
Anisotropic Mechanical Properties of Orthorhombic SiP2 Monolayer: A First-Principles Study




Abstract
:
1. Introduction
2. Results and Discussion
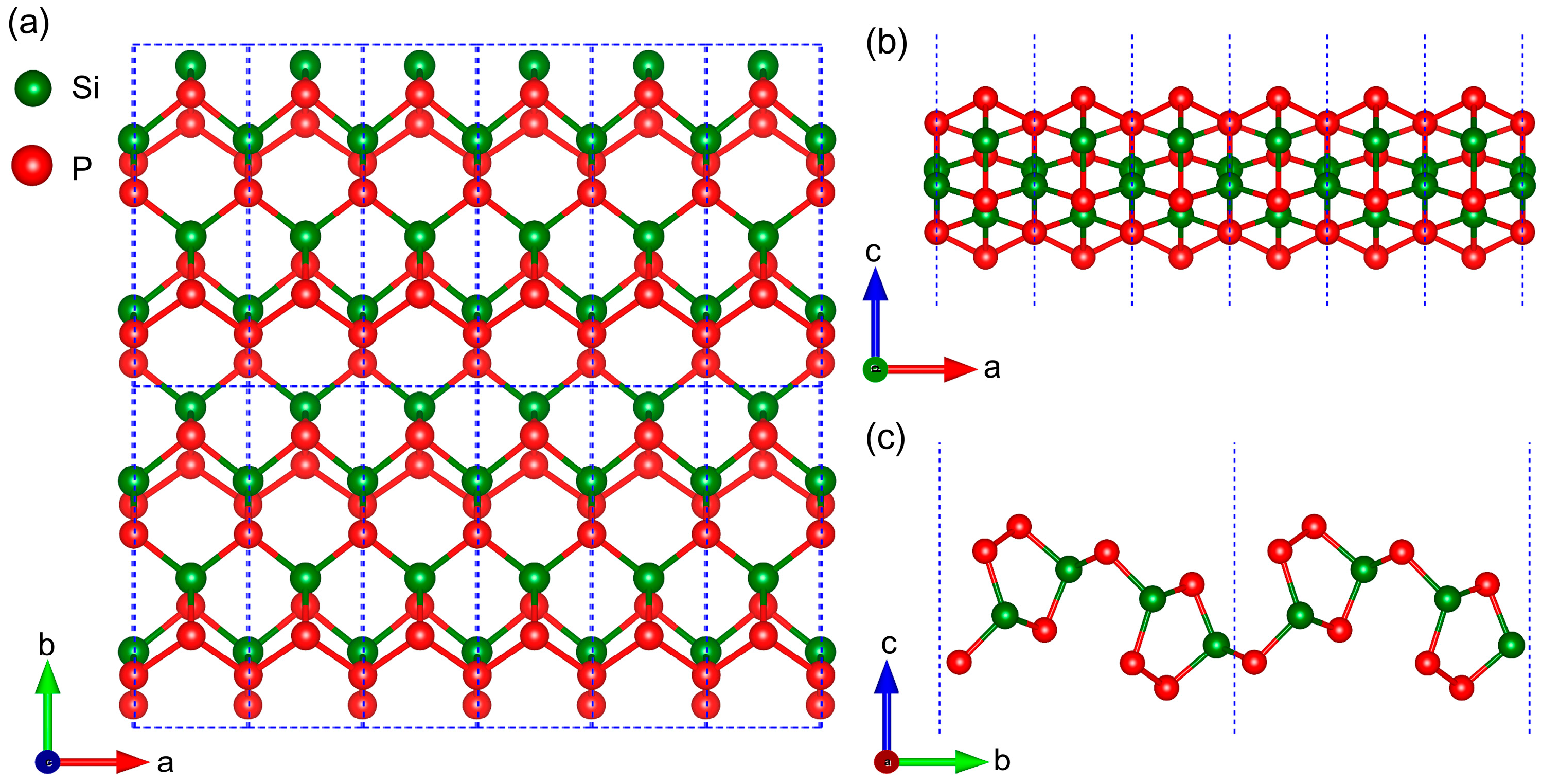
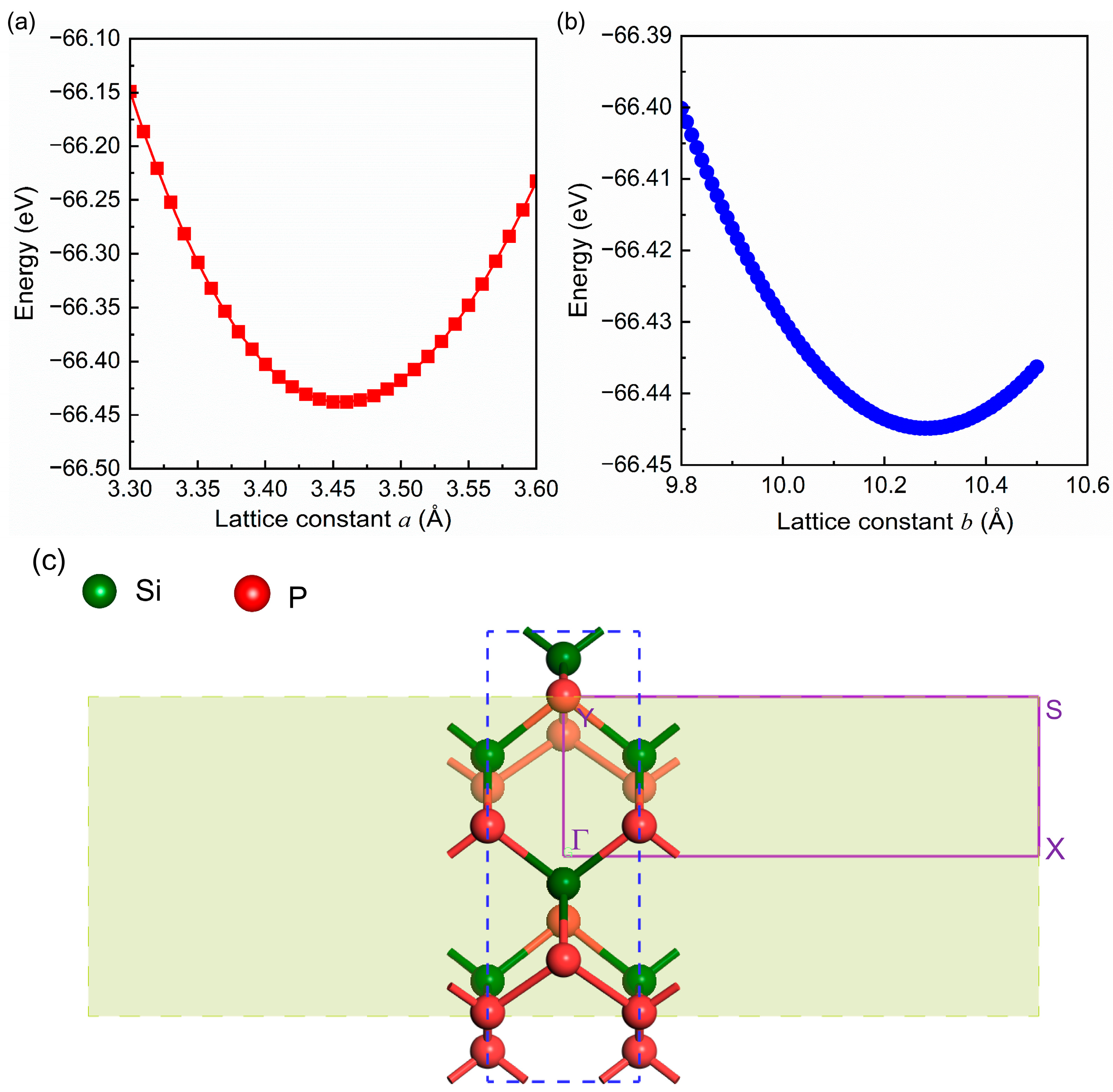
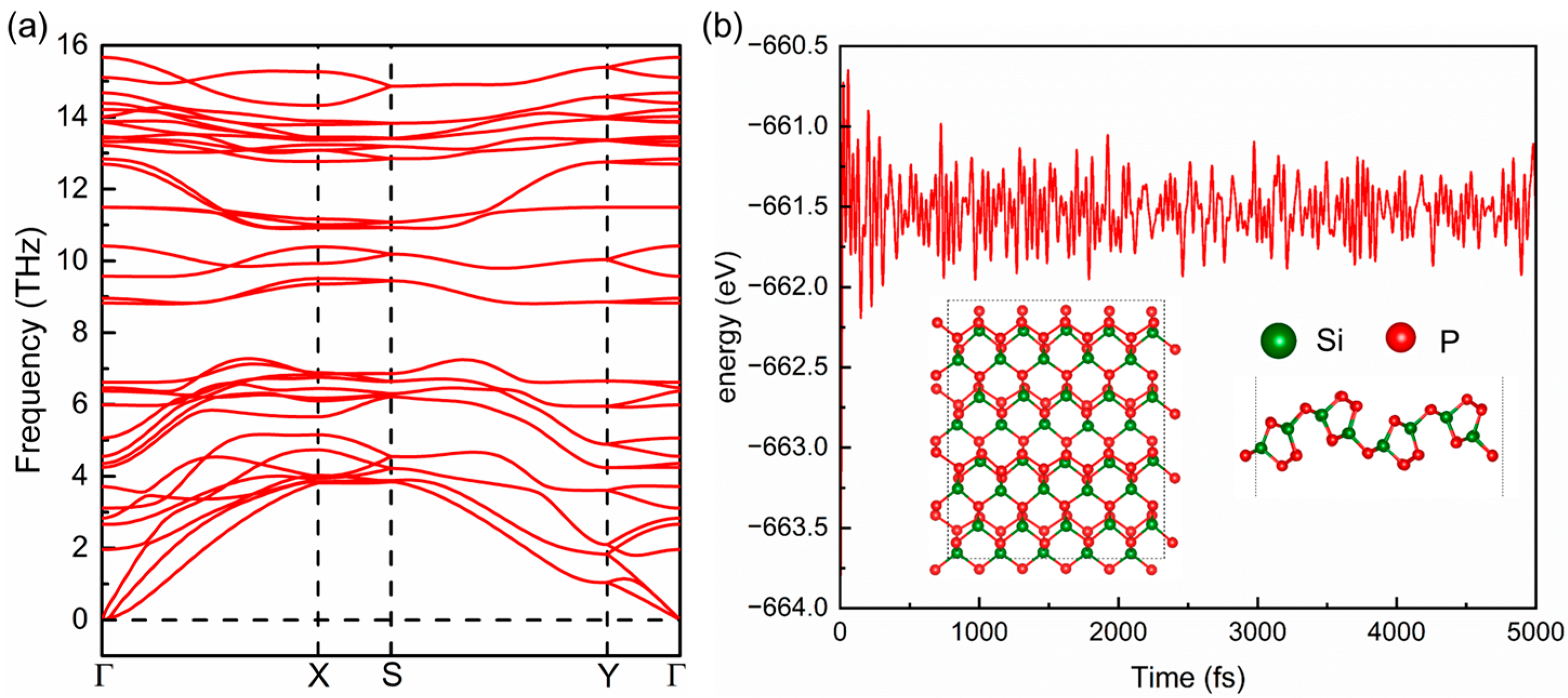
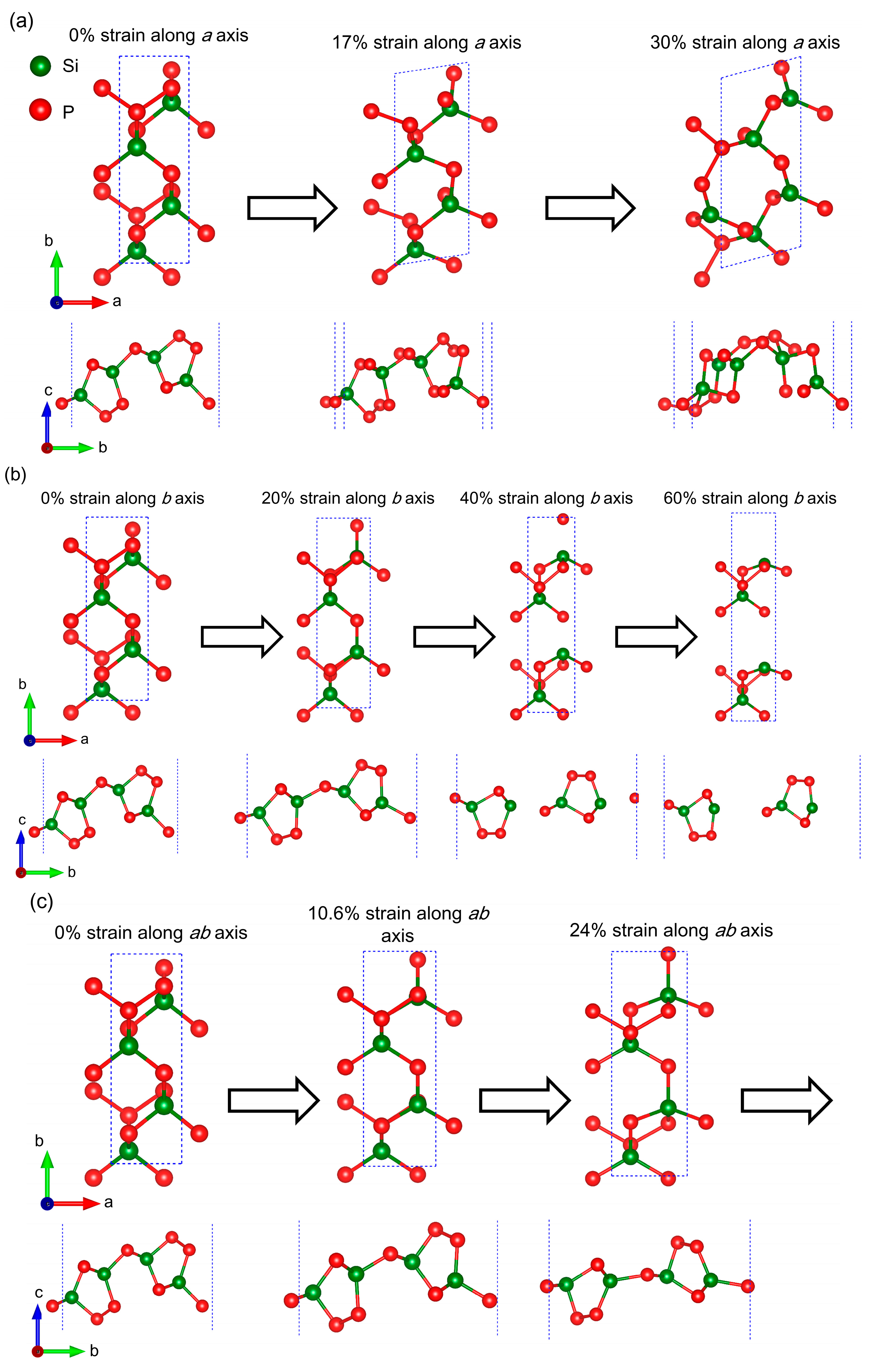
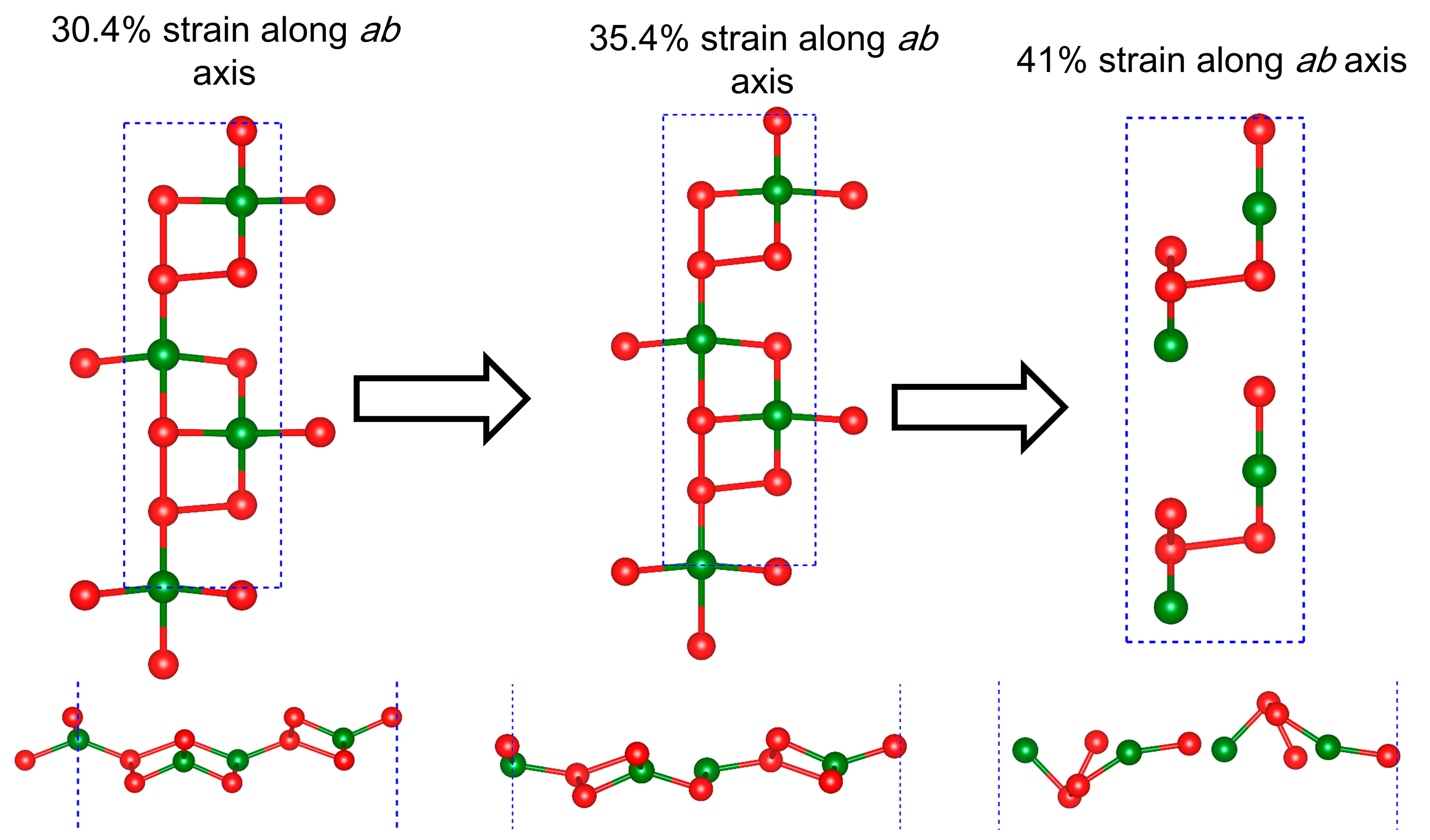
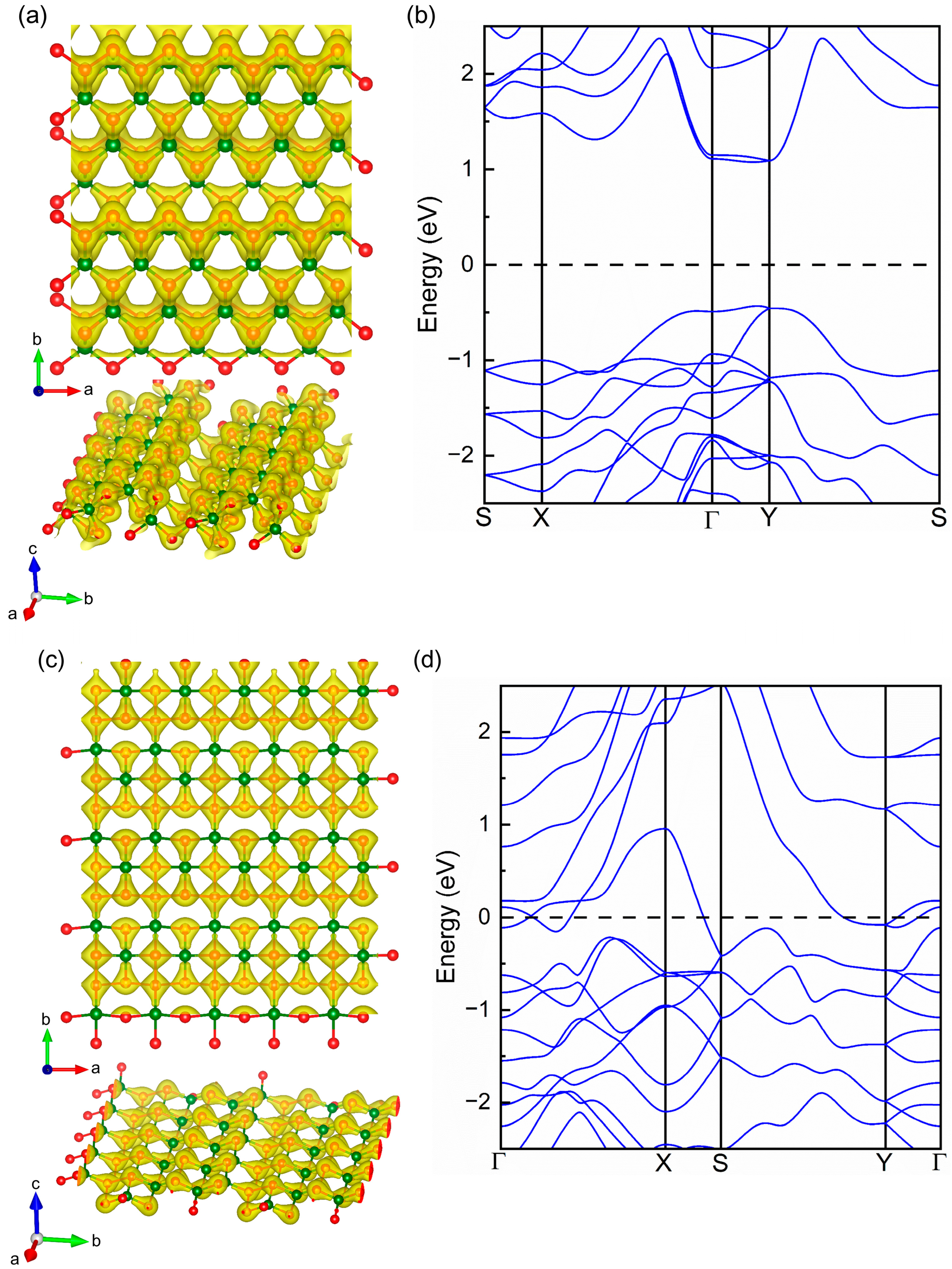
2.1. Geometrical Structure and Stability
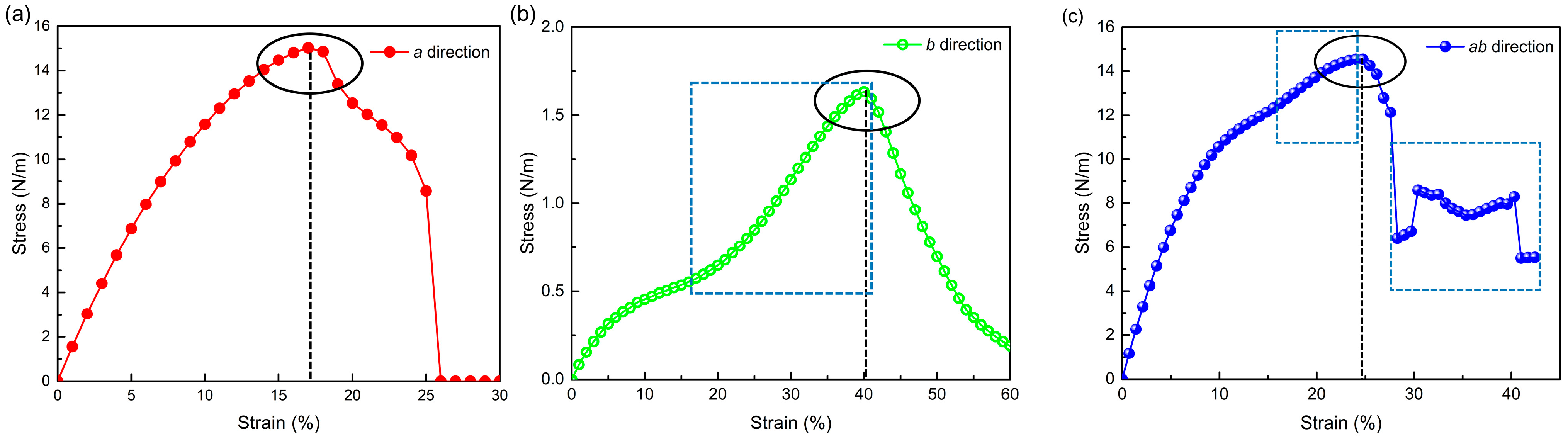
2.2. Anisotropic Mechanical Properties
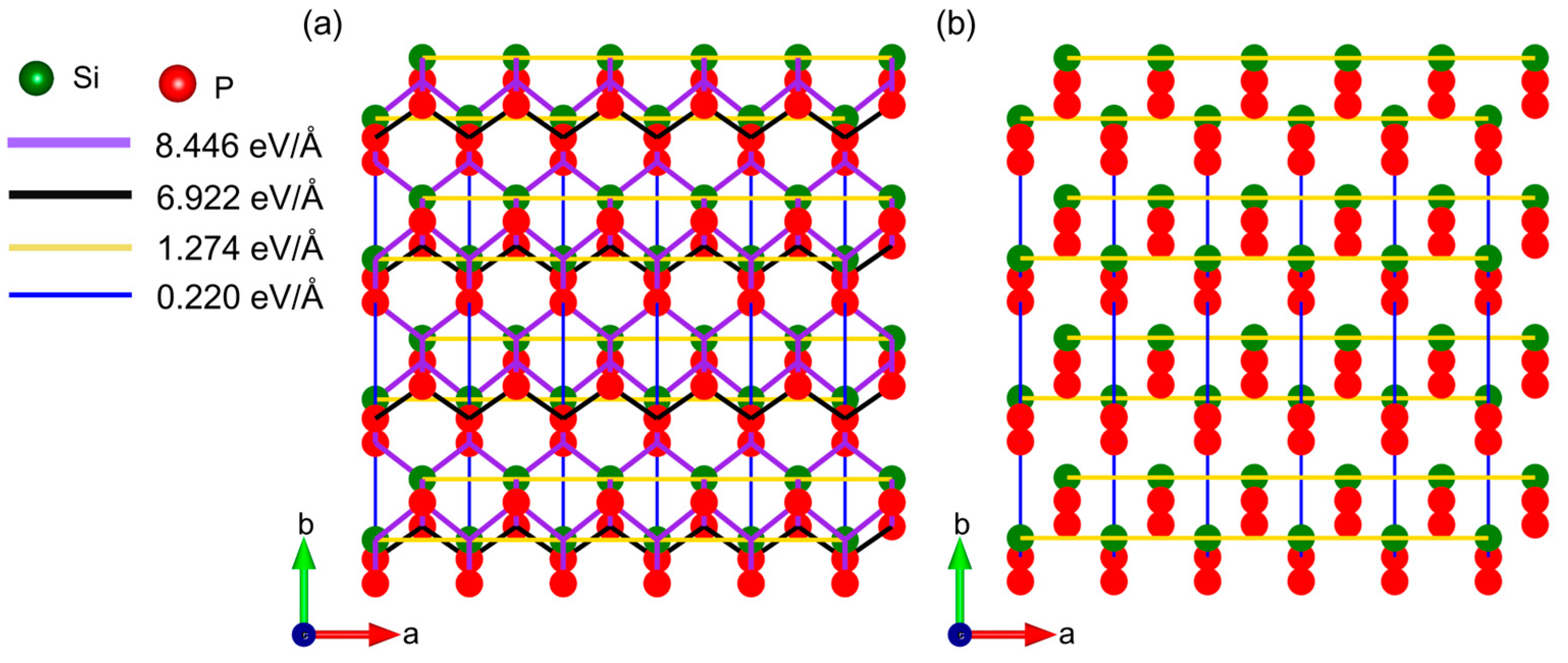
2.3. Origin of Mechanical In-Plane Anisotropy
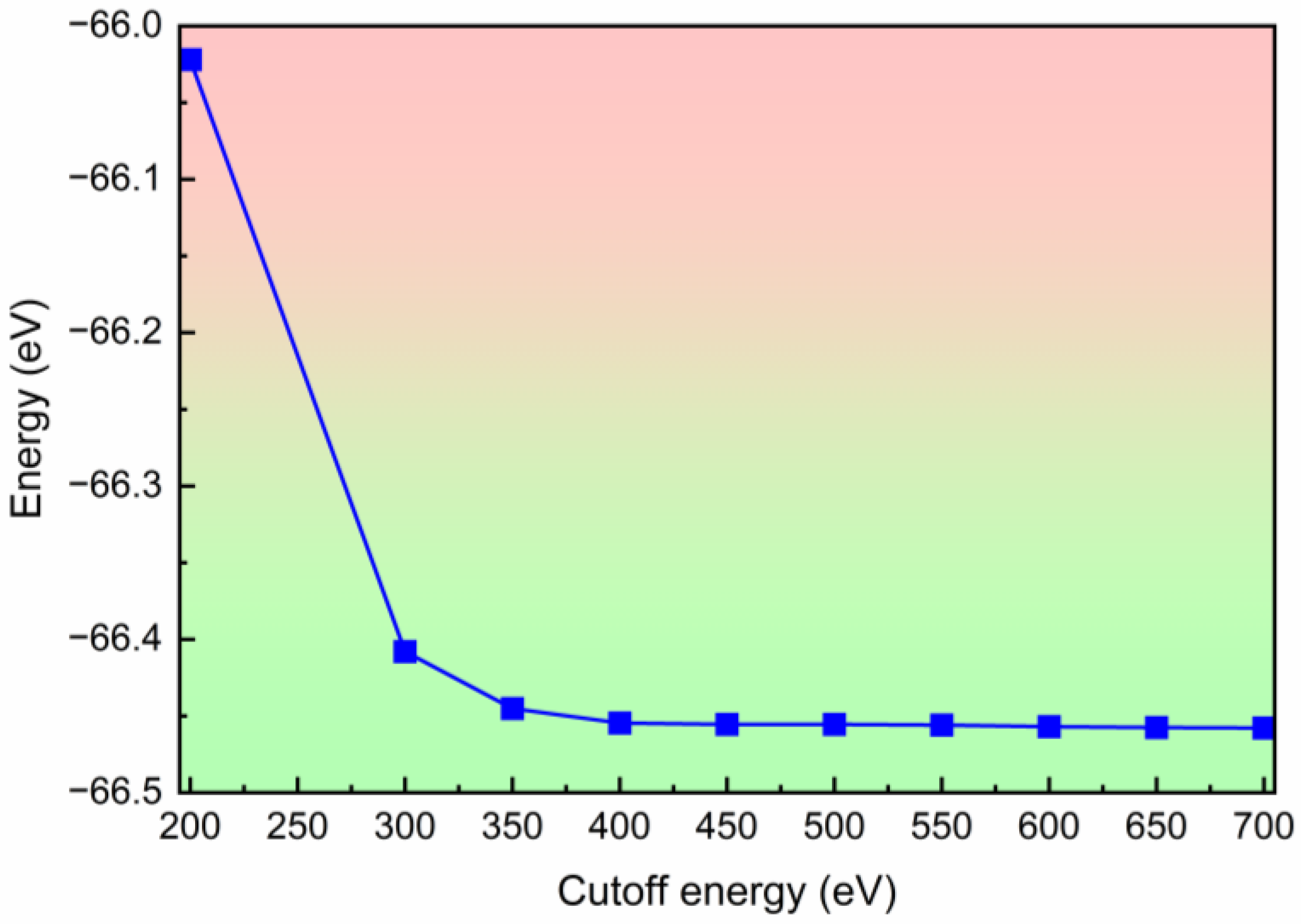
3. Computational Details
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Guo, S.-D.; Mu, W.-Q.; Wang, J.-H.; Yang, Y.-X.; Wang, B.; Ang, Y.-S. Strain effects on the topological and valley properties of the Janus monolayer VSiGeN4. Phys. Rev. B 2022, 106, 064416. [Google Scholar] [CrossRef]
- Guo, S.-D.; Mu, W.-Q.; Xiao, X.-B.; Liu, B.-G. Intrinsic room-temperature piezoelectric quantum anomalous hall insulator in Janus monolayer Fe2IX (X = Cl and Br). Nanoscale 2021, 13, 12956–12965. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.-D.; Zhu, Y.-T.; Qin, K.; Ang, Y.-S. Large out-of-plane piezoelectric response in ferromagnetic monolayer NiClI. Appl. Phys. Lett. 2022, 120, 232403. [Google Scholar] [CrossRef]
- Yankowitz, M.; Chen, S.; Polshyn, H.; Zhang, Y.; Watanabe, K.; Taniguchi, T.; Graf, D.; Young, A.F.; Dean, C.R. Tuning superconductivity in twisted bilayer graphene. Science 2019, 363, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Mielke, C.; Das, D.; Yin, J.X.; Liu, H.; Gupta, R.; Jiang, Y.X.; Medarde, M.; Wu, X.; Lei, H.C.; Chang, J.; et al. Time-reversal symmetry-breaking charge order in a kagome superconductor. Nature 2022, 602, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Górnicka, K.; Gui, X.; Wiendlocha, B.; Nguyen, L.T.; Xie, W.; Cava, R.J.; Klimczuk, T. NbIr2B2 and TaIr2B2—New Low Symmetry Noncentrosymmetric Superconductors with Strong Spin–Orbit Coupling. Adv. Funct. Mater. 2021, 31, 2007960. [Google Scholar] [CrossRef]
