 91.4 | - | 81.9 | - | 147.7 |
| 5 | 2.77 (1H, m) | 39.2 | 2.83 (1H, dd, 13.0, 2.6) | 40.3 | 2.37 (1H, dd, 13.0, 2.6) | 42.7 | 2.79 (1H, dd, 13.0, 3.4) | 43.9 |
| 6 | 2.15 (1H, m)/ 1.87 (1H, m) | 27.6 | 2.49 (1H, m)/ 1.99 (1H, m) | 28.8 | 2.29 (1H, m)/ 1.68 (1H, m) | 28.1 | 2.07 (1H, m)/ 1.76 (1H, dt, 14.5, 3.4) | 28.4 |
| 7 | 3.75 (1H, m) | 70.7 | 3.91 (1H, t, 3.1) | 73 | 3.85 (1H, t, 3.1) | 73 | 4.03 (1H, dd, 3.7, 1.5) | 78.4 |
| 8 | - | 42.8 | - | 44.5 | - | 44.6 | - | 44.3 |
| 9 | 2.40 (1H, m) | 33.3 | 2.52 (1H, m) | 34.8 | 2.29 (1H, m) | 35.3 | 2.30 (1H, m) | 35.5 |
| 10 | - | 43.6 | - | 45.2 | - | 47.1 | - | 45.5 |
| 11 | 1.22 (1H, m)/ 1.40 (1H, m) | 16.4 | 1.47 (1H, m)/ 1.52 (1H, m) | 17.8 | 1.68 (1H, m)/ 2.01 (1H, m) | 20.3 | 1.63 (1H, m)/ 1.94 (1H, m) | 20.1 |
| 12 | 1.62 (1H, m)/ 1.40 (1H, m) | 34.4 | 1.84 (1H, m)/ 1.49 (1H, m) | 35.6 | 1.50 (1H, dt, 13.1, 9.3)/ 1.92 (1H, m) | 36.1 | 1.57 (1H, m)/ 1.87 (1H, m) | 37.3 |
| 13 | - | 46 | - | 47.7 | - | 47.3 | - | 47.3 |
| 14 | - | 160.2 | - | 162 | - | 161.8 | - | 159.5 |
| 15 | 5.32 (1H, m) | 119.1 | 5.46 (1H, dd, 3.5, 1.5) | 121 | 5.45 (1H, dd, 3.8, 1.5) | 120.7 | 5.45 (1H, dd, 3.7, 1.7) | 121.6 |
| 16 | 2.02 (1H, m)/ 2.15 (1H, m) | 34.6 | 2.18 (1H, m)/ 2.34 (1H, m) | 35.9 | 2.18 (1H, m)/ 2.29 (1H, m) | 35.9 | 2.08 (1H, m)/ 2.30 (1H, m) | 36.2 |
| 17 | 1.57 (1H, m) | 55.9 | 1.66 (1H, td, 10.4, 7.2) | 56.2 | 1.68 (1H, m) | 57.9 | 1.63 (1H, m) | 58.2 |
| 18 | 0.90 (3H, s) | 18.4 | 1.04 (3H, s) | 19 | 1.10 (3H, s) | 19.4 | 1.12 (3H, s) | 21.1 |
| 19 | 0.94 (3H, s) | 13.1 | 1.14 (3H, s) | 14.1 | 1.04 (3H, s) | 14.8 | 0.97 (3H, s) | 15.7 |
| 20 | 1.68 (1H, m) | 39.5 | 1.84 (1H, m) | 41.7 | 1.19 (1H, m) | 41.8 | 1.87 (1H, m) | 42.1 |
| 21 | 3.23 (1H, m)/ 3.59 (1H, m) | 63.9 | 3.34 (1H, m)/3.84 (1H, m) | 66 | 3.36 (1H, m)/ 3.89 (1H, dd, 10.6, 3.6) | 66.1 | 3.34 (1H, m)/ 3.89 (1H, dd, 10.5, 3.6) | 66.3 |
| 22 | 1.94 (1H, m)/ 1.22 (1H, m) | 36.9 | 1.36 (1H, m)/ 2.18 (1H, m) | 38.5 | 1.37 (1H, m)/ 2.18 (1H, m) | 38.7 | 1.35 (1H, ddd, 14.7, 9.3, 7.3)/ 2.22 (1H, dt, 14.8, 2.9) | 38.8 |
| 23 | 3.49 (1H, m) | 72.9 | 3.66 (1H, td, 8.9,2.1) | 74.7 | 3.67 (1H, td, 8.9, 2.1) | 74.8 | 3.67 (1H, td, 9.0, 2.1) | 74.9 |
| 24 | 2.89 (1H, d, 8.1) | 78.4 | 3.09 (1H, d, 8.2) | 79.9 | 3.08 (1H, d, 8.2) | 79.9 | 3.08 (1H, m) | 79.9 |
