

Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
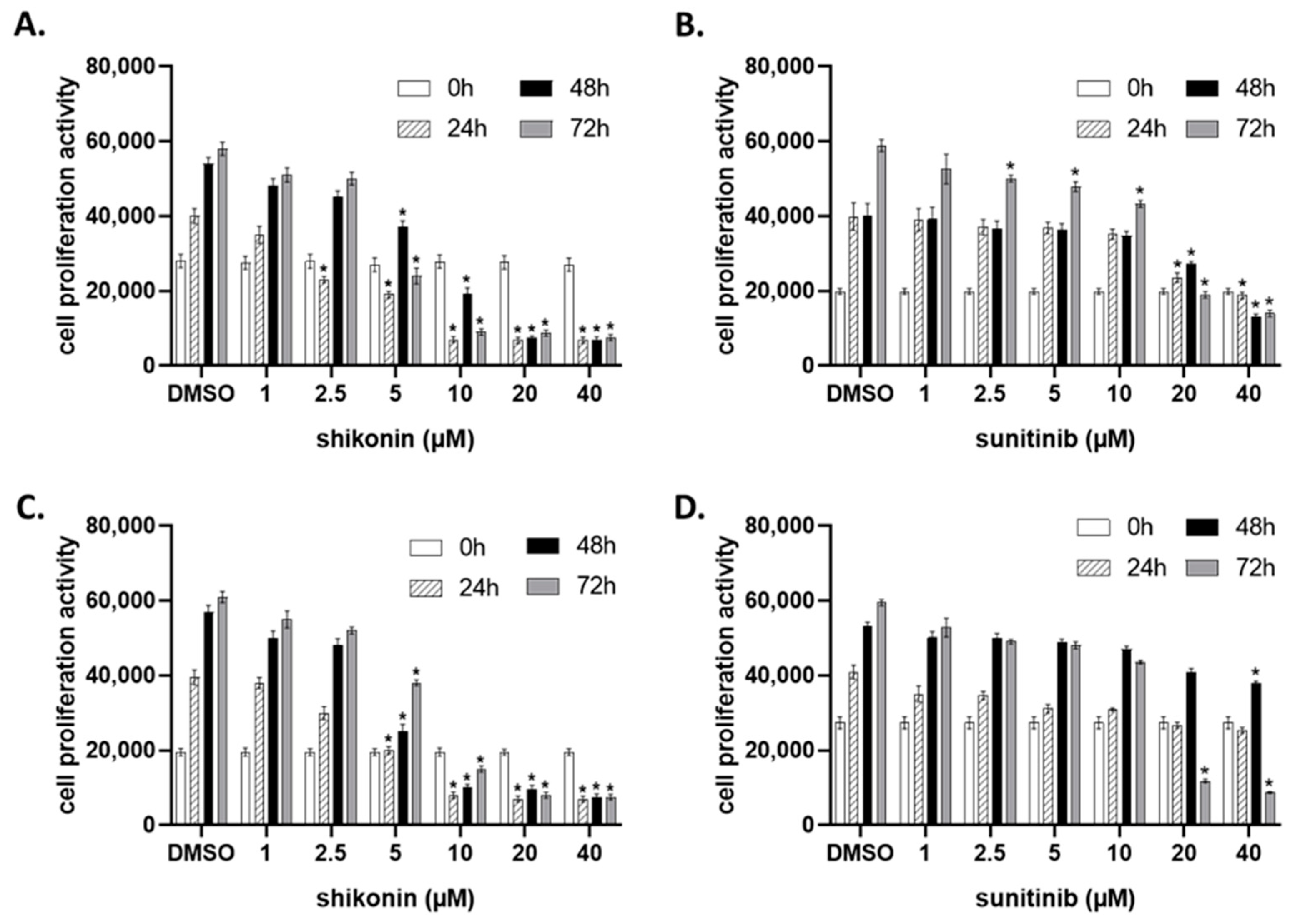
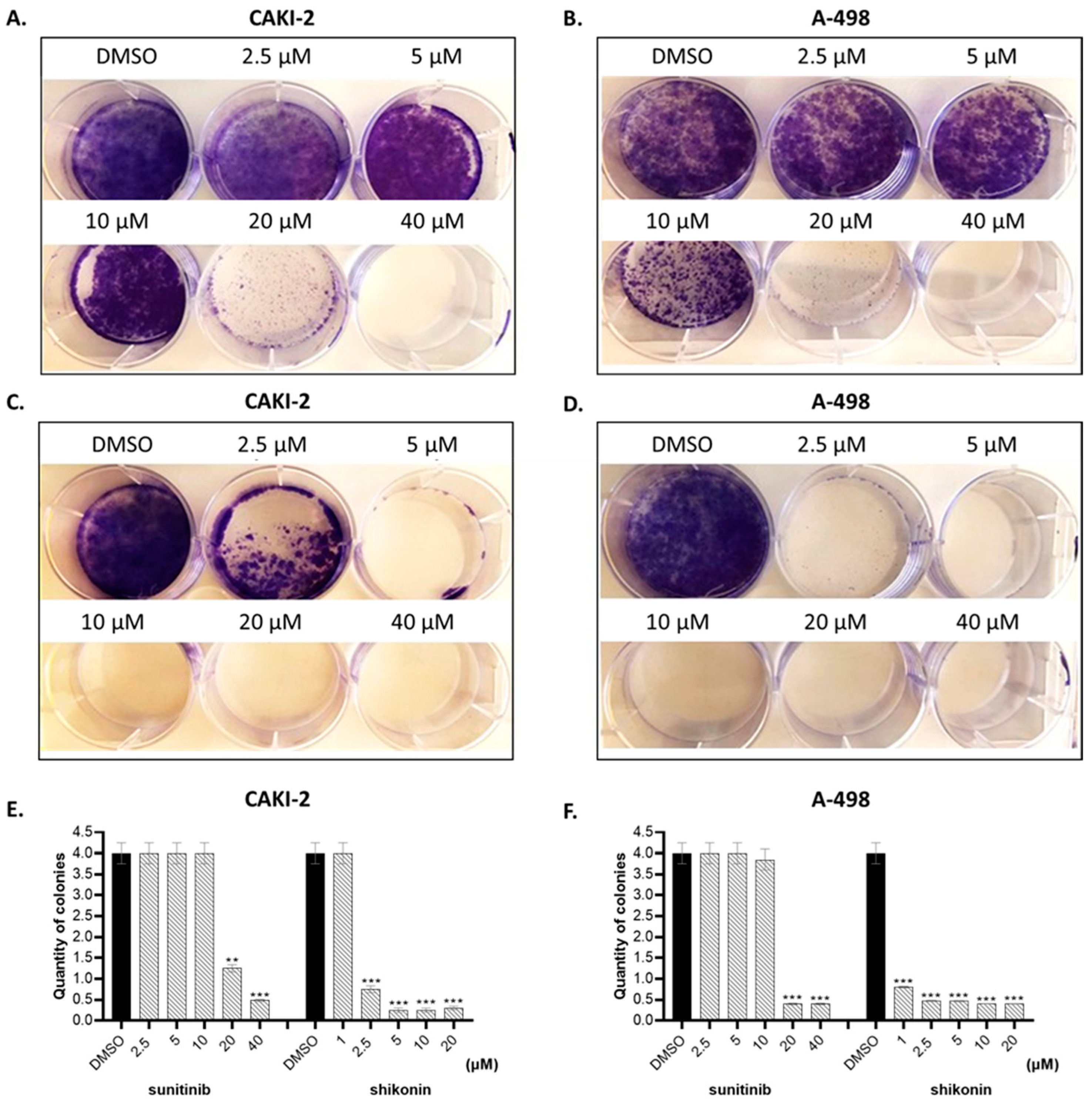
2.1. Shikonin Inhibits Cell Proliferation in a Dose- and Time-Dependent Manner
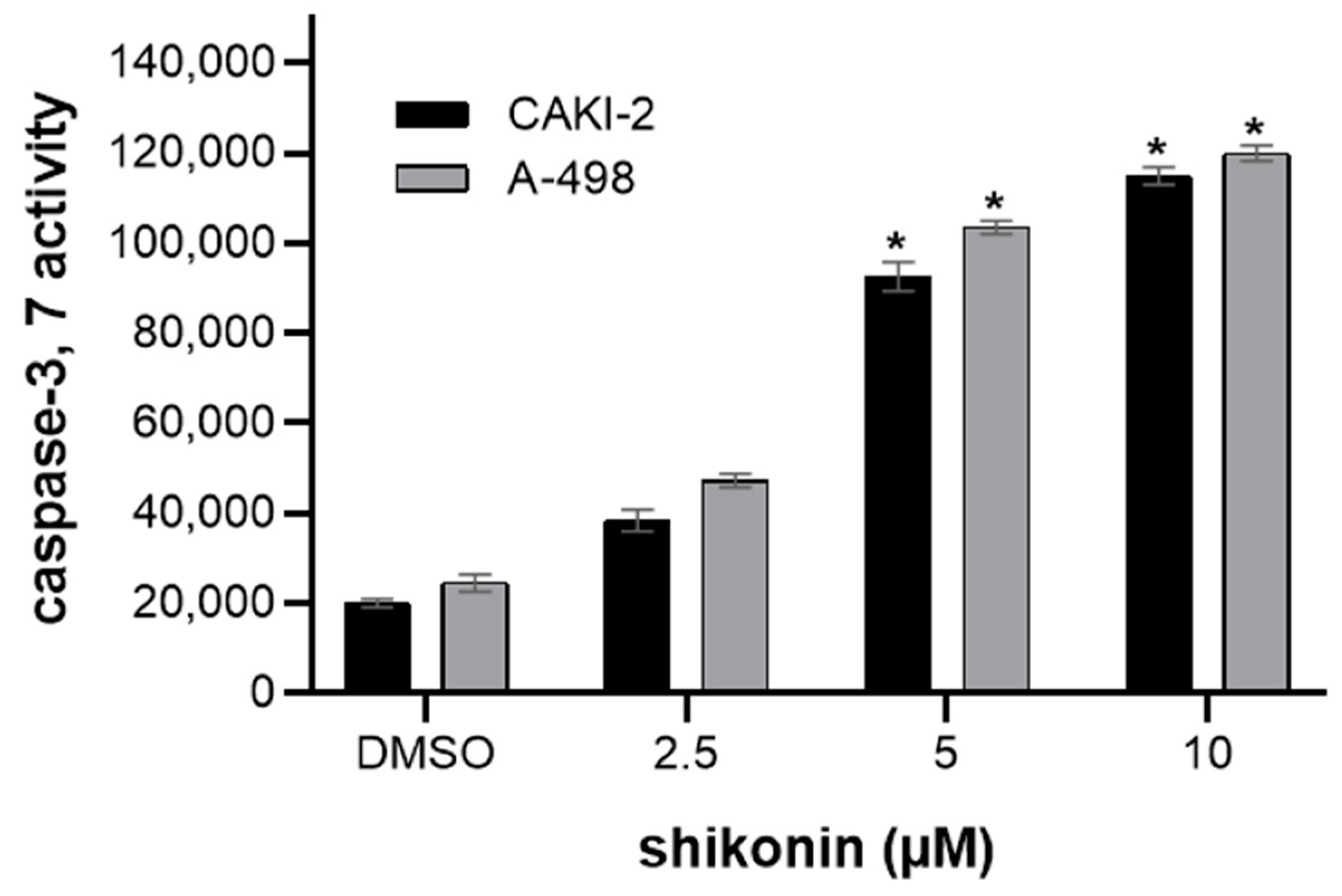
2.2. Shikonin-Induced Apoptosis in Kidney Cancer Cells
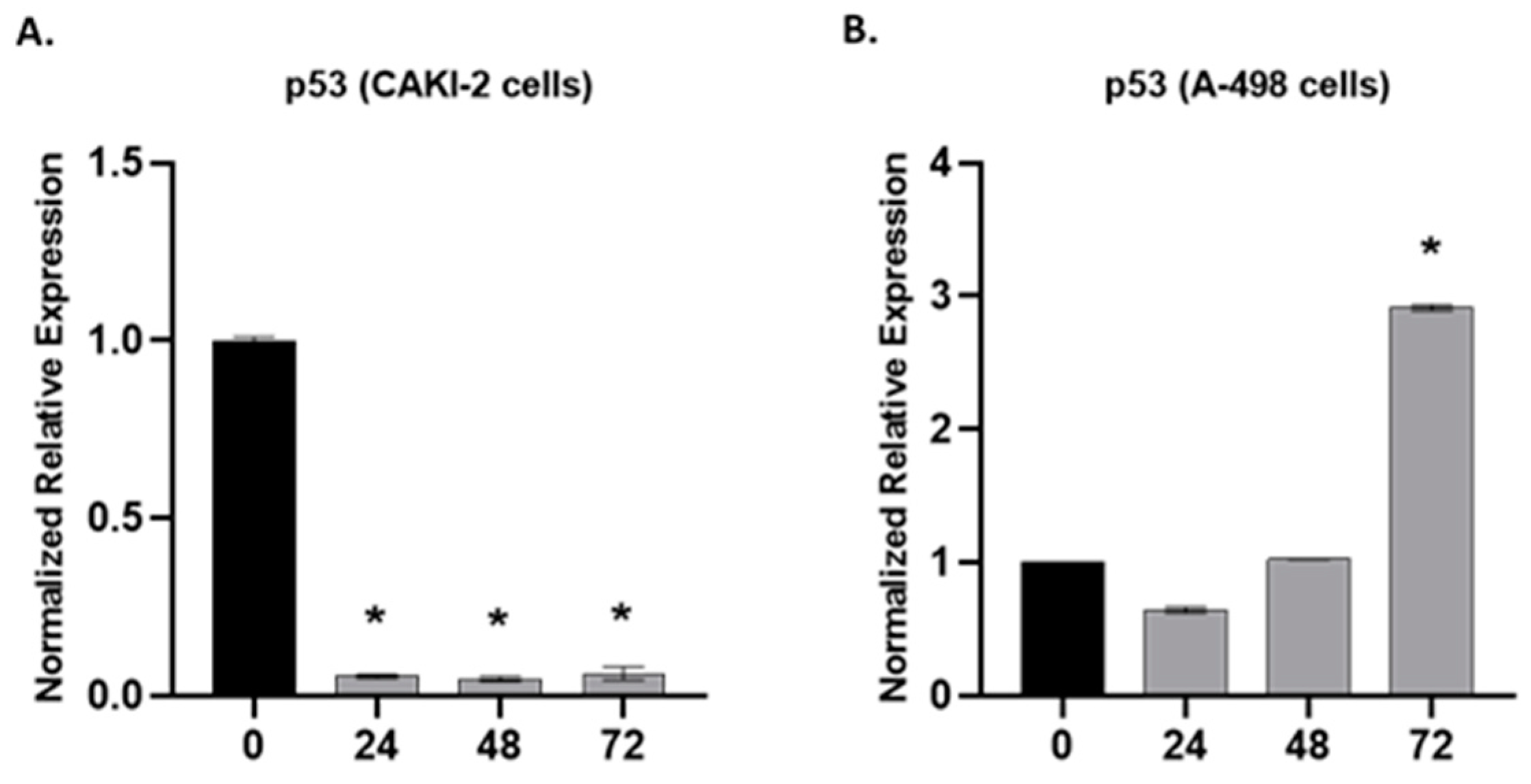
2.3. The Effect of Shikonin on the Expression of Apoptotic and Tumorsuppressor Genes
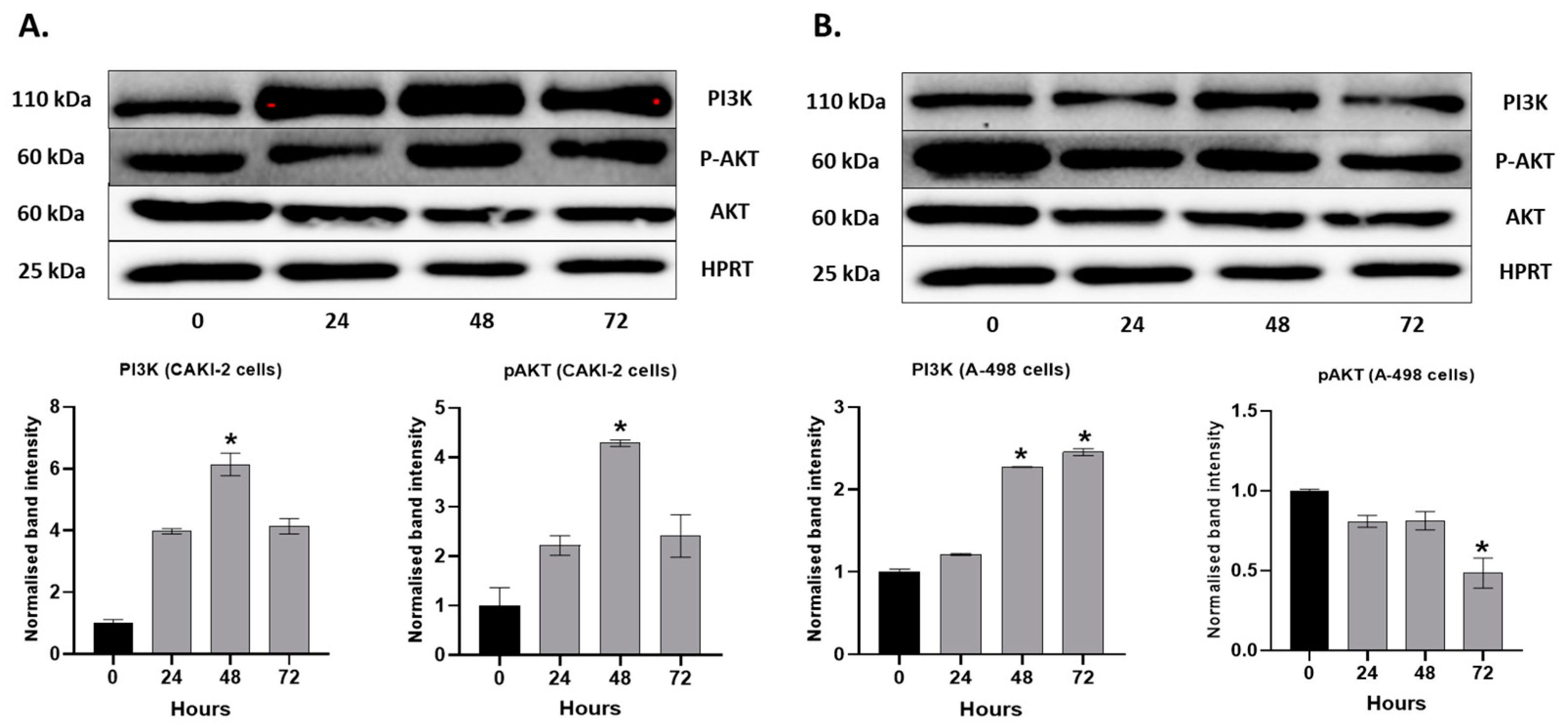
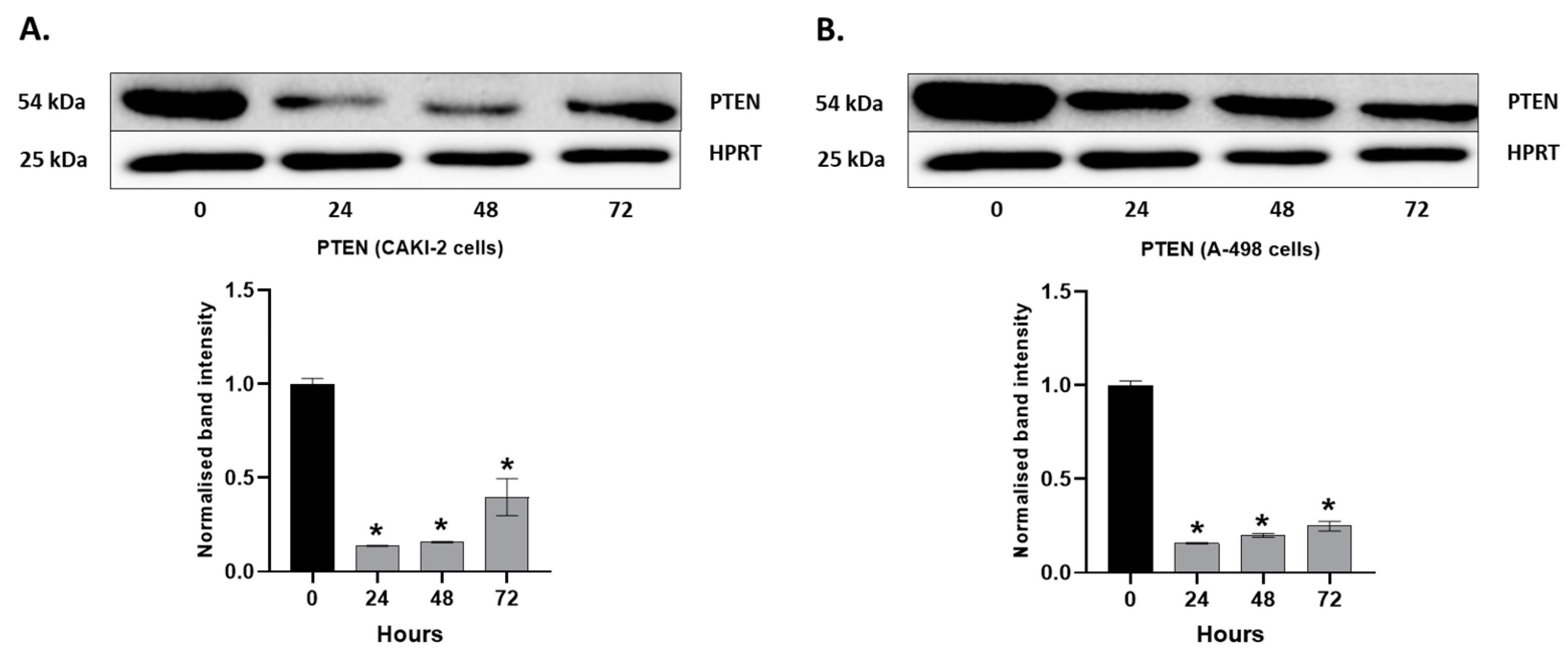
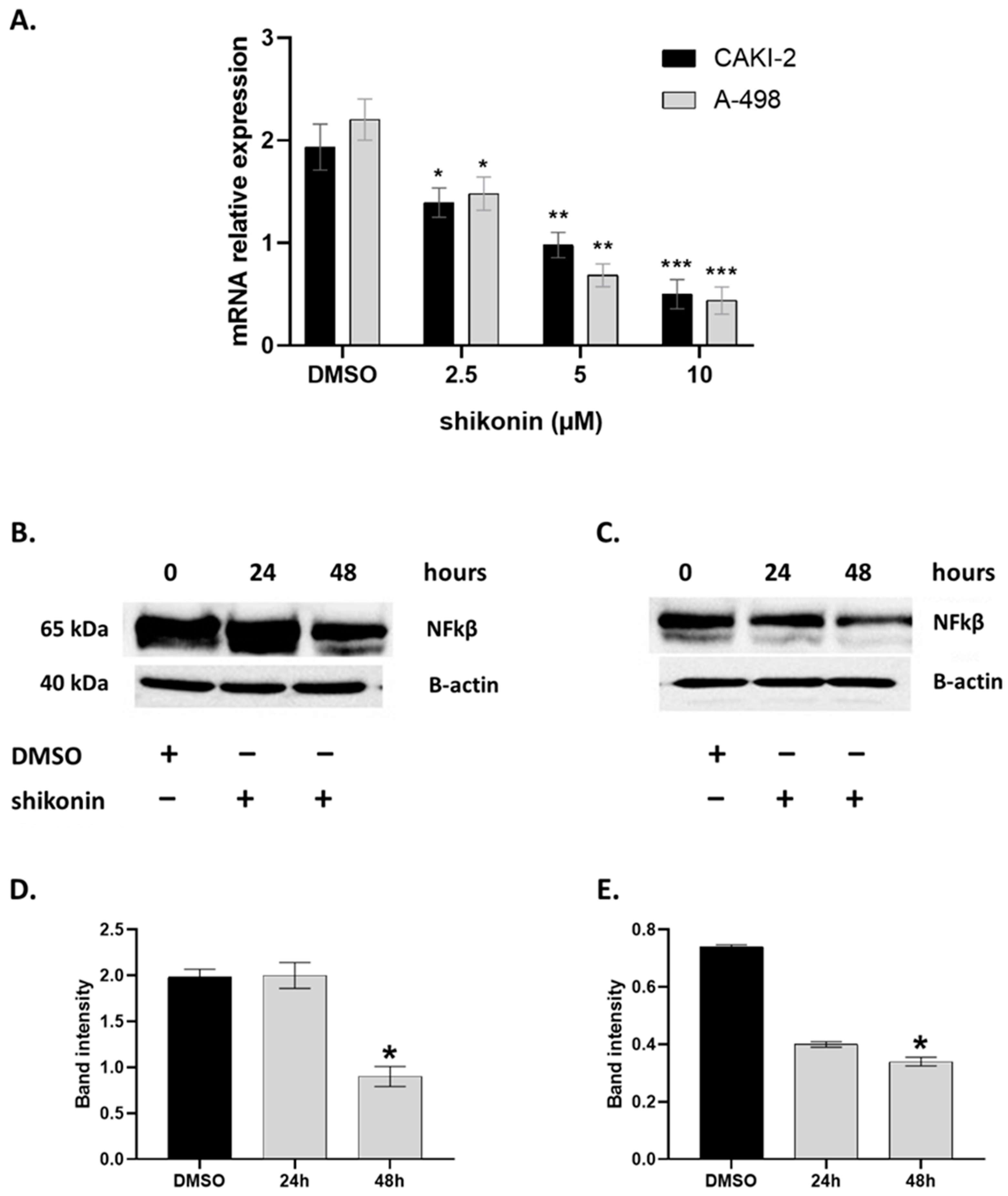
2.4. MAPKs/PI3K Pathways Might Be Associated with Shikonin-Induced Cell Apoptosis
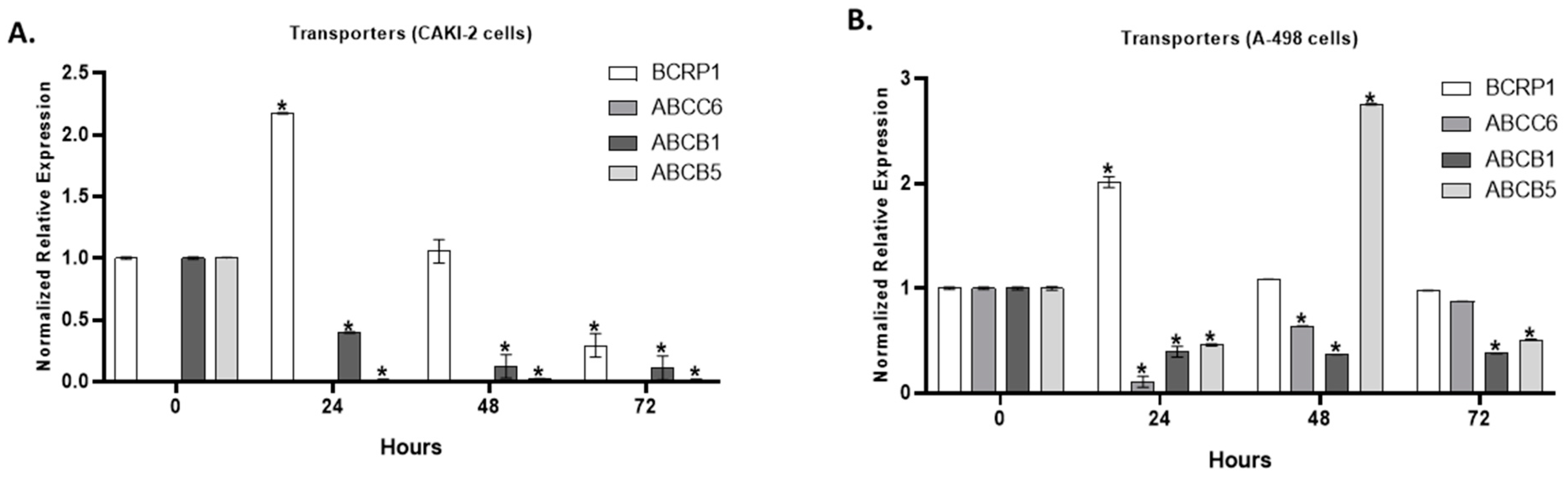
2.5. The Effect of Shikonin on the Expression of Multidrug Transporter Genes
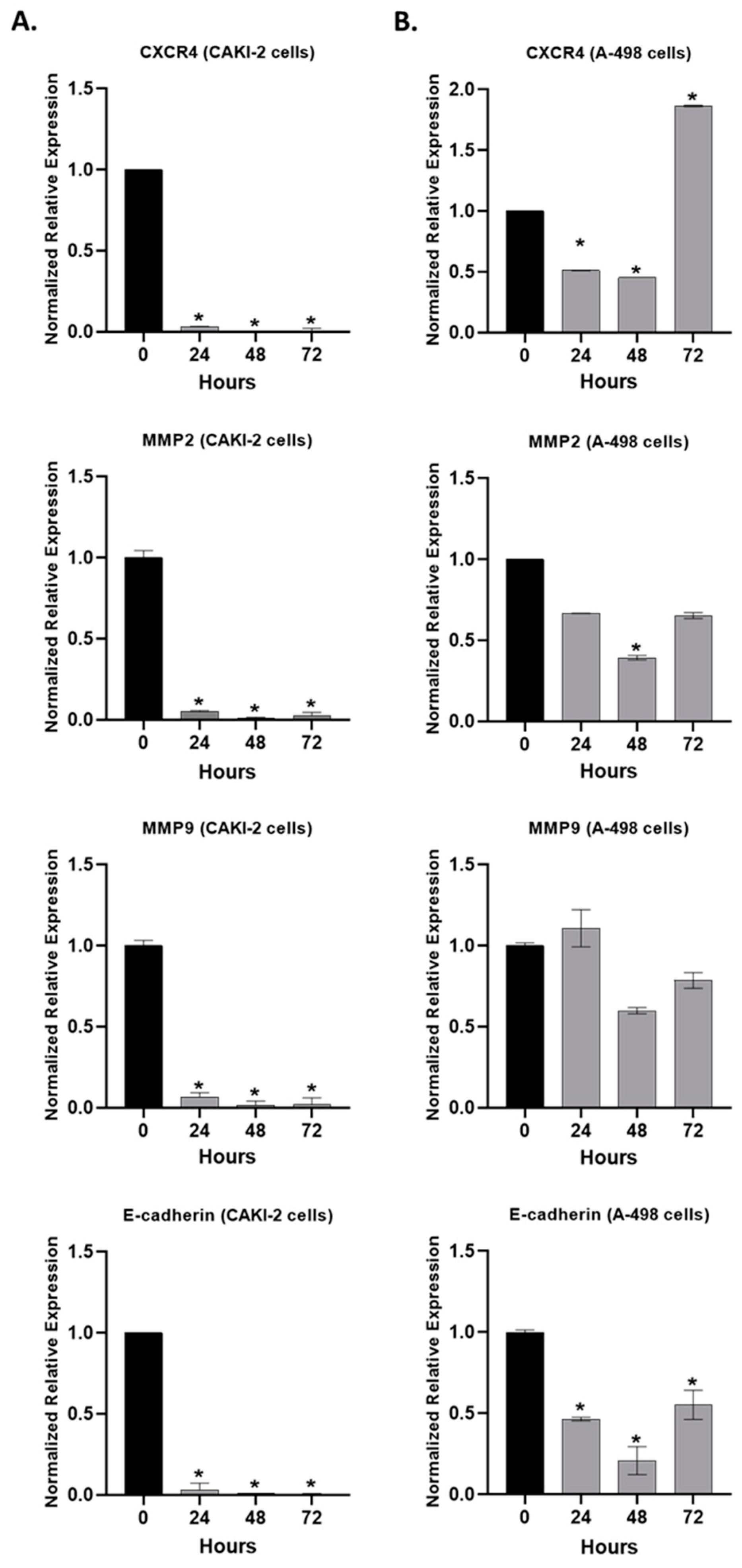
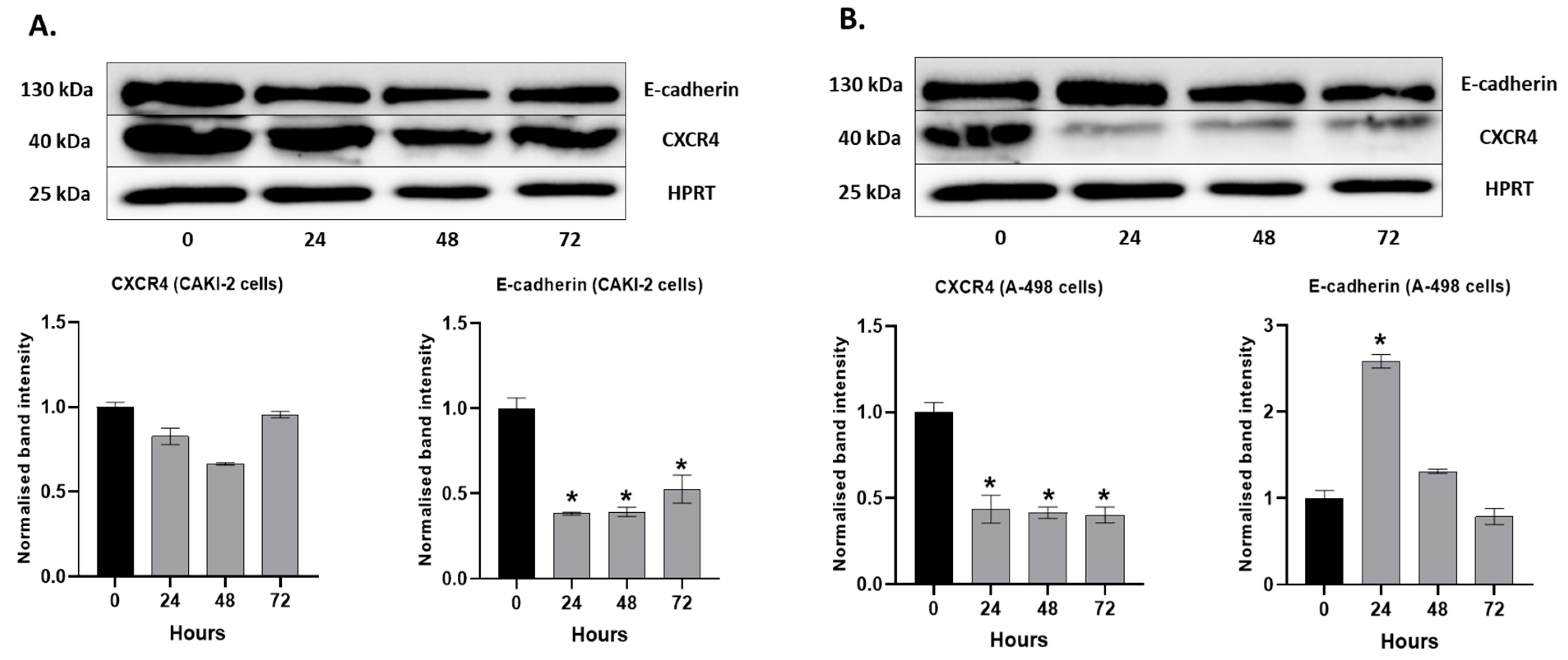
2.6. The Effect of Shikonin on the Expression of the Extracellular Matrix Proteins
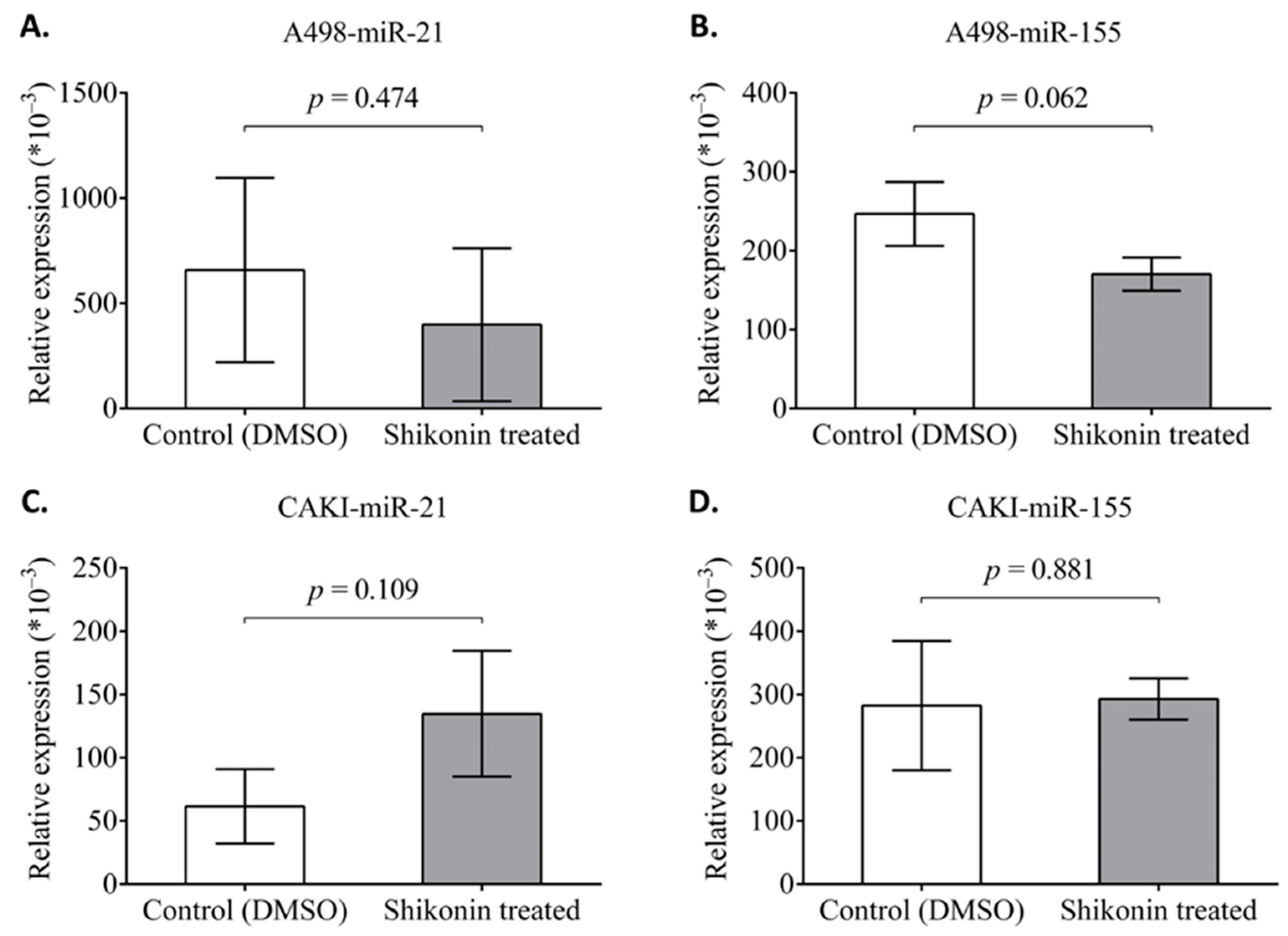
2.7. The Effect of Shikonin on the Expression of miR-21 and miR-155
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Cultures
4.3. Detection of Cell Proliferation Activity
4.4. Clonogenic Cell Survival Assay
4.5. Caspase-3 and -7 Activity Assay
4.6. Isolation of RNA and RT-PCR
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. Western Blot Detection of Proteins
4.9. MicroRNA (miRNA) Specific Stem-Loop RT-qPCR Analysis
- (1)
- miRNAs transcription into cDNA via reverse transcription was performed from total RNA (10 ng) using miRNA-specific stem loop-RT primer (500 nM, Integrated DNA Technologies, Leuven, Belgium) and TaqMan® MicroRNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA).
- (2)
- miRNA quantification was performed by RT-qPCR using designed miRNA-specific forward primer (100 μM, Integrated DNA Technologies), universal reverse primer (100 μM, Integrated DNA Technologies), and UPL probe #21 (10 μM, Roche Diagnostics, Mannheim, Germany) with Taq polymerase (5 U/μL) and dNTPs (2.5 mM) (Thermo Scientific, Wilmington, DE, USA).