- Mortazavi, B.; Shapeev, A.V. Anisotropic mechanical response, high negative thermal expansion, and outstanding dynamical stability of biphenylene monolayer revealed by machine-learning interatomic potentials. FlatChem 2022, 32, 100347. [Google Scholar] [CrossRef]
- Wang, K.; Ren, K.; Cheng, Y.; Chen, S.; Zhang, G. The impacts of molecular adsorption on antiferromagnetic MnPS3 monolayers: Enhanced magnetic anisotropy and intralayer Dzyaloshinskii–Moriya interaction. Mater. Horiz. 2022, 9, 2384–2392. [Google Scholar] [CrossRef]
- Wu, W.; Wang, L.; Li, Y.; Zhang, F.; Lin, L.; Niu, S.; Chenet, D.; Zhang, X.; Hao, Y.; Heinz, T.F.; et al. Piezoelectricity of single-atomic-layer MoS2 for energy conversion and piezotronics. Nature 2014, 514, 470–474. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Demir, A.; Fang, S.; Tomarken, S.L.; Luo, J.Y.; Sanchez-Yamagishi, J.D.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; et al. Correlated insulator behaviour at half-filling in magic-angle graphene superlattices. Nature 2018, 556, 80–84. [Google Scholar] [CrossRef]
- Cao, Y.; Fatemi, V.; Fang, S.; Watanabe, K.; Taniguchi, T.; Kaxiras, E.; Jarillo-Herrero, P. Unconventional superconductivity in magic-angle graphene superlattices. Nature 2018, 556, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sarma, S.D.; Adam, S.; Hwang, E.; Rossi, E. Electronic transport in two-dimensional graphene. Rev. Mod. Phys. 2011, 83, 407. [Google Scholar] [CrossRef]
- Balandin, A.A.; Ghosh, S.; Bao, W.; Calizo, I.; Teweldebrhan, D.; Miao, F.; Lau, C.N. Superior Thermal Conductivity of Single-Layer Graphene. Nano Lett. 2008, 8, 902–907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Ma, L.; Fan, F.; Zeng, Z.; Peng, C.; Loya, P.E.; Liu, Z.; Gong, Y.; Zhang, J.; Zhang, X.; et al. Fracture toughness of graphene. Nat. Commun. 2014, 5, 3782. [Google Scholar] [CrossRef]