| 25 | - | 73.2 | - | 74.9 | - | 75 | - | 75 |
| 26 | 1.08 (3H, s) | 28 | 1.22 (3H, s) | 27.9 | 1.22 (3H, s) | 28 | 1.24 (3H, s) | 23.9 |
| 27 | 1.09 (3H, s) | 24 | 1.24 (3H, s) | 24.1 | 1.24 (3H, s) | 23.9 | 1.22 (3H, s) | 28 |
| 28 | 3.62 (1H, m)/ 3.81 (1H, d, 10.9) | 172.2 | 3.78 (1H, d, 14.7)/ 4.01 (1H, d, 11.1) | 177.6 | 3.78 (1H, d, 10.4)/ 4.01 (1H, d, 10.4) | 178.7 | 4.96 (1H, d, 2.4)/ 4.88 (1H, d, 2.4) | 116.1 |
| 29 | - | 70.7 | - | 70.9 | - | 69.4 | 1.83 (3H, s) | 23.7 |
| 30 | 1.03 (3H, s) | 27.4 | 1.16 (3H, s) | 28.3 | 1.08 (3H, s) | 27.6 | 1.17 (3H, s) | 27.5 |
| 1-OAc | - | 169.5 | - | 171.6 | - | 172.5 | - | 172.4 |
| 1.99 (3H, s) | 20.7 | 2.06 (3H, s) | 20.9 | 2.00 (3H, s) | 21.2 | 2.00 (3H, s) | 21.1 | |
| 1′ | - | - | - | - | - | - | 4.30 d (7.8) | 100.3 |
| 2′ | - | - | - | - | - | - | 3.12 (1H, m) | 75.4 |
| 3′ | - | - | - | - | - | - | 3.33 (1H, m) | 78.8 |
| 4′ | - | - | - | - | - | - | 3.17 (1H, m) | 72.3 |
| 5′ | - | - | - | - | - | - | 3.18 (1H, dd, 9.7, 2.4) | 77.6 |
| 6′ | - | - | - | - | - | - | 3.59 (1H, dd, 11.5, 5.9)/ 3.88 (1H, dd, 11.4, 2.3) | 63.5 |
| Position | 5 | 6 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 7.28 (1H, d, 10.2) | 161.3 | 7.28(1H, d, 10.2) | 161.3 |
| 2 | 5.77 (1H, d, 10.2) | 125.6 | 5.77(1H, d, 10.2) | 125.6 |
| 3 | 206.3 | 206.3 | ||
| 4 | 51.7 | 51.7 | ||
| 5 | 2.63 (1H, dd, 10.2, 5.2) | 46 | 2.63(1H, dd, 10.2, 5.4) | 46 |
| 6 | 2.03 (1H, m) | 23.3 | 2.03 (1H, m) | 23.3 |
| 7 | 4.14 (1H, t, 2.8, 2.8) | 78.3 | 4.14 (1H, t, 2.8, 2.8) | 78.4 |
| 8 | 44.9 | 44.9 | ||
| 9 | 2.25 (1H, m) | 40.1 | 2.25 (1H, m) | 40.1 |
| 10 | 41.1 | 41 | ||
| 11 | 1.68 (1H, m)/1.87 (1H, m) | 18.7 | 1.66 (1H, m)/1.87 (1H, m) | 18.7 |
| 12 | 1.68 (1H, m)/1.87 (1H, m) | 37.3 | 1.66 (1H, m)/1.87 (1H, m) | 37.5 |
| 13 | 47.6 | 47.6 | ||
| 14 | 158.7 | 159.1 | ||
| 15 | 5.49 (1H, dd, 3.7, 1.7) | 122.2 | 5.49 (1H, dd, 3.7, 1.7) | 122.2 |
| 16 | 2.03(1H, m)/2.25(1H, m) | 36.1 | 2.07 (1H, m)/2.30 (1H, ddd, 15.1, 7.2, 3.5) | 36.3 |
| 17 | 1.72(1H, m) | 57.5 | 1.66 (1H, m) | 58.1 |
| 18 | 1.06 (3H, s) | 21.2 | 1.06 (3H, s) | 21.1 |
| 19 | 1.24 (3H, s) | 19.7 | 1.23 (3H, s) | 19.7 |
| 20 | 1.80 (1H, m) | 40.9 | 1.86 (1H, m) | 42 |
| 21 | 3.44 (1H, dd, 10.6, 6.8)/3.79 (1H, dd, 10.6, 6.8) | 65.3 | 3.35 (1H, m)/3.87 (1H, m) | 66.2 |