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Capitanio, U.; Bensalah, K.; Bex, A.; Boorjian, S.A.; Bray, F.; Coleman, J.; Gore, J.L.; Sun, M.; Wood, C.; Russo, P. Epidemiology of Renal Cell Carcinoma. Eur. Urol. 2019, 75, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.M.; Eisen, T. Renal cell carcinoma and the use of sorafenib. Ther. Clin. Risk Manag. 2006, 2, 87–98. [Google Scholar] [PubMed]
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Patruno, R.; Ruggieri, E.; Montemurro, S.; Valerio, P.; Ribatti, D. Vascular Endothelial Growth Factor (VEGF) as a Target of Bevacizumab in Cancer: From the Biology to the Clinic. Curr. Med. Chem. 2006, 13, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Piao, J.-L.; Jin, Y.-J.; Li, M.-L.; Zakki, S.A.; Sun, L.; Feng, Q.-W.; Zhou, D.; Kondo, T.; Cui, Z.-G.; Inadera, H. Excessive Oxidative Stress in the Synergistic Effects of Shikonin on the Hyperthermia-Induced Apoptosis. Curr. Mol. Med. 2018, 18, 322–334. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Z.; Li, X.; Zhao, R.; Pu, Y.; Wu, H.; Guan, W. Shikonin causes apoptosis by disrupting intracellular calcium homeostasis and mitochondrial function in human hepatoma cells. Exp. Ther. Med. 2018, 15, 1484–1492. [Google Scholar] [CrossRef]
- Wang, Z.; Yin, J.; Li, M.; Shen, J.; Xiao, Z.; Zhao, Y.; Huang, C.; Zhang, H.; Zhang, Z.; Cho, C.H.; et al. Combination of shikonin with paclitaxel overcomes multidrug resistance in human ovarian carcinoma cells in a P-gp-independent manner through enhanced ROS generation. Chin. Med. 2019, 14, 7. [Google Scholar] [CrossRef]
- Boulos, J.C.; Rahama, M.; Hegazy, M.-E.F.; Efferth, T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019, 459, 248–267. [Google Scholar] [CrossRef]
- Yadav, S.; Sharma, A.; Nayik, G.A.; Cooper, R.; Bhardwaj, G.; Sohal, H.S.; Mutreja, V.; Kaur, R.; Areche, F.O.; AlOudat, M.; et al. Review of Shikonin and Derivatives: Isolation, Chemistry, Biosynthesis, Pharmacology and Toxicology. Front. Pharmacol. 2022, 13, 905755. [Google Scholar] [CrossRef]