- Conley, H.J.; Wang, B.; Ziegler, J.I.; Haglund, R.F., Jr.; Pantelides, S.T.; Bolotin, K.I. Bandgap Engineering of Strained Monolayer and Bilayer MoS2. Nano Lett. 2013, 13, 3626–3630. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.P.; Shen, Z.X. Structural and electronic properties of h-BN. Phys. Rev. B 2003, 68, 104102. [Google Scholar] [CrossRef]
- Cai, Y.; Ke, Q.; Zhang, G.; Feng, Y.P.; Shenoy, V.B.; Zhang, Y.-W. Giant Phononic Anisotropy and Unusual Anharmonicity of Phosphorene: Interlayer Coupling and Strain Engineering. Adv. Funct. Mater. 2015, 25, 2230–2236. [Google Scholar] [CrossRef]
- Island, J.O.; Barawi, M.; Biele, R.; Almazán, A.; Clamagirand, J.M.; Ares, J.R.; Sánchez, C.; van der Zant, H.S.J.; Álvarez, J.V.; D’Agosta, R.; et al. TiS3 Transistors with Tailored Morphology and Electrical Properties. Adv. Mater. 2015, 27, 2595–2601. [Google Scholar] [CrossRef]
- Fan, Q.; Yan, L.; Tripp, M.W.; Krejčí, O.; Dimosthenous, S.; Kachel, S.R.; Chen, M.; Foster, A.S.; Koert, U.; Liljeroth, P.; et al. Biphenylene network: A nonbenzenoid carbon allotrope. Science 2021, 372, 852–856. [Google Scholar] [CrossRef]
- Wang, K.; Ren, K.; Zhang, D.; Cheng, Y.; Zhang, G. Phonon properties of biphenylene monolayer by first-principles calculations. Appl. Phys. Lett. 2022, 121, 042203. [Google Scholar] [CrossRef]
- Ling, X.; Huang, S.; Hasdeo, E.H.; Liang, L.; Parkin, W.M.; Tatsumi, Y.; Nugraha, A.R.T.; Puretzky, A.A.; Das, P.M.; Sumpter, B.G.; et al. Anisotropic Electron-Photon and Electron-Phonon Interactions in Black Phosphorus. Nano Lett. 2016, 16, 2260–2267. [Google Scholar] [CrossRef] [PubMed]
- Yarmohammadi, M.; Mortezaei, M.; Mirabbaszadeh, K. Anisotropic basic electronic properties of strained black phosphorene. Phys. E 2020, 124, 114323. [Google Scholar] [CrossRef]
