


A Combined Experimental and Computational Study of Novel Benzotriazinone Carboxamides as Alpha-Glucosidase Inhibitors

 , , ,
, , ,  , ,
, , 
Abstract
:1. Introduction
2. Result and Discussion
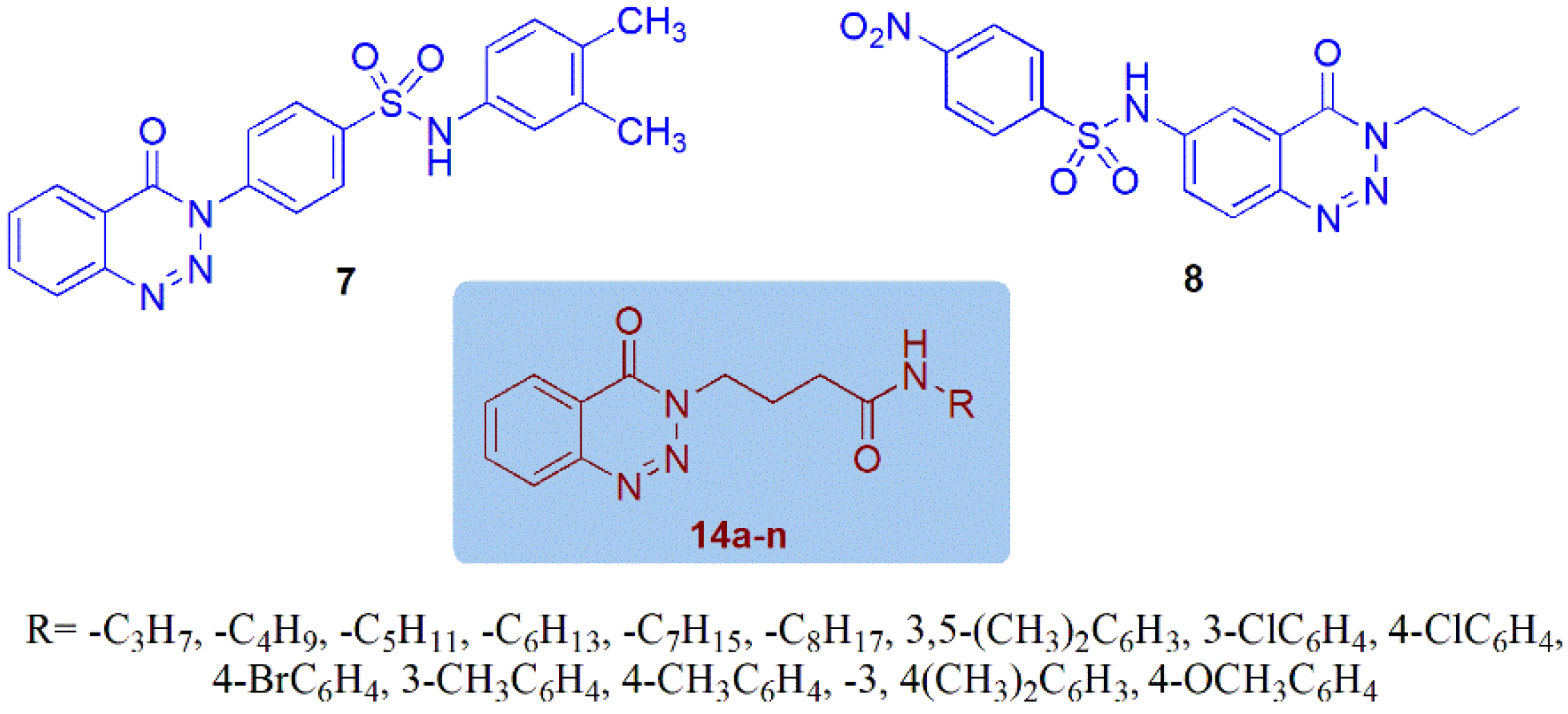
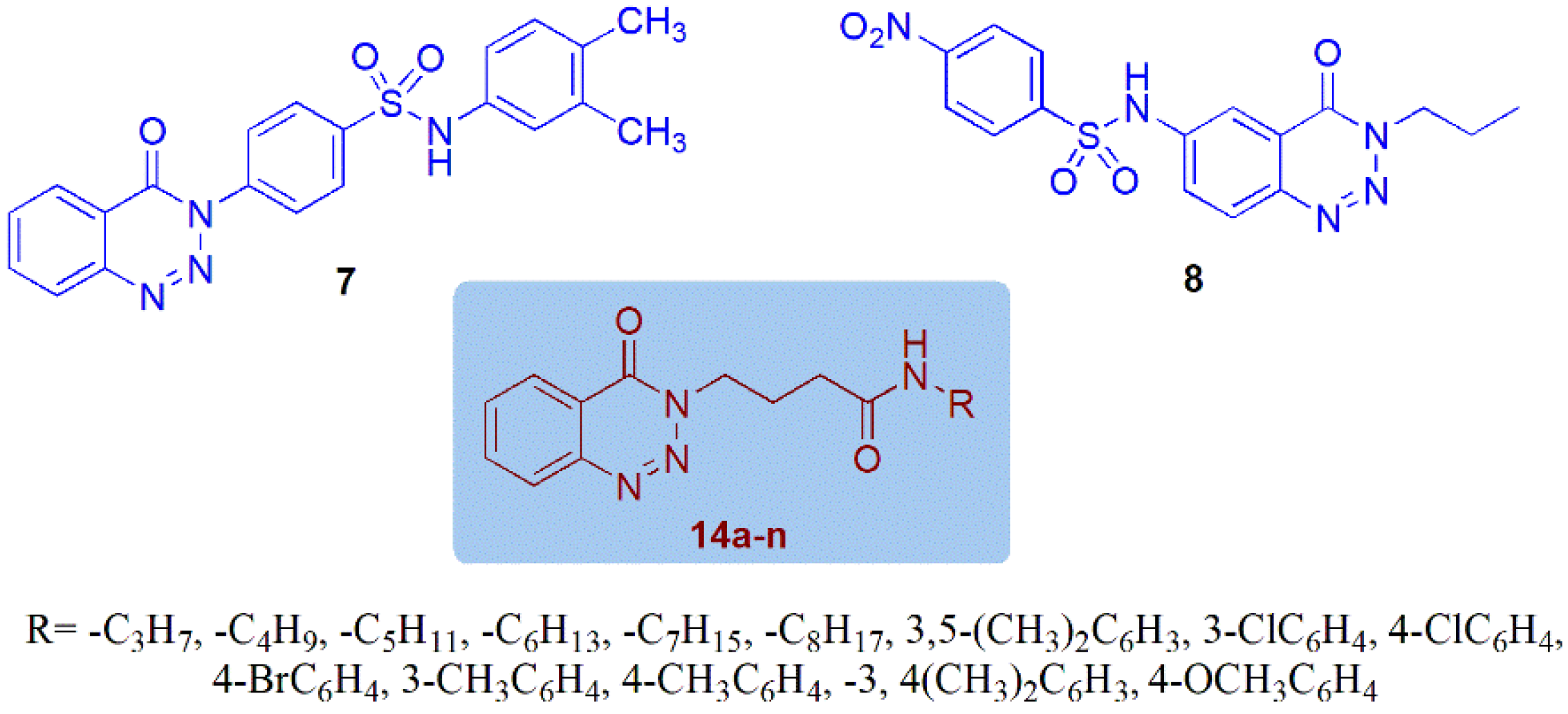
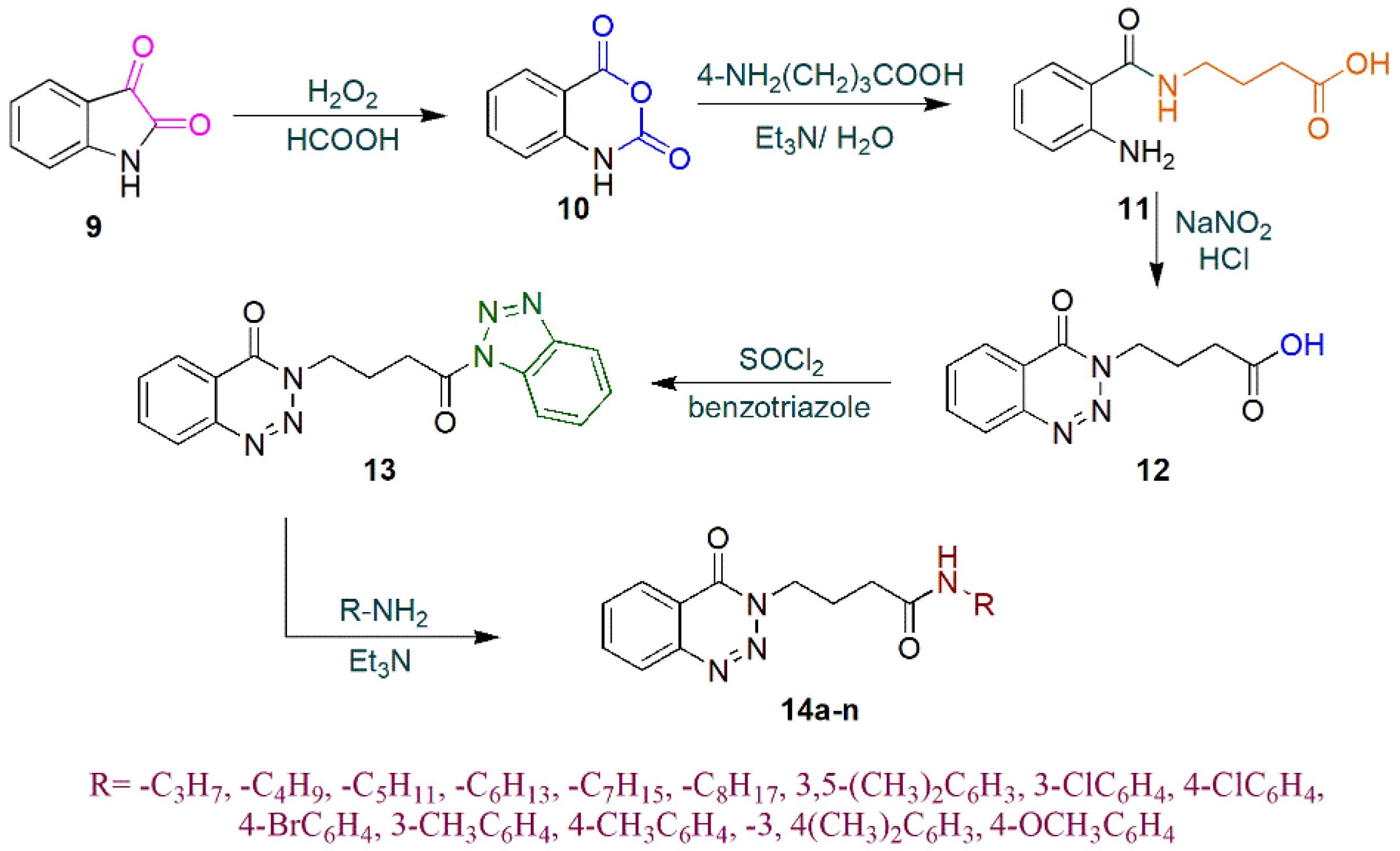
2.1. Chemistry
2.2. Spectroscopic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Codes | Molecular Structures | Time | Temperature (°C) | Yield (%) |
|---|---|---|---|---|---|
| 1. | 14a | 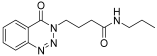 | 10 min | 25 | 91 |
| 2. | 14b | 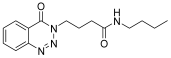 | 10 min | 25 | 85 |
| 3. | 14c | 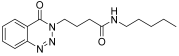 | 15 min | 25 | 86 |
| 4. | 14d | 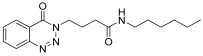 | 20 min | 25 | 85 |
| 5. | 14e |  | 25 min | 25 | 83 |
| 6. | 14f |  | 30 min | 25 | 84 |
| 7. | 14g |  | 24 h | 25 | 75 |
| 8. | 14h | 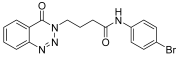 | 24 h | 25 | 71 |
| 9. | 14i | 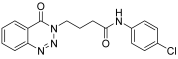 | 24 h | 25 | 69 |
| 10. | 14j |  | 24 h | 25 | 71 |
| 11. | 14k | 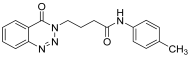 | 24 h | 25 | 73 |
| 12. | 14l | 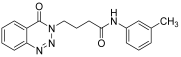 | 24 h | 25 | 72 |
| 13. | 14m | 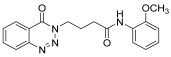 | 24 h | 25 | 76 |
| 14. | 14n | 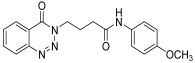 | 24 h | 25 | 80 |
2.3. Enzyme Inhibition Assay
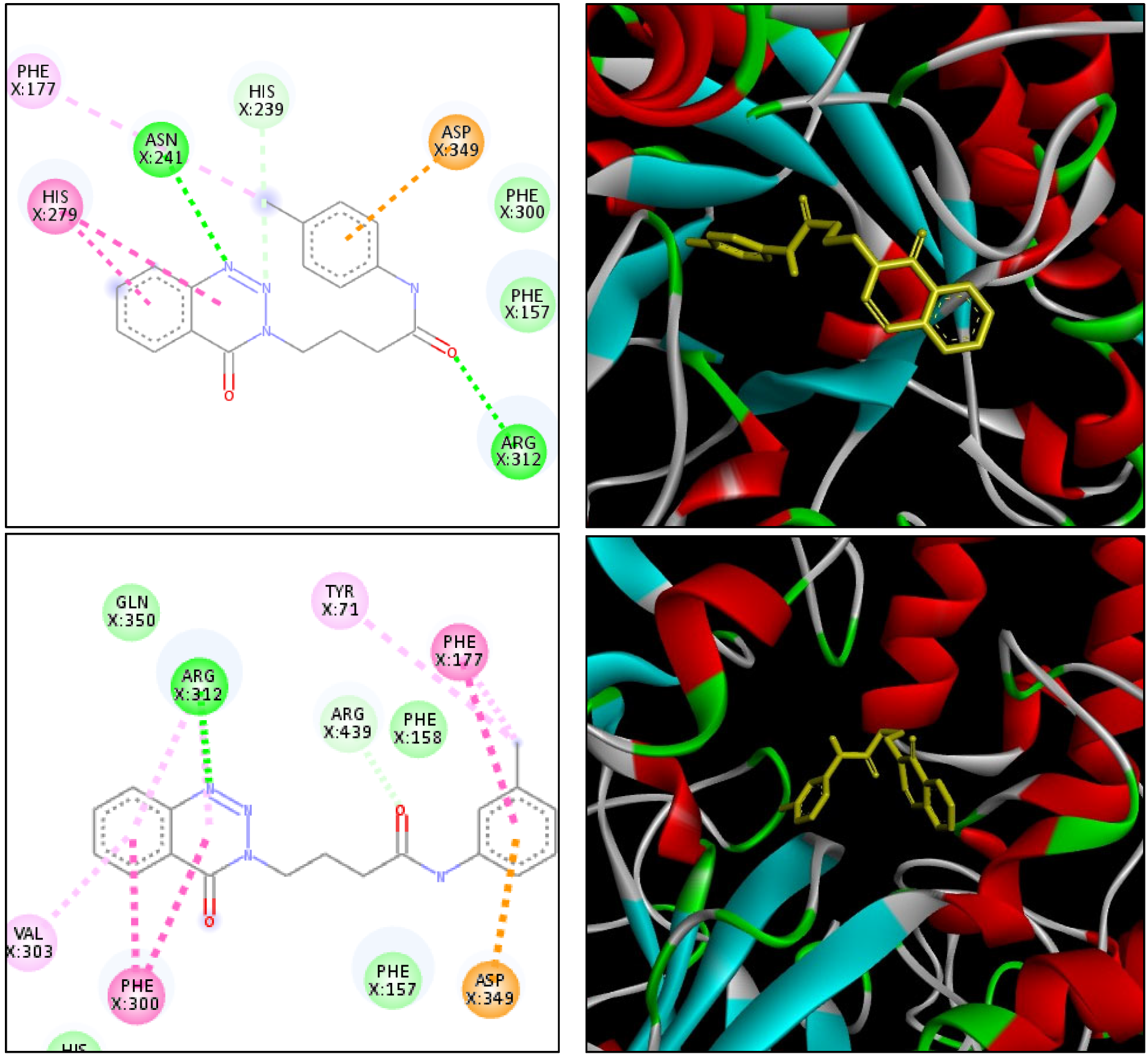
2.4. Molecular Docking
2.5. Computational Study
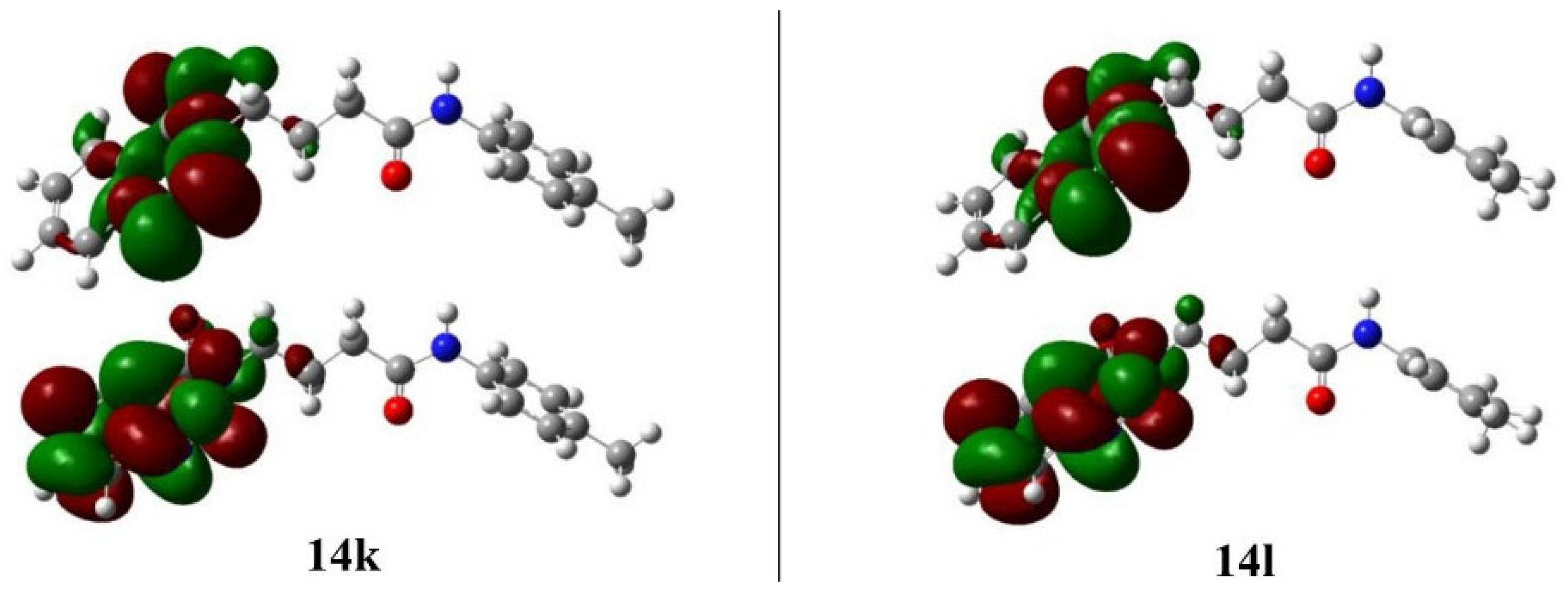
2.5.1. Frontier Molecular Orbital Analysis
2.5.2. Molecular Electrostatic Potential
3. Materials and Methods
3.1. General
3.1.1. Isatoic Anhydride (11)
3.1.2. 4-(4-Oxobenzo[1,2,3]triazin-3(4H)-yl)butanoic Acid (12)
3.1.3. 3-(4-(1H-Benzotriazol-1-yl)-4-oxobutyl)benzo[1,2,3]triazin-4(3H)-one (13)
3.1.4. N-alkyl/aryl-4-N-alkyl/aryl-4-(4-Oxobenzo[1,2,3]triazin-3(4H)-yl)butanamide(14a–14n)
3.2. Procedure for In Vitro Study
3.3. Molecular Docking Protocol
3.4. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Raut, S.K.; Khullar, M. Oxidative stress in metabolic diseases: Current scenario and therapeutic relevance. Mol. Cell. Biochem. 2023, 478, 185–196. [Google Scholar] [CrossRef]
