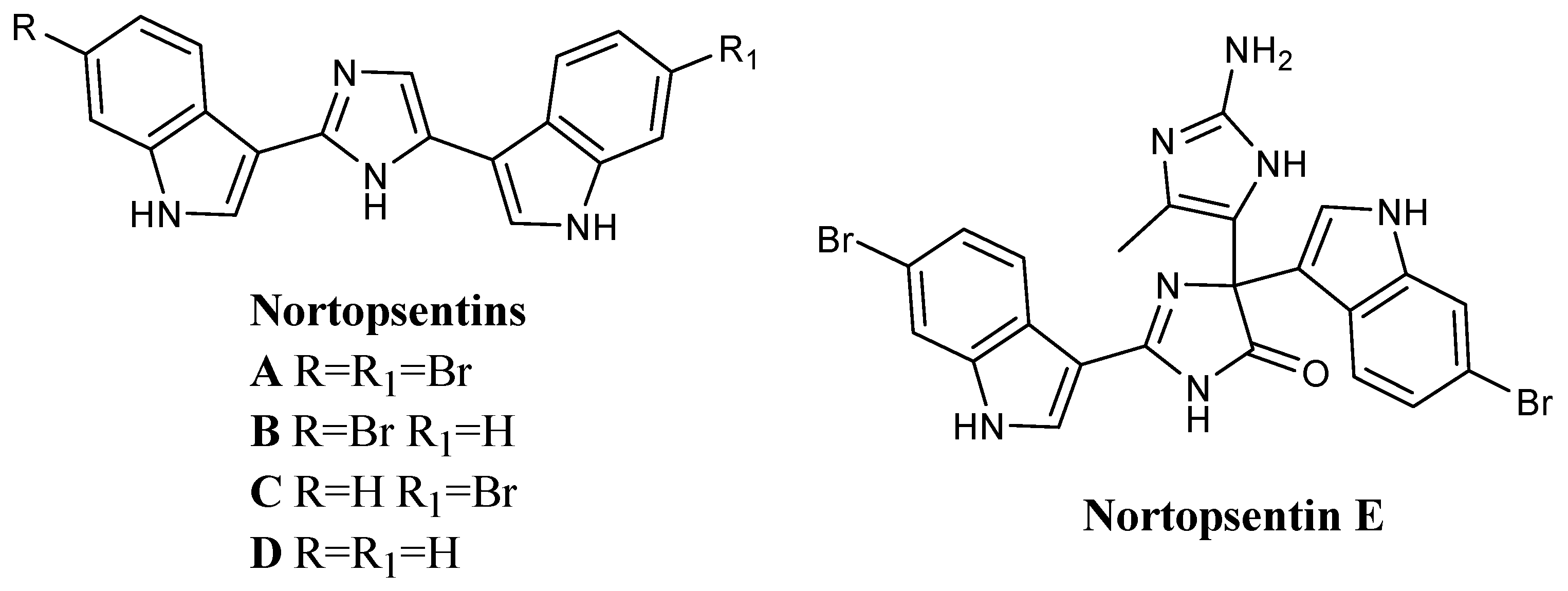
2.1.1. Thiazoles
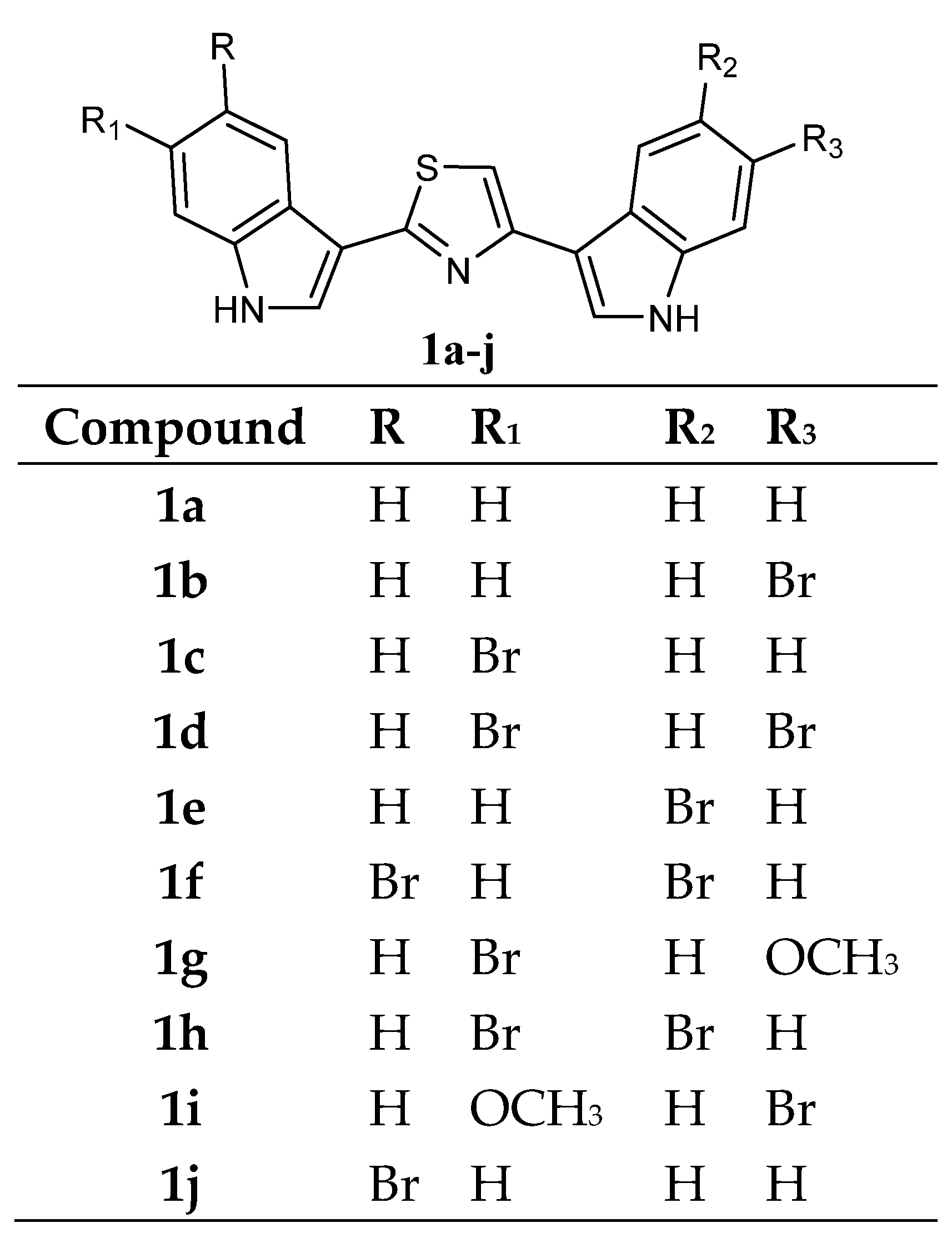
Several bis-indolyl-thiazole compounds
1 (
Figure 2) were synthesised and tested against the National Cancer Institute (NCI, Bethesda, MD 20892, USA) full panel of 60 human cancer cell lines derived from nine cancer cell types and grouped into disease subpanels including leukaemia, non-small cell lung, colon, central nervous system, melanoma, ovarian, renal, prostate, and breast cancers. Many compounds showed GI
50 values in the micromolar-submicromolar range. In particular, compounds
1a–
j exhibited cytotoxic activities against a variety of human cancer cell lines. The compound
1a exhibited highly selective
in vitro cytotoxicity against leukaemia (GI
50 of 3.27 µM in K562, 5.31 µM in Molt-4) and ovarian cancer cell lines (GI
50 8.14 µM in IGROV1) (
Table 1). In many other human tumour cell lines, the GI
50 of compound
1a exceeded 100 µM. It is worth noting that unlike the unsubstituted compound
1a, the bis-indolyl-thiazoles
1b–
j showed broad effects on leukaemia, colon, CNS, and breast cancer panels, suggesting that substituents in the indole ring might result in a potency increase (
Table 2) [
57,
58].