- Pearce, A.K.; Wilks, T.R.; Arno, M.C.; O’Reilly, R.K. Synthesis and applications of anisotropic nanoparticles with precisely defined dimensions. Nat. Rev. Chem. 2021, 5, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Roper, S.W.K.; Lee, H.; Huh, M.; Kim, I.Y. Simultaneous isotropic and anisotropic multi-material topology optimization for conceptual-level design of aerospace components. Struct. Multidiscip. O 2021, 64, 441–456. [Google Scholar] [CrossRef]
- Chen, Q.; Mangadlao, J.D.; Wallat, J.; De Leon, A.; Pokorski, J.K.; Advincula, R.C. 3D Printing Biocompatible Polyurethane/Poly(lactic acid)/Graphene Oxide Nanocomposites: Anisotropic Properties. ACS Appl. Mater. Inter. 2017, 9, 4015–4023. [Google Scholar] [CrossRef]
- Yan, D.; Chang, J.; Zhang, H.; Liu, J.; Song, H.; Xue, Z.; Zhang, F.; Zhang, Y. Soft three-dimensional network materials with rational bio-mimetic designs. Nat. Commun. 2020, 11, 1180. [Google Scholar] [CrossRef]
- Rokoš, O.; Ameen, M.M.; Peerlings, R.H.J.; Geers, M.G.D. Micromorphic computational homogenization for mechanical metamaterials with patterning fluctuation fields. J. Mech. Phys. Solids 2019, 123, 119–137. [Google Scholar] [CrossRef]
- Matta, S.K.; Zhang, C.; Jiao, Y.; O’Mullane, A.; Du, A. Versatile two-dimensional silicon diphosphide (SiP2) for photocatalytic water splitting. Nanoscale 2018, 10, 6369–6374. [Google Scholar] [CrossRef]
- Xu, Y.; Li, Z.; He, C.; Li, J.; Ouyang, T.; Zhang, C.; Tang, C.; Zhong, J. Intrinsic piezoelectricity of monolayer group IV–V MX2: SiP2, SiAs2, GeP2, and GeAs2. Appl. Phys. Lett. 2020, 116, 023103. [Google Scholar] [CrossRef]
- Shojaei, F.; Mortazavi, B.; Zhuang, X.; Azizi, M. Silicon diphosphide (SiP2) and silicon diarsenide (SiAs2): Novel stable 2D semiconductors with high carrier mobilities, promising for water splitting photocatalysts. Mater. Today Energy 2020, 16, 100377. [Google Scholar] [CrossRef]