| 22 | 1.65 (1H, m)/1.77 (1H, m) | 38.2 | 1.34 (1H, m)/2.20 (1H, m) | 38.8 |
| 23 | 4.09 (1H, m) | 71.2 | 3.66 (1H, m) | 74.9 |
| 24 | 3.17 (1H, m) | 78.7 | 3.08(1H, m) | 79.9 |
| 25 | 74.7 | 75 | ||
| 26 | 1.27 (3H, s) | 27.2 | 1.22 (3H, s) | 28 |
| 27 | 1.24 (3H, s) | 26.5 | 1.24 (3H, s) | 23.9 |
| 28 | 3.62 (1H, d, 11.3)/3.74 (1H, d, 11.3) | 66.2 | 3.62 (1H, m)/3.74 (1H, d, 11.4) | 66.2 |
| 29 | 1.25 (3H, s) | 21.6 | 1.25 (3H, s) | 21.6 |
| 30 | 1.21 (3H, s) | 28.3 | 1.20 (3H, s) | 28.2 |
| 1′ | 4.32 (1H, d, 7.6) | 100.7 | 4.32 (1H, d, 7.6) | 100.7 |
| 2′ | 3.08 (1H, m) | 75.3 | 3.09 (1H, m) | 75.3 |
| 3′ | 3.34 (1H, m) | 78.4 | 3.32 (1H, m) | 74.9 |
| 4′ | 3.15 (1H, m) | 72.3 | 3.15 (1H, m) | 72.3 |
| 5′ | 3.23 (1H, ddd, 9.2, 6.4, 2.5) | 77.6 | 3.22 (1H, ddd, 9.2, 6.6, 2.4) | 77.6 |
| 6′ | 3.35 (1H, m)/3.88 (1H, m) | 63.6 | 3.58 (1H, m)/3.87 (1H, m) | 63.6 |
| Compounds | HCT116 | A549 | HepG2 | MCF7 | MDAMB |
|---|---|---|---|---|---|
| 1 | 19.13 | >160 | >160 | >160 | >160 |
| 2 | 40.9 | >160 | >160 | >160 | >160 |
| 3 | >160 | >160 | >160 | >160 | >160 |
| 4 | 17.66 | >160 | >160 | >160 | >160 |
| 5 | 57.9 | >160 | >160 | >160 | >160 |
| 6 | 32.62 | >160 | >160 | >160 | >160 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Liu, P.; Wei, Y.; Song, J.; Yan, X.; Jiang, X.; Li, J.; Li, X.; Li, D.; Lu, F. The Triterpenoids from Munronia pinnata and Their Anti-Proliferative Effects. Molecules 2023, 28, 6839. https://doi.org/10.3390/molecules28196839
Yang X, Liu P, Wei Y, Song J, Yan X, Jiang X, Li J, Li X, Li D, Lu F. The Triterpenoids from Munronia pinnata and Their Anti-Proliferative Effects. Molecules. 2023; 28(19):6839. https://doi.org/10.3390/molecules28196839
Chicago/Turabian StyleYang, Xuerong, Peiyuan Liu, Yulu Wei, Jingru Song, Xiaojie Yan, Xiaohua Jiang, Jianxing Li, Xiangqin Li, Dianpeng Li, and Fenglai Lu. 2023. "The Triterpenoids from Munronia pinnata and Their Anti-Proliferative Effects" Molecules 28, no. 19: 6839. https://doi.org/10.3390/molecules28196839
APA StyleYang, X., Liu, P., Wei, Y., Song, J., Yan, X., Jiang, X., Li, J., Li, X., Li, D., & Lu, F. (2023). The Triterpenoids from Munronia pinnata and Their Anti-Proliferative Effects. Molecules, 28(19), 6839. https://doi.org/10.3390/molecules28196839