- Borges, L.D.; Dias, H.H.; de Souza Ferreira, E.; Alves, P.M.; Silva, B.O.; de Paiva Santos, K.; da Costa, G.D.; Moreira, T.R.; Santos, D.S.; Cotta, R.M.M. Prevalence of Diabetes Mellitus among individuals with chronic kidney disease: Systematic review and meta-analysis. J. Evid. Based Healthc. 2023, 5, e4060. [Google Scholar] [CrossRef]
- Hussain, F.; Hafeez, J.; Khalifa, A.S.; Naeem, M.; Ali, T.; Eed, E.M. In vitro and in vivo study of inhibitory potentials of α-glucosidase and acetylcholinesterase and biochemical profiling of M. charantia in alloxan-induced diabetic rat models. Am. J. Transl. Res. 2022, 14, 3824. [Google Scholar]
- Farwa, U.; Raza, M.A. Heterocyclic compounds as a magic bullet for diabetes mellitus: A review. RSC Adv. 2022, 12, 22951–22973. [Google Scholar] [CrossRef]
- Khalid, Z.; Alnuwaiser, M.A.; Ahmad, H.A.; Shafqat, S.S.; Munawar, M.A.; Kamran, K.; Abbas, M.M.; Kalam, M.A.; Ewida, M.A. Experimental and Computational Analysis of Newly Synthesized Benzotriazinone Sulfonamides as Alpha-Glucosidase Inhibitors. Molecules 2022, 27, 6783. [Google Scholar] [CrossRef]
- Gondaliya, P.; Sayyed, A.A.; Bhat, P.; Mali, M.; Arya, N.; Khairnar, A.; Kalia, K. Mesenchymal stem cell-derived exosomes loaded with miR-155 inhibitor ameliorate diabetic wound healing. Mol. Pharm. 2022, 19, 1294–1308. [Google Scholar] [CrossRef]
- Elahabaadi, E.; Salarian, A.A.; Nassireslami, E. Design, synthesis, and molecular docking of novel hybrids of coumarin-dithiocarbamate alpha-glucosidase inhibitors targeting type 2 diabetes mellitus. Polycycl. Aromat. Compd. 2022, 42, 4317–4327. [Google Scholar] [CrossRef]
- Pałasz, A.; Cież, D.; Trzewik, B.; Miszczak, K.; Tynor, G.; Bazan, B. In the search of glycoside-based molecules as antidiabetic agents. Top. Curr. Chem. 2019, 377, 19. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, N.; Asemanipoor, N.; Mousavianfard, S.A.; Hoseini, S.; Faramarzi, M.A.; Adib, M.; Biglar, M.; Larijani, B.; Hamedifar, H. New acridine-9-carboxamide linked to 1,2,3-triazole-N-phenylacetamide derivatives as potent α-glucosidase inhibitors: Design, synthesis, in vitro, and in silico biological evaluations. Med. Chem. Res. 2020, 29, 1836–1845. [Google Scholar] [CrossRef]