- Wang, Z.; Luo, P.; Han, B.; Zhang, X.; Zhao, S.; Wang, S.; Chen, X.; Wei, L.; Yang, S.; Zhou, X.; et al. Strong In-Plane Anisotropic SiP2 as a IV–V 2D Semiconductor for Polarized Photodetection. ACS Nano 2021, 15, 20442–20452. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Huang, J.; Windgaetter, L.; Ong, C.S.; Zhao, X.; Zhang, C.; Tang, M.; Li, Z.; Qiu, C.; Latini, S.; et al. Unconventional excitonic states with phonon sidebands in layered silicon diphosphide. Nat. Mater. 2022, 21, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Huang, J.; Qin, F.; Zhou, L.; Dai, X.; Bi, X.; Zhang, C.; Li, Z.; Tang, M.; Qiu, C.; et al. Strain engineering of anisotropic light–matter interactions in one-dimensional P-P chain of SiP2. Nano Res. 2022, 15, 7378–7383. [Google Scholar] [CrossRef]
- Zhang, P.; Jiang, E.; Ouyang, T.; Tang, C.; He, C.; Li, J.; Zhang, C.; Zhong, J. Potential thermoelectric candidate monolayer silicon diphosphide (SiP2) from a first-principles calculation. Comp. Mater. Sci. 2021, 188, 110154. [Google Scholar] [CrossRef]
- Yang, J.; Zhou, W.; Chen, C.; Zhang, J.; Qu, H.; Ji, Y.; Tao, L.; Wu, Z.; Zeng, H.; Zhang, S. Extreme Anisotropic Dispersion and One-Dimensional Confined Electrons in 2-D SiP2 FETs With High Transmission Coefficients. IEEE Trans. Electron Devices 2023, 70, 1330–1337. [Google Scholar] [CrossRef]
- Studart, A.R. Biological and Bioinspired Composites with Spatially Tunable Heterogeneous Architectures. Adv. Funct. Mater. 2013, 23, 4423–4436. [Google Scholar] [CrossRef]
- Wadsten, T. Crystal structures of SiP2 SiAs2 and gep. Acta Chem. Scand. 1967, 21, 593. [Google Scholar] [CrossRef]
- Bryden, J. The crystal structures of the germanium–arsenic compounds: I. Germanium diarsenide, GeAs2. Acta Crystallogr. 1962, 15, 167–171. [Google Scholar] [CrossRef]
- Li, P.; Zhang, W.; Li, D.; Liang, C.; Zeng, X.C. Multifunctional Binary Monolayers GexPy: Tunable Band Gap, Ferromagnetism, and Photocatalyst for Water Splitting. ACS Appl. Mater. Inter. 2018, 10, 19897–19905. [Google Scholar] [CrossRef]