- Li, Y.; Hu, X.; Li, Q.; Wang, F.; Zhang, B.; Ding, K.; Tan, B.; Lin, N.; Zhang, C. Shikonin sensitizes wild-type EGFR NSCLC cells to erlotinib and gefitinib therapy. Mol. Med. Rep. 2018, 18, 3882–3890. [Google Scholar] [CrossRef]
- Su, Y.; Lu, S.; Li, J.; Deng, L. Shikonin-mediated up-regulation of miR-34a and miR-202 inhibits retinoblastoma proliferation. Toxicol. Res. 2018, 7, 907–912. [Google Scholar] [CrossRef]
- Wei, Y.; Li, M.; Cui, S.; Wang, D.; Zhang, C.-Y.; Zen, K.; Li, L. Shikonin Inhibits the Proliferation of Human Breast Cancer Cells by Reducing Tumor-Derived Exosomes. Molecules 2016, 21, 777. [Google Scholar] [CrossRef]
- Gara, R.K.; Srivastava, V.K.; Duggal, S.; Bagga, J.K.; Bhatt, M.; Sanyal, S.; Mishra, D.P. Shikonin selectively induces apoptosis in human prostate cancer cells through the endoplasmic reticulum stress and mitochondrial apoptotic pathway. J. Biomed. Sci. 2015, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-T.; Li, Z.-L.; Wu, J.-Y.; Lu, F.-J.; Chen, C.-H. An Oxidative Stress Mechanism of Shikonin in Human Glioma Cells. PLoS ONE 2014, 9, e94180. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Hu, G. Shikonin suppresses proliferation and induces apoptosis in endometrioid endometrial cancer cells via modulating miR-106b/PTEN/AKT/mTOR signaling pathway. Biosci. Rep. 2018, 38, BSR20171546. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.-L.; Zhong, L.; Xiao, C.-L.; Gan, L.-G.; Xu, T.; Song, H.; Yang, R.; Li, L.; Liu, B.-Z. Shikonin suppresses proliferation and induces apoptosis in human leukemia NB4 cells through modulation of MAPKs and c-Myc. Mol. Med. Rep. 2017, 16, 3055–3060. [Google Scholar] [CrossRef]
- Yang, Q.; Li, S.; Fu, Z.; Lin, B.; Zhou, Z.; Wang, Z.; Hua, Y.; Cai, Z. Shikonin promotes adriamycin-induced apoptosis by upregulating caspase-3 and caspase-8 in osteosarcoma. Mol. Med. Rep. 2017, 16, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kang, X.; Niu, G.; He, S.; Zhang, T.; Bai, Y.; Li, Y.; Hao, H.; Chen, C.; Shou, Z.; et al. Shikonin induces apoptosis and prosurvival autophagy in human melanoma A375 cells via ROS-mediated ER stress and p38 pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gao, Q.; Li, W.; Zhu, L.; Shang, Q.; Feng, S.; Jia, J.; Jia, Q.; Shen, S.; Su, Z. Shikonin inhibits cancer cell cycling by targeting Cdc25s. BMC Cancer 2019, 19, 20. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, Y.; Shen, Y.; Xu, Y.; Li, X. miR-21 Modulates Cell Apoptosis by Targeting Multiple Genes in Renal Cell Carcinoma. Urology 2011, 78, 474.e13–474.e19. [Google Scholar] [CrossRef]
- Ji, H.; Tian, D.; Zhang, B.; Zhang, Y.; Yan, D.; Wu, S. Overexpression of miR-155 in clear-cell renal cell carcinoma and its oncogenic effect through targeting FOXO3a. Exp. Ther. Med. 2017, 13, 2286–2292. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Jin, J.; Zhang, Z.; Zuo, L.; Jiang, M.; Xie, C. Shikonin exerts antitumor activity by causing mitochondrial dysfunction in hepatocellular carcinoma through PKM2–AMPK–PGC1α signaling pathway. Biochem. Cell Biol. 2019, 97, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Huang, X.; Chen, Z.; Qian, W.; Tong, X. Shikonin exerts antitumor activity in Burkitt’s lymphoma by inhibiting C-MYC and PI3K/AKT/mTOR pathway and acts synergistically with doxorubicin. Sci. Rep. 2018, 8, 3317. [Google Scholar] [CrossRef] [PubMed]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Liu, P.; Wang, S.; Zhao, G.; Zhang, T.; Guo, S.; Jiang, K.; Wu, H.; Deng, G. Shikonin exerts anti-inflammatory effects in LPS-induced mastitis by inhibiting NF-κB signaling pathway. Biochem. Biophys. Res. Commun. 2018, 505, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Schinkel, A.H. Multidrug resistance and pharmacological protection mediated by the breast cancer resistance protein (BCRP/ABCG2). Mol. Cancer Ther. 2002, 1, 427–434. [Google Scholar]
- Klein, T.; Bischoff, R. Physiology and pathophysiology of matrix metalloproteases. Amino Acids 2010, 41, 271–290. [Google Scholar] [CrossRef]
- Lokeshwar, S.D.; Jordan, A.R.; Lahorewala, S.S.; Lokeshwar, V.B. Molecular Characterization of Renal Cell Carcinoma: A Potential Three-MicroRNA Prognostic Signature. Cancer Epidemiol. Biomark. Prev. 2018, 27, 464–472. [Google Scholar] [CrossRef]
- Bautista-Sánchez, D.; Arriaga-Canon, C.; Pedroza-Torres, A.; De La Rosa-Velázquez, I.A.; González-Barrios, R.; Contreras-Espinosa, L.; Montiel-Manríquez, R.; Castro-Hernández, C.; Fragoso-Ontiveros, V.; Álvarez-Gómez, R.M.; et al. The Promising Role of miR-21 as a Cancer Biomarker and Its Importance in RNA-Based Therapeutics. Mol. Ther. Nucleic Acids 2020, 20, 409–420. [Google Scholar] [CrossRef]
- Li, S.; Chen, T.; Zhong, Z.; Wang, Y.; Li, Y.; Zhao, X. microRNA-155 silencing inhibits proliferation and migration and induces apoptosis by upregulating BACH1 in renal cancer cells. Mol. Med. Rep. 2012, 5, 949–954. [Google Scholar] [CrossRef]
- Bahadoram, S.; Davoodi, M.; Hassanzadeh, S.; Bahadoram, M.; Barahman, M.; Mafakher, L. Renal cell carcinoma: An overview of the epidemiology, diagnosis, and treatment. G. Ital. Nefrol. 2022, 3, 1–16. [Google Scholar]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-Y.; Hu, Y.; Que, Z.-Y.; Wang, P.; Liu, Y.-H.; Wang, Z.-H.; Xue, Y.-X. Shikonin Inhibits the Migration and Invasion of Human Glioblastoma Cells by Targeting Phosphorylated β-Catenin and Phosphorylated PI3K/Akt: A Potential Mechanism for the Anti-Glioma Efficacy of a Traditional Chinese Herbal Medicine. Int. J. Mol. Sci. 2015, 16, 23823–23848. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Z.-Y.; Chen, L.; Zhang, J.-Y.; Fu, L.-Y.; Tao, L.; Zhang, Y.; Hu, X.-X.; Shen, X.-C. Shikonin inhibits triple-negative breast cancer-cell metastasis by reversing the epithelial-to-mesenchymal transition via glycogen synthase kinase 3β-regulated suppression of β-catenin signaling. Biochem. Pharmacol. 2019, 166, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, X.; Cao, Z. Shikonin-induced necroptosis in nasopharyngeal carcinoma cells via ROS overproduction and upregulation of RIPK1/RIPK3/MLKL expression. Onco Targets Ther. 2019, 12, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Markowitsch, S.D.; Vakhrusheva, O.; Schupp, P.; Akele, Y.; Kitanovic, J.; Slade, K.S.; Efferth, T.; Thomas, A.; Tsaur, I.; Mager, R.; et al. Shikonin Inhibits Cell Growth of Sunitinib-Resistant Renal Cell Carcinoma by Activating the Necrosome Complex and Inhibiting the AKT/mTOR Signaling Pathway. Cancers 2022, 14, 1114. [Google Scholar] [CrossRef]
- Markowitsch, S.D.; Juetter, K.M.; Schupp, P.; Hauschulte, K.; Vakhrusheva, O.; Slade, K.S.; Thomas, A.; Tsaur, I.; Cinatl, J.; Michaelis, M.; et al. Shikonin Reduces Growth of Docetaxel-Resistant Prostate Cancer Cells Mainly through Necroptosis. Cancers 2021, 13, 882. [Google Scholar] [CrossRef]
- Guo, N.; Miao, R.; Huang, D.; Hu, Z.; Ji, N.; Nan, Y.; Jiang, F.; Gou, X. Shikonin inhibits proliferation and induces apoptosis in glioma cells via downregulation of CD147. Mol. Med. Rep. 2019, 19, 4335–4343. [Google Scholar] [CrossRef]
- Boswell-Casteel, R.C.; Fukuda, Y.; Schuetz, J.D. ABCB6, an ABC Transporter Impacting Drug Response and Disease. AAPS J. 2017, 20, 8. [Google Scholar] [CrossRef]
- Liu, X.; Sun, G. Shikonin enhances Adriamycin antitumor effects by inhibiting efflux pumps in A549 cells. Oncol. Lett. 2017, 14, 4270–4276. [Google Scholar] [CrossRef]
- Volpicelli, E.R.; Lezcano, C.; Zhan, Q.; Girouard, S.D.; Kindelberger, D.W.; Frank, M.H.; Frank, N.Y.; Crum, C.P.; Murphy, G.F. The Multidrug-Resistance Transporter ABCB5 is Expressed in Human Placenta. Int. J. Gynecol. Pathol. 2014, 33, 45–51. [Google Scholar] [CrossRef]
- Kim, H.-J.; Hwang, K.-E.; Park, D.-S.; Oh, S.-H.; Jun, H.Y.; Yoon, K.-H.; Jeong, E.-T.; Kim, H.-R.; Kim, Y.-S. Shikonin-induced necroptosis is enhanced by the inhibition of autophagy in non-small cell lung cancer cells. J. Transl. Med. 2016, 15, 123. [Google Scholar] [CrossRef]
- Royds, J.A.; Dower, S.K.; Qwarnstrom, E.E.; Lewis, C.E. Response of tumour cells to hypoxia: Role of p53 and NFkB. Mol. Pathol. 1998, 51, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.M.; Trougakos, I.P.; Erin, N.; Papageorgis, P. Apoptosis Deregulation and the Development of Cancer Multi-Drug Resistance. Cancers 2021, 13, 4363. [Google Scholar] [CrossRef]
- Zhao, Q.; Assimopoulou, A.N.; Klauck, S.M.; Damianakos, H.; Chinou, I.; Kretschmer, N.; Rios, J.-L.; Papageorgiou, V.P.; Bauer, R.; Efferth, T. Inhibition of c-MYC with involvement of ERK/JNK/MAPK and AKT pathways as a novel mechanism for shikonin and its derivatives in killing leukemia cells. Oncotarget 2015, 6, 38934–38951. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, T.; Du, J.; Li, Y.; Wang, X.; Zhou, Y.; Yu, X.; Fan, W.; Zhu, Q.; Tong, X.; et al. The Critical Role of PTEN/PI3K/AKT Signaling Pathway in Shikonin-Induced Apoptosis and Proliferation Inhibition of Chronic Myeloid Leukemia. Cell. Physiol. Biochem. 2018, 47, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Akhtarkhavari, T.; Bahrami, A.R.; Matin, M.M. Downregulation of miR-21 as a promising strategy to overcome drug resistance in cancer. Eur. J. Pharmacol. 2022, 932, 175233. [Google Scholar] [CrossRef] [PubMed]
- Bayraktar, R. and K. Van Roosbroeck, miR-155 in cancer drug resistance and as target for miRNA-based therapeutics. Cancer Metastasis Rev. 2018, 37, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Hengartner, M.O. miRNAs and apoptosis: RNAs to die for. Oncogene 2006, 25, 6176–6187. [Google Scholar] [CrossRef]
- Li, M.; Wang, Y.; Song, Y.; Bu, R.; Yin, B.; Fei, X.; Guo, Q.; Wu, B. MicroRNAs in renal cell carcinoma: A systematic review of clinical implications (Review). Oncol. Rep. 2015, 33, 1571–1578. [Google Scholar] [CrossRef]
- Saini, S.; Yamamura, S.; Majid, S.; Shahryari, V.; Hirata, H.; Tanaka, Y.; Dahiya, R. MicroRNA-708 induces apoptosis and suppresses tumorigenicity in renal cancer cells. Cancer Res. 2011, 71, 6208–6219. [Google Scholar] [CrossRef] [PubMed]
- Fejes, Z.; Póliska, S.; Czimmerer, Z.; Káplár, M.; Penyige, A.; Szabó, G.G.; Debreceni, I.B.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B. Hyperglycaemia suppresses microRNA expression in platelets to increase P2RY12 and SELP levels in type 2 diabetes mellitus. Thromb. Haemost. 2017, 117, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Fejes, Z.; Czimmerer, Z.; Szük, T.; Póliska, S.; Horváth, A.; Balogh, E.; Jeney, V.; Váradi, J.; Fenyvesi, F.; Balla, G.; et al. Endothelial cell activation is attenuated by everolimus via transcriptional and post-transcriptional regulatory mechanisms after drug-eluting coronary stenting. PLoS ONE 2018, 13, e0197890. [Google Scholar] [CrossRef] [PubMed]
- Czimmerer, Z.; Hulvely, J.; Simandi, Z.; Varallyay, E.; Havelda, Z.; Szabo, E.; Varga, A.; Dezso, B.; Balogh, M.; Horvath, A.; et al. A Versatile Method to Design Stem-Loop Primer-Based Quantitative PCR Assays for Detecting Small Regulatory RNA Molecules. PLoS ONE 2013, 8, e55168. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Király, J.; Szabó, E.; Fodor, P.; Fejes, Z.; Nagy, B., Jr.; Juhász, É.; Vass, A.; Choudhury, M.; Kónya, G.; Halmos, G.; et al. Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways. Molecules 2023, 28, 6725. https://doi.org/10.3390/molecules28186725
Király J, Szabó E, Fodor P, Fejes Z, Nagy B Jr., Juhász É, Vass A, Choudhury M, Kónya G, Halmos G, et al. Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways. Molecules. 2023; 28(18):6725. https://doi.org/10.3390/molecules28186725
Chicago/Turabian StyleKirály, József, Erzsébet Szabó, Petra Fodor, Zsolt Fejes, Béla Nagy, Jr., Éva Juhász, Anna Vass, Mahua Choudhury, Gábor Kónya, Gábor Halmos, and et al. 2023. "Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways" Molecules 28, no. 18: 6725. https://doi.org/10.3390/molecules28186725
APA StyleKirály, J., Szabó, E., Fodor, P., Fejes, Z., Nagy, B., Jr., Juhász, É., Vass, A., Choudhury, M., Kónya, G., Halmos, G., & Szabó, Z. (2023). Shikonin Causes an Apoptotic Effect on Human Kidney Cancer Cells through Ras/MAPK and PI3K/AKT Pathways. Molecules, 28(18), 6725. https://doi.org/10.3390/molecules28186725