- Popović-Djordjević, J.B.; Jevtić, I.I.; Grozdanić, N.D.; Šegan, S.B.; Zlatović, M.V.; Ivanović, M.D.; Stanojković, T.P. α-Glucosidase inhibitory activity and cytotoxic effects of some cyclic urea and carbamate derivatives. J. Enzyme Inhib. Med. Chem. 2017, 32, 298–303. [Google Scholar] [CrossRef]
- Zhang, J.-P.; Li, Q.; Zhang, C.; Li, P.; Chen, L.-J.; Wang, Y.-H.; Ruan, X.H.; Xiao, W.; Xue, W. Synthesis, antibacterial, and antiviral activities of novel penta-1,4-dien-3-one derivatives containing a benzotriazin-4(3H)-one moiety. Chem. Pap. 2018, 72, 2193–2202. [Google Scholar] [CrossRef]
- Komet, M. Microwave synthesis and anticonvulsant activity of new 3-benzyl-1,2,3-benzotriazin-4(3H)-ones. J. Heterocycl. Chem. 1997, 34, 1391–1393. [Google Scholar] [CrossRef]
- Selim, Y.A.; Abd El-Azim, M.H.; El-Farargy, A.F. Synthesis and Anti-inflammatory Activity of Some New 1,2,3-Benzotriazine Derivatives Using 2-(4-Oxo-6-phenylbenzo[d][1,2,3]triazin-3(4H)-yl)acetohydrazide as a Starting Material. J. Heterocycl. Chem. 2018, 55, 1756–1764. [Google Scholar] [CrossRef]
- El Rayes, S.; Ali, I.; Fathalla, W.; Mahmoud, M. Synthesis and Biological Activities of Some New Benzotriazinone Derivatives Based on Molecular Docking; Promising HepG2 Liver Carcinoma Inhibitors. ACS Omega 2020, 5, 6781–6791. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, E.S.; Macedo, A.B.; Resop, R.S.; Howard, J.N.; Nell, R.; Sarabia, I.; Newman, D.; Ren, Y.; Jones, R.B.; Planelles, V.; et al. Structure-activity relationship analysis of benzotriazine analogues as HIV-1 latency-reversing agents. Antimicrob. Agents Chemother. 2020, 64, e00888-20. [Google Scholar] [CrossRef]
- Zhang, F.; Wu, D.; Wang, G.-L.; Hou, S.; Ou-Yang, P.; Huang, J.; Xu, X.Y. Synthesis and biological evaluation of novel 1,2,3-benzotriazin-4-one derivatives as leukotriene A4 hydrolase aminopeptidase inhibitors. Chin. Chem. Lett. 2017, 28, 1044–1048. [Google Scholar] [CrossRef]