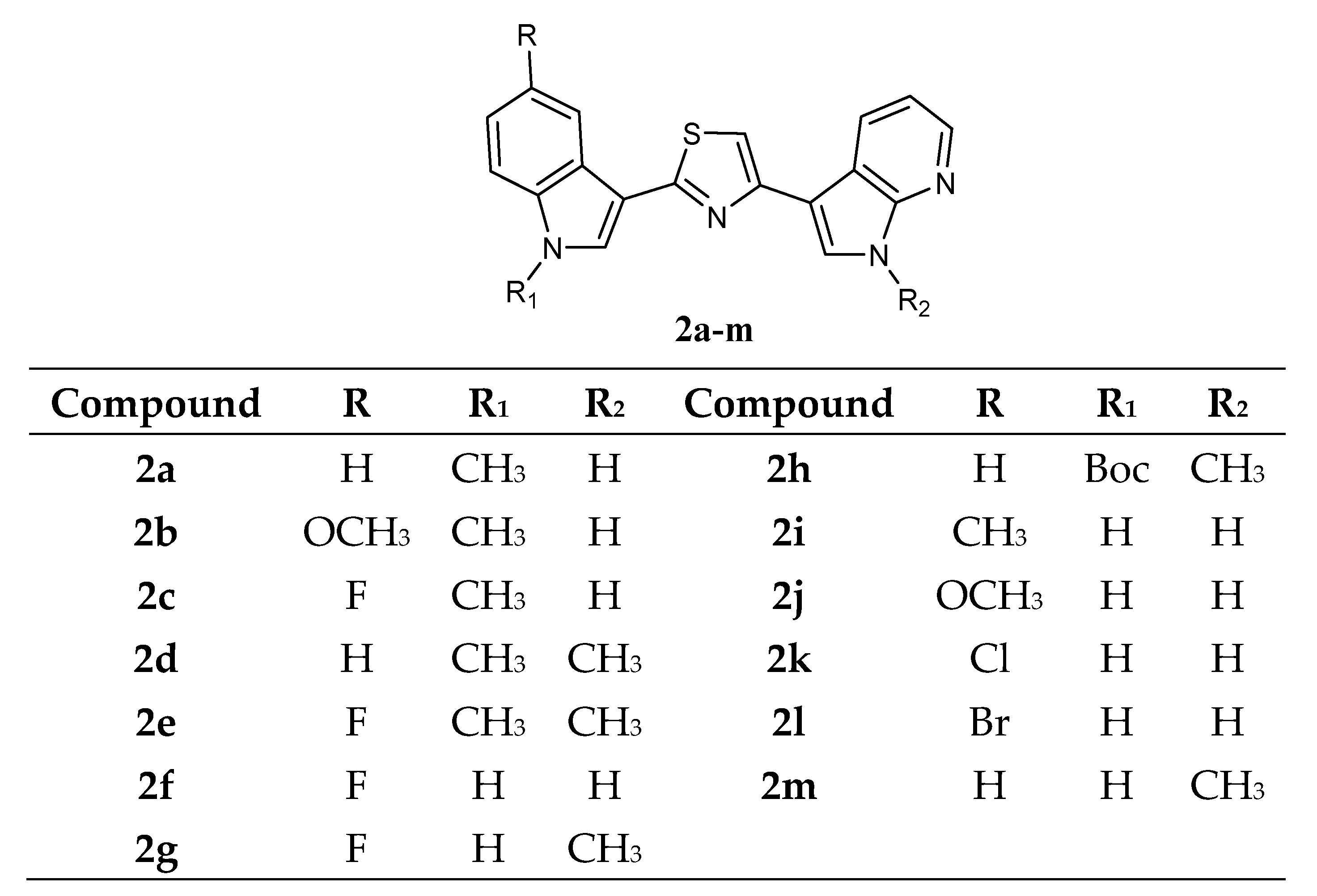
Another thiazole series
2a–
m (
Figure 3), bearing an indole and a 7-azaindole moiety, has been reported. These derivatives were tested by NCI against a panel of 60 human cancer cell lines.
Data revealed that these compounds
2a–
m showed GI
50 values in the micromolar–submicromolar range. The five most active compounds,
2c,
2d,
2e,
2g, and
2m, which did not show selectivity against any of the tumour subpanels, were further investigated in two additional cell lines, STO and MesoII, derived from human diffuse malignant peritoneal mesothelioma (DMPM), a tumour type not included in the NCI panel. Seventy-two hours of exposure to increasing concentrations of each compound resulted in dose-dependent cell proliferation inhibition in both cellular models. Compounds
2c,
2d,
2e,
2g, and
2m, exhibited comparable activity in STO cells with IC
50 values ranging from 0.33 to 0.61 μM. By contrast, a variable growth inhibitory effect was induced by the different compounds in MesoII cells (IC
50 values ranging from 4.11 to 25.12 μM). In addition, compounds
2c,
2e, and
2m, did not interfere with the growth of normal cells (
Table 3). The anti-tumour activity of
2c,
2e, and
2m derivatives was then evaluated on STO cells xenotransplanted in athymic nude mice. The treatment with the different compounds resulted in marked tumour growth inhibition. Specifically, at the end of the experiment, a statistically significant tumour volume inhibition (TVI) compared with the control (73%, 75%, and 58%, for
2c,
2e, and
2m derivatives, respectively) was observed, and two complete responses (disappearance of tumour) were also identified in each treatment group (
Table 4). Moreover, the compounds
2c,
2e, and
2m were well tolerated without any appreciable sign of toxicity.
In vitro kinase assays revealed CDK1 inhibition exerted by the compounds with IC
50 values of 0.89, 0.75, and 0.86 μM, respectively, for the derivatives
2c,
2e, and
2m (
Table 5). These results were comparable to those reported for two well-known CDK1 inhibitors, roscovitine and purvanalol A. In addition, derivatives
2c,
2e, and
2m were able to inhibit GSK3β, but only at higher concentrations (IC
50 values of 42.18, 40.18, and 35.68 μM, respectively) (
Table 5). Further investigations revealed a marked time-dependent cell cycle arrest at the G2/M phase and an increase in the apoptotic rate by reducing the phosphorylated form of the antiapoptotic protein survivin. Moreover, the addition of compound
2m to paclitaxel-treated cells resulted in a synergistic cytotoxic effect due to an increased apoptotic response [
20].
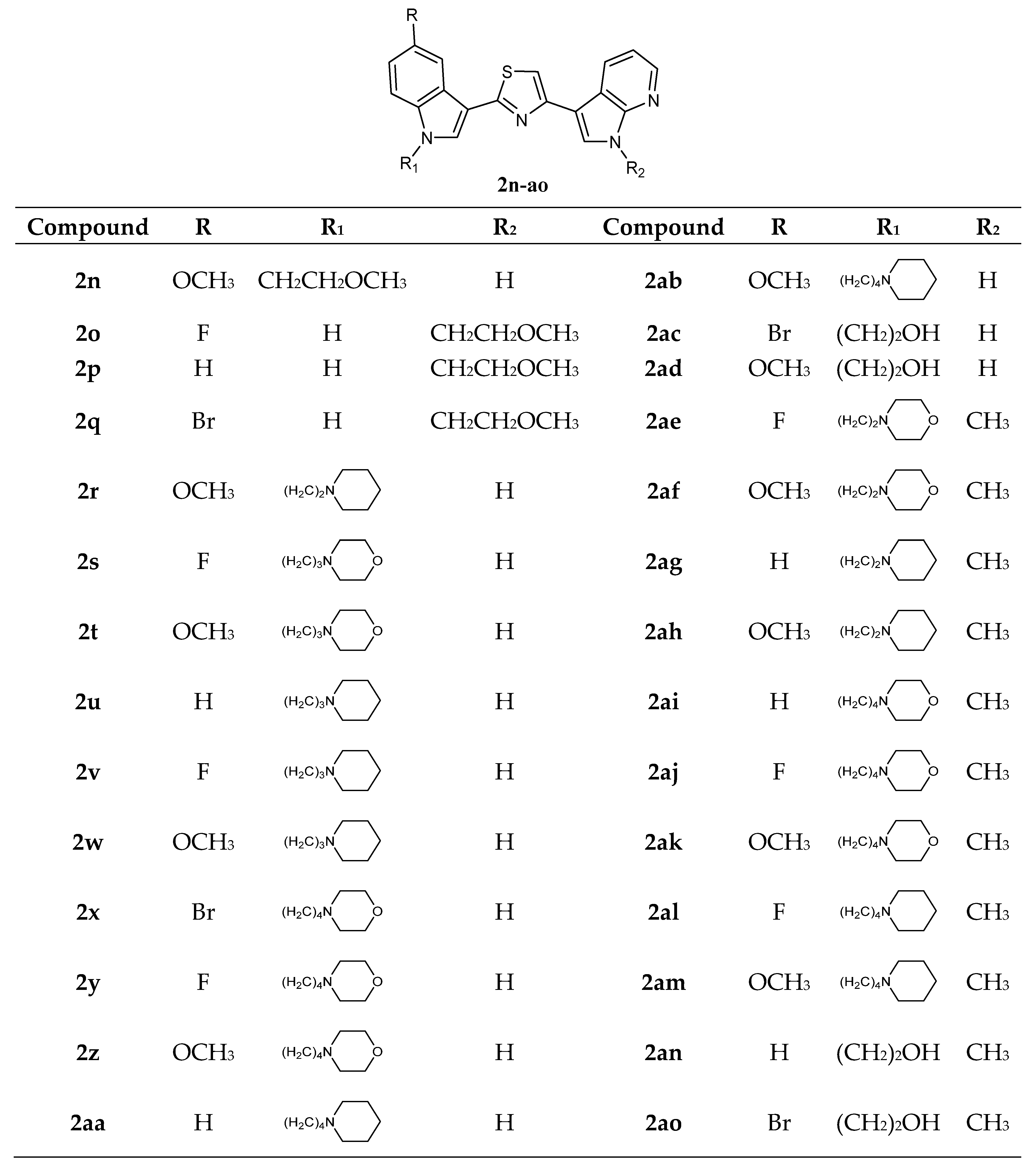
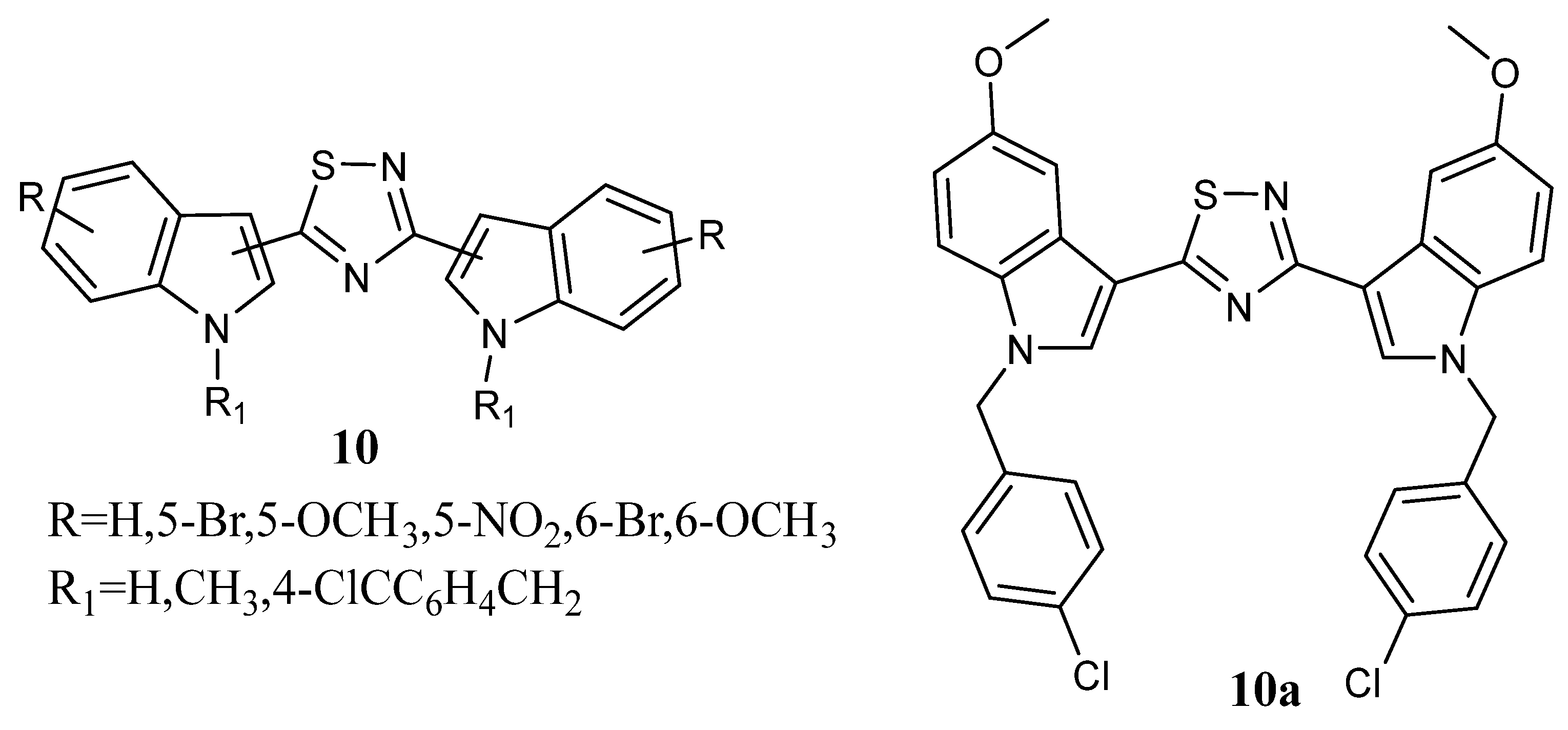
Thiazole Nortopsentin analogues of type
2n–
q (
Figure 4), in which the nitrogen atom of the indole and/or 7-azaindole moiety is substituted with a 2-methoxyethyl chain, and analogues
2r–
2ao, in which indole nitrogen is substituted with alkylmorpholine or alkylpiperidine, were synthesized. Derivatives
2n–
2q,
2t–
w,
2z,
2ab,
2ae, and
2ak–
am were tested by NCI on the full panel of approximately 60 human cancer cell lines and showed good antiproliferative activity with GI
50 in the micromolar–nanomolar range. Compounds
2n,
2q,
2t–
w,
2z,
2ab,
2ae, and
2ak–
am were active against the total number of cell lines investigated, whereas compounds
2o and
2p were cytotoxic against a very high percentage of the tested cell lines (96% and 93%, respectively). Their action mechanism, investigated on human breast cancer MCF-7 cells, was pro-apoptotic, being associated with externalisation of plasma membrane phosphatidylserine and DNA fragmentation, accompanied by perturbation of the cell cycle progression. It was found that the derivatives
2n–
p confined viable cells in the G2/M phase. Derivative
2n showed the most interesting
in vitro anticancer activity, expressing lower GI
50 values (0.03–12.6 µM) and, markedly,
in vitro inhibited CDK1 activity with an IC
50 value of 1.14 ± 0.09 µM, comparable to that reported for other indolyl-thiazolyl-7-azaindole derivatives or well-known CDK1 inhibitors, roscovitine and purvanalol A. Moreover, cytotoxicity assays on intestinal normal-like differentiated Caco-2 cells after treatment with compounds
2n–
q in the 25–100 µM range revealed that these compounds were selectively cytotoxic to cancer cells (
Table 6) [
20].
Among the derivatives
2r–
2ao, compound
2ak was the most active of the series, showing selectivity against leukaemia and colon cancer subpanels. In addition,
2ak was effective against the A498 cell line of the renal cancer subpanel (GI
50 value of 20 nM) (
Table 7). Cytotoxicity and selectivity experiments were performed for compounds
2r–
2ao using the HepG2 cell line of human hepatoma, the MCF-7 cell line of human breast cancer, and the non-tumorigenic MCF 10A cell line by single-dose administration (10 µM). Most of the molecules
2r–
2ao showed antiproliferative effects on both Hep G2 and MCF-7 cells. Some of these molecules (
2r,
2ac,
2aa,
2ad,
2ae,
2am,
2an, and
2ao) showed cytotoxicity against the tumour cell lines without compromising non-tumorigenic MCF-10A cell viability. The N-alkylpiperidine substituted compounds (
2u,
2v,
2w,
2ag, and
2ah) were very active against the MCF-7 cell line but were also cytotoxic over MCF-10A cell line. Among the alkylmorpholino derivatives, propyl- and ethyl-morpholino derivatives (
2t and
2ae) were active over both Hep G2 and MCF-7 cells and non-toxic over non-tumorigenic cells, while compound
2af was active on cancer and non-cancer cell lines examined. On the other hand, butylmorpholino derivatives gave different results, being
2s,
2ai,
2aj, and
2ak cytotoxic, while
2x and
2y did not impair non-tumorigenic cells; in particular, the compound
2y showed selective toxicity on the Hep G2 cancer cell line (EC
50 values of 3.25, 23.05, and 29.09 µM for Hep G2, MCF-7, and MCF-10A cell lines, respectively). Moreover, considering the overexpression of the enzyme glutaminase-1 (GLS-1) in hepatic cancer cell lines and its low expression in the MCF-7 cell line, enzymatic assays were also performed, revealing good inhibitory potency of the compound
2y over GLS-1 (IC
50 value of 3.96 µM). This could explain the selective cytotoxicity shown by
2y on Hep G2 as opposed to MCF-7 and MCF-10A. Additional experiments on aggressive cancer cell lines with GLS-1 overexpression, like glioblastoma (U-87 MG), pancreatic cancer (MIA PaCa-2), osteosarcoma (Saos2), melanoma (A-375), and non-small lung cancer (A549) confirmed the cell growth inhibition potency for the compound
2y, with EC
50 values in the micromolar range (
Table 8). Data suggest that the decoration of the nitrogen atom of the indole and/or 7-azaindole moiety with 2-methoxyethyl, alkylmorpholine, or alkylpiperidine chain led to interesting biological results [
20,
59].
A series of thiazole Nortopsentin analogues of type
3 (
Figure 5), in which the imidazole moiety of Nortopsentins was replaced by a thiazole ring and one indole unit by a 5-azaindole ring, was synthesized. Derivatives
3a–
p were active against the NCI full panel, showing good antiproliferative activity in the micro–submicromolar range. Thiazoles
3e,
3f, and
3p were particularly cytotoxic against the leukaemia subpanel (GI
50 in the range 0.24–1.71 µM, 0.24–1.57 µM, and 0.35–2.13 µM, respectively) (
Table 9 and
Table 10). Compound
3b turned out to be the most active against the breast cancer subpanel (GI
50 in the range 0.27–2.16 µM). Moreover, compounds
3b and
3f proved to be selective against the HCT-116 cell line of the colon cancer subpanel (GI
50 of 0.93 µM and 0.18 µM, respectively) [
60].