- Sun, S.; Meng, F.; Xu, Y.; He, J.; Ni, Y.; Wang, H. Flexible, auxetic and strain-tunable two dimensional penta-X2C family as water splitting photocatalysts with high carrier mobility. J. Mater. Chem. A 2019, 7, 7791–7799. [Google Scholar] [CrossRef]
- Shojaei, F.; Hahn, J.R.; Kang, H.S. Electronic structure and photocatalytic band offset of few-layer GeP2. J. Mater. Chem. A 2017, 5, 22146–22155. [Google Scholar] [CrossRef]
- Tan, Z.W.; Wang, J.-S.; Gan, C.K. First-Principles Study of Heat Transport Properties of Graphene Nanoribbons. Nano Lett. 2011, 11, 214–219. [Google Scholar] [CrossRef]
- Wang, K.; Zhou, W.; Cheng, Y.; Zhang, M.; Wang, H.; Zhang, G. Magnetic order-dependent phonon properties in 2D magnet CrI3. Nanoscale 2021, 13, 10882–10890. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; He, J.; Zhang, M.; Wang, H.; Zhang, G. Magnon–phonon interaction in antiferromagnetic two-dimensional MXenes. Nanotechnology 2020, 31, 435705. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, F.; Coudert, F.-X. Necessary and sufficient elastic stability conditions in various crystal systems. Phys. Rev. B 2014, 90, 224104. [Google Scholar] [CrossRef]
- Cadelano, E.; Palla, P.L.; Giordano, S.; Colombo, L. Elastic properties of hydrogenated graphene. Phys. Rev. B 2010, 82, 235414. [Google Scholar] [CrossRef]
- Wei, Q.; Peng, X. Superior mechanical flexibility of phosphorene and few-layer black phosphorus. Appl. Phys. Lett. 2014, 104, 251915. [Google Scholar] [CrossRef]
- Xu, X.; Ma, Y.; Zhang, T.; Lei, C.; Huang, B.; Dai, Y. Prediction of two-dimensional antiferromagnetic ferroelasticity in an AgF2 monolayer. Nanoscale Horiz. 2020, 5, 1386–1393. [Google Scholar] [CrossRef]
- Liu, K.; Yan, Q.; Chen, M.; Fan, W.; Sun, Y.; Suh, J.; Fu, D.; Lee, S.; Zhou, J.; Tongay, S.; et al. Elastic Properties of Chemical-Vapor-Deposited Monolayer MoS2, WS2, and Their Bilayer Heterostructures. Nano Lett. 2014, 14, 5097–5103. [Google Scholar] [CrossRef]
- Wang, Y.; Qiao, M.; Li, Y.; Chen, Z. A two-dimensional CaSi monolayer with quasi-planar pentacoordinate silicon. Nanoscale Horiz. 2018, 3, 327–334. [Google Scholar] [CrossRef]
- Qin, G.; Qin, Z. Negative Poisson’s ratio in two-dimensional honeycomb structures. NPJ Comput. Mater. 2020, 6, 51. [Google Scholar] [CrossRef]
- Du, Y.; Maassen, J.; Wu, W.; Luo, Z.; Xu, X.; Ye, P.D. Auxetic Black Phosphorus: A 2D Material with Negative Poisson’s Ratio. Nano Lett. 2016, 16, 6701–6708. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.-W.; Park, H.S. Negative poisson’s ratio in single-layer black phosphorus. Nat. Commun. 2014, 5, 4727. [Google Scholar] [CrossRef]
- Liu, S.; Liu, B.; Shi, X.; Lv, J.; Niu, S.; Yao, M.; Li, Q.; Liu, R.; Cui, T.; Liu, B. Two-dimensional Penta-BP5 Sheets: High-stability, Strain-tunable Electronic Structure and Excellent Mechanical Properties. Sci. Rep. 2017, 7, 2404. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Yang, H.; Fu, L.; He, C.; Huo, J.; Guo, J.; Li, L. Prediction of semiconducting SiP2 monolayer with negative Possion’s ratio, ultrahigh carrier mobility and CO2 capture ability. Chin. Chem. Lett. 2021, 32, 1089–1094. [Google Scholar] [CrossRef]
- Xu, B.; Zhu, J.; Xiao, F.; Jiao, C.; Liang, Y.; Wen, T.; Wu, S.; Zhang, Z.; Lin, L.; Pei, S.; et al. Identifying, Resolving, and Quantifying Anisotropy in ReS2 Nanomechanical Resonators. Small 2023, 19, 2300631. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Wang, K.; Yu, Y.; Zhang, D.; Zhang, G.; Cheng, Y. Tuning the mechanical anisotropy of biphenylene by boron and nitrogen doping. Comp. Mater. Sci. 2023, 222, 112119. [Google Scholar] [CrossRef]
- Peng, X.; Wei, Q.; Copple, A. Strain-engineered direct-indirect band gap transition and its mechanism in two-dimensional phosphorene. Phys. Rev. B 2014, 90, 085402. [Google Scholar] [CrossRef]
- Zhou, H.; Cai, Y.; Zhang, G.; Zhang, Y.-W. Superior lattice thermal conductance of single-layer borophene. NPJ 2D Mater. Appl. 2017, 1, 14. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Gonze, X. Adiabatic density-functional perturbation theory. Phys. Rev. A 1995, 52, 1096. [Google Scholar] [CrossRef] [PubMed]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio simulation of the metal/nonmetal transition in expanded fluid mercury. Phys. Rev. B 1997, 55, 7539. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Lattice Constants (Å) | Layer Thickness (Å) | Bond Length (Å) | ||
|---|---|---|---|---|---|
| a | b | P-P | P-Si | ||
| 0% | 3.46 | 10.28 | 5.58 | 2.269 | 2.281 |
| 17%-a | 4.06 | 10.32 | 5.43 | 2.369 | 2.313 |
| 20%-b | 3.46 | 12.34 | 5.41 | 2.269 | 2.326 |
| 40%-b | 3.46 | 14.39 | 4.65 | 2.266 | 2.362 |
| 10.6%-ab | 3.83 | 11.37 | 4.85 | 2.369 | 2.305 |
| 24%-ab | 4.29 | 12.75 | 4.80 | 2.478 | 2.297 |
| 30.4%-ab | 4.51 | 13.41 | 2.76 | 2.441 | 2.251 |
| 35.4%-ab | 4.68 | 13.92 | 1.79 | 2.285 | 2.287 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, Y.; Ren, K.; Wei, Y.; Yang, D.; Cui, Z.; Wang, K. Anisotropic Mechanical Properties of Orthorhombic SiP2 Monolayer: A First-Principles Study. Molecules 2023, 28, 6514. https://doi.org/10.3390/molecules28186514
Hou Y, Ren K, Wei Y, Yang D, Cui Z, Wang K. Anisotropic Mechanical Properties of Orthorhombic SiP2 Monolayer: A First-Principles Study. Molecules. 2023; 28(18):6514. https://doi.org/10.3390/molecules28186514
Chicago/Turabian StyleHou, Yinlong, Kai Ren, Yu Wei, Dan Yang, Zhen Cui, and Ke Wang. 2023. "Anisotropic Mechanical Properties of Orthorhombic SiP2 Monolayer: A First-Principles Study" Molecules 28, no. 18: 6514. https://doi.org/10.3390/molecules28186514
APA StyleHou, Y., Ren, K., Wei, Y., Yang, D., Cui, Z., & Wang, K. (2023). Anisotropic Mechanical Properties of Orthorhombic SiP2 Monolayer: A First-Principles Study. Molecules, 28(18), 6514. https://doi.org/10.3390/molecules28186514