- Moghimi, S.; Goli-Garmroodi, F.; Pilali, H.; Mahdavi, M.; Firoozpour, L.; Nadri, H.; Moradi, A.; Asadipour, A.; Shafiee, A.; Foroumadi, A. Synthesis and anti-acetylcholinesterase activity of benzotriazinone-triazole systems. J. Chem. Sci. 2016, 128, 1445–1449. [Google Scholar] [CrossRef]
- Reddy, G.S.; Snehalatha, A.V.; Edwin, R.K.; Hossain, K.A.; Giliyaru, V.B.; Hariharapura, R.C.; Shenoy, G.G.; Misra, P.; Pal, M. Synthesis of 3-indolylmethyl substituted (pyrazolo/benzo) triazinone derivatives under Pd/Cu-catalysis: Identification of potent inhibitors of chorismate mutase (CM). Bioorg. Chem. 2019, 91, 103155. [Google Scholar] [CrossRef]
- Yan, Y.-C.; Wu, W.; Huang, G.-Y.; Yang, W.-C.; Chen, Q.; Qu, R.-Y.; Lin, H.Y.; Yang, G.F. Pharmacophore-Oriented Discovery of Novel 1,2,3-Benzotriazine-4-one Derivatives as Potent 4-Hydroxyphenylpyruvate Dioxygenase Inhibitors. J. Agric. Food Chem. 2022, 70, 6644–6657. [Google Scholar] [CrossRef]
- Roca-Paixão, L.; Correia, N.T.; Danède, F.; Guerain, M.; Affouard, F. Carbamazepine/tartaric acid cocrystalline forms: When stoichiometry and synthesis method matter. Cryst. Growth Des. 2023, 23, 3. [Google Scholar] [CrossRef]
- Beydoun, A.; Kutluay, E. Oxcarbazepine. Expert Opin. Pharmacother. 2002, 3, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Vendrame, M.; Loddenkemper, T.; Gooty, V.D.; Takeoka, M.; Rotenberg, A.; Bergin, A.M.; Eksioglu, Y.Z.; Poduri, A.; Duffy, F.H.; Libenson, M.; et al. Experience with rufinamide in a pediatric population: A single center’s experience. Pediatr. Neurol. 2010, 43, 155–158. [Google Scholar] [CrossRef]
- Fleischmann, R.; Iqbal, I.; Slobodin, G. Meloxicam. Expert Opin. Pharmacother. 2002, 3, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, M.; Kanazawa, A.; Hirose, T.; Yoshihara, T.; Kobayashi-Kimura, S.; Nakanishi, R.; Tosaka, Y.; Sasaki-Omote, R.; Kudo-Fujimaki, K.; Komiya, K.; et al. Comparison of sitagliptin with nateglinide on postprandial glucose and related hormones in drug-naïve Japanese patients with type 2 diabetes mellitus: A pilot study. J. Diabetes Investig. 2015, 6, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Abe, M.; Ito, H. Anagliptin in the treatment of type 2 diabetes: Safety, efficacy, and patient acceptability. Diabetes Metab. Syndr. Obes. 2015, 8, 163–171. [Google Scholar] [CrossRef]
- Peytam, F.; Adib, M.; Shourgeshty, R.; Firoozpour, L.; Rahmanian-Jazi, M.; Jahani, M.; Moghimi, S.; Divsalar, K.; Faramarzi, M.A.; Mojtabavi, S.; et al. An efficient and targeted synthetic approach towards new highly substituted 6-amino-pyrazolo [1,5-a]pyrimidines with α-glucosidase inhibitory activity. Sci. Rep. 2020, 10, 2595. [Google Scholar] [CrossRef]
- Fershtat, L.L.; Makhova, N.N. Molecular hybridization tools in the development of furoxan-based NO-donor prodrugs. Chem. Eur. J. 2017, 12, 622–638. [Google Scholar] [CrossRef]
- Harrison, J.R.; Brand, S.; Smith, V.; Robinson, D.A.; Thompson, S.; Smith, A.; Davies, K.; Mok, N.; Torrie, L.S.; Collie, I.; et al. A molecular hybridization approach for the design of potent, highly selective, and brain-penetrant N-myristoyltransferase inhibitors. J. Med. Chem. 2018, 61, 8374–8389. [Google Scholar] [CrossRef]
- Khalid, Z.; Shafqat, S.S.; Ahmad, H.A.; Rehman, H.M.; Munawar, M.A.; Ahmad, M.; Asiri, A.M.; Ashraf, M. Synthesis of 1,2,3-Benzotriazin-4(3H)-one derivatives as α-glucosidase inhibitor and their in-silico study. Med. Chem. Res. 2022, 31, 819–831. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef]
- Batool, M.; Tajammal, A.; Farhat, F.; Verpoort, F.; Khattak, Z.A.; Shahid, M.; Ahmad, H.A.; Munawar, M.A.; Zia-ur-Rehman, M.; Asim Raza Basra, M. Molecular docking, computational, and antithrombotic studies of novel 1,3,4-oxadiazole derivatives. Int. J. Mol. Sci. 2018, 19, 3606. [Google Scholar] [CrossRef]
- Kazachenko, A.S.; Tomilin, F.N.; Pozdnyakova, A.A.; Vasilyeva, N.Y.; Malyar, Y.N.; Kuznetsova, S.A.; Avramov, P.V. Theoretical DFT interpretation of infrared spectra of biologically active arabinogalactan sulphated derivatives. Chem. Pap. 2020, 74, 4103–4113. [Google Scholar] [CrossRef]
- Reyes-Chaparro, A.; Flores-Lopez, N.; Quintanilla-Guerrero, F.; Nicolás-Álvarez, D.E.; Hernandez-Martinez, A. Design of new reversible and selective inhibitors of monoamine oxidase A and a comparison with drugs already approved. Bull. Natl. Res. Cent. 2023, 47, 46. [Google Scholar] [CrossRef]
- Sayed, D.S.E.; Abdelrehim, E.-S.M. Spectroscopic details on the molecular structure of pyrimidine-2-thiones heterocyclic compounds: Computational and antiviral activity against the main protease enzyme of SARS-CoV-2. BMC Chem. 2022, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Channar, P.A.; Arshad, N.; Larik, F.A.; Farooqi, S.I.; Saeed, A.; Hökelek, T.; Batool, B.; Ujan, R.; Ali, H.S.; Flörke, U. 4-(4-Bromophenyl)thiazol-2-amine: Crystal structure determination, DFT calculations, visualizing intermolecular interactions using Hirshfeld surface analysis, and DNA binding studies. J. Phys. Org. Chem. 2019, 32, e3968. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Taha, M.; Rahim, F.; Sarfraz, M.; Iqbal, R.; Iqbal, N.; Hussain, R.; Ali Shah, S.A.; Ayub, K.; et al. Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors. Molecules 2023, 28, 559. [Google Scholar] [CrossRef] [PubMed]
- Elkolli, M.; Chafai, N.; Chafaa, S.; Kadi, I.; Bensouici, C.; Hellal, A.J. New phosphinic and phosphonic acids: Synthesis, antidiabetic, anti-Alzheimer, antioxidant activity, DFT study and SARS-CoV-2 inhibition. J. Mol. Struct. 2022, 1268, 133701. [Google Scholar] [CrossRef]
- Kanaani, A.; Ajloo, D.; Kiyani, H.; Ghasemian, H.; Vakili, M.; Feizabadi, M. Molecular structure, spectroscopic investigations and computational study on the potential molecular switch of (E)-1-(4-(2-hydroxybenzylideneamino)phenyl)ethanone. Mol. Phys. 2016, 114, 2081–2097. [Google Scholar] [CrossRef]
- Kosar, B.; Albayrak, C.; Spectroscopy, B. Spectroscopic investigations and quantum chemical computational study of (E)-4-methoxy-2-[(p-tolylimino)methyl]phenol. Spectrochim. Acta A Mol. Biomol. 2011, 78, 160–167. [Google Scholar] [CrossRef]
- Akram, N.; Mansha, A.; Premkumar, R.; Franklin Benial, A.M.; Asim, S.; Iqbal, S.Z.; Ali, H.S. Spectroscopic, quantum chemical and molecular docking studies on 2,4-dimethoxy-1,3,5-triazine: A potent inhibitor of protein kinase CK2 for the development of breast cancer drug. Mol. Simul. 2020, 46, 1340–1353. [Google Scholar] [CrossRef]
- Chaudhry, F.; Naureen, S.; Ashraf, M.; Al-Rashida, M.; Jahan, B.; Munawar, M.A.; Khan, M.A. Imidazole-pyrazole hybrids: Synthesis, characterization and in-vitro bioevaluation against α-glucosidase enzyme with molecular docking studies. Bioorg. Chem. 2019, 82, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]

| Sr. No. | Codes | Inhibition (%) at 0.5 mM | IC50 (μM) |
|---|---|---|---|
| 1. | 14a | 39.13 ± 0.19 | - |
| 2. | 14b | 67.53 ± 0.14 | 243.41 ± 0.18 |
| 3. | 14c | 42.34 ± 0.25 | - |
| 4. | 14d | 66.20 ± 0.11 | 335.04 ± 0.07 |
| 5. | 14e | 40.23 ± 0.21 | - |
| 6. | 14f | 30.57 ± 0.16 | - |
| 7. | 14g | 78.32 ± 0.18 | 196.52 ± 0.08 |
| 8. | 14h | 74.25 ± 0.27 | 238.15 ± 0.15 |
| 9. | 14i | 82.77 ± 0.20 | 163.29 ± 0.13 |
| 10. | 14j | 89.35 ± 0.21 | 52.01 ± 0.07 |
| 11. | 14k | 94.18 ± 0.23 | 27.13 ± 0.12 |
| 12. | 14l | 92.63 ± 0.12 | 32.14 ± 0.11 |
| 13. | 14m | 84.31 ± 0.18 | 121.26 ± 0.15 |
| 14. | 14n | 87.46 ± 0.15 | 75.37 ± 0.14 |
| 15. | Acarbose | 92.23 ± 0.16 | 37.38 ± 0.12 |
| Ligand | Amino Acid | Distance (Å) | Attractive Forces |
|---|---|---|---|
| 14k | Asn241 | 2.38 | Hydrogen bond |
| 14k | Arg312 | 2.57 | Hydrogen bond |
| 14k | His239 | 2.94 | Hydrogen bond |
| 14k | Asp349 | 4.75 | Electrostatic |
| 14k | His279 | 4.14 | Hydrophobic |
| 14k | His279 | 5.15 | Hydrophobic |
| 14k | Phe177 | 5.37 | Hydrophobic |
| 14l | Arg312 | 2.75 | Hydrogen bond |
| 14l | Arg439 | 2.53 | Hydrogen bond |
| 14l | Asp349 | 4.14 | Electrostatic |
| 14l | Phe300 | 4.44 | Hydrophobic |
| 14l | Phe300 | 4.98 | Hydrophobic |
| 14l | Phe177 | 4.99 | Hydrophobic |
| 14l | Tyr71 | 4.19 | Hydrophobic |
| 14l | Val303 | 5.04 | Hydrophobic |
| 14l | Arg313 | 5.37 | Hydrophobic |
| 14l | Tyr71 | 5.11 | Hydrophobic |
| 14l | Phe177 | 4.61 | Hydrophobic |
| Compounds | EHOMO | ELUMO | Egap | I | A | η | S | μ | χ | ω | D |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 14a | −6.24 | −1.92 | 4.33 | 6.24 | 1.92 | 2.16 | 0.23 | −4.08 | 4.08 | 3.85 | 1.58 |
| 14b | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.16 | 0.23 | −4.08 | 4.08 | 3.84 | 1.52 |
| 14c | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.16 | 0.23 | −4.08 | 4.08 | 3.84 | 1.58 |
| 14d | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.16 | 0.23 | −4.07 | 4.07 | 3.84 | 1.54 |
| 14e | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.16 | 0.23 | −4.07 | 4.07 | 3.84 | 1.57 |
| 14f | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.16 | 0.23 | −4.07 | 4.07 | 3.84 | 1.51 |
| 14g | −6.25 | −1.92 | 4.33 | 6.25 | 1.92 | 2.16 | 0.23 | −4.08 | 4.08 | 3.85 | 1.02 |
| 14h | −6.25 | −1.92 | 4.34 | 6.25 | 1.92 | 2.17 | 0.23 | −4.08 | 4.08 | 3.85 | 2.99 |
| 14i | −6.26 | −1.92 | 4.34 | 6.26 | 1.92 | 2.17 | 0.23 | −4.09 | 4.09 | 3.85 | 3.16 |
| 14j | −6.25 | −1.92 | 4.33 | 6.25 | 1.92 | 2.16 | 0.23 | −4.09 | 4.09 | 3.86 | 2.80 |
| 14k | −6.22 | −1.93 | 4.29 | 6.22 | 1.93 | 2.14 | 0.23 | −4.08 | 4.08 | 3.88 | 1.10 |
| 14l | −6.25 | −1.93 | 4.32 | 6.25 | 1.93 | 2.16 | 0.23 | −4.09 | 4.09 | 3.87 | 1.20 |
| 14m | −6.25 | −1.92 | 4.33 | 6.25 | 1.92 | 2.17 | 0.23 | −4.08 | 4.08 | 3.85 | 2.06 |
| 14n | −6.24 | −1.91 | 4.33 | 6.24 | 1.91 | 2.17 | 0.23 | −4.07 | 4.07 | 3.83 | 2.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khalid, Z.; Shafqat, S.S.; Ahmad, H.A.; Munawar, M.A.; Mutahir, S.; Elkholi, S.M.; Shafqat, S.R.; Huma, R.; Asiri, A.M. A Combined Experimental and Computational Study of Novel Benzotriazinone Carboxamides as Alpha-Glucosidase Inhibitors. Molecules 2023, 28, 6623. https://doi.org/10.3390/molecules28186623
Khalid Z, Shafqat SS, Ahmad HA, Munawar MA, Mutahir S, Elkholi SM, Shafqat SR, Huma R, Asiri AM. A Combined Experimental and Computational Study of Novel Benzotriazinone Carboxamides as Alpha-Glucosidase Inhibitors. Molecules. 2023; 28(18):6623. https://doi.org/10.3390/molecules28186623
Chicago/Turabian StyleKhalid, Zunera, Syed Salman Shafqat, Hafiz Adnan Ahmad, Munawar Ali Munawar, Sadaf Mutahir, Safaa M. Elkholi, Syed Rizwan Shafqat, Rahila Huma, and Abdullah Mohammed Asiri. 2023. "A Combined Experimental and Computational Study of Novel Benzotriazinone Carboxamides as Alpha-Glucosidase Inhibitors" Molecules 28, no. 18: 6623. https://doi.org/10.3390/molecules28186623
APA StyleKhalid, Z., Shafqat, S. S., Ahmad, H. A., Munawar, M. A., Mutahir, S., Elkholi, S. M., Shafqat, S. R., Huma, R., & Asiri, A. M. (2023). A Combined Experimental and Computational Study of Novel Benzotriazinone Carboxamides as Alpha-Glucosidase Inhibitors. Molecules, 28(18), 6623. https://doi.org/10.3390/molecules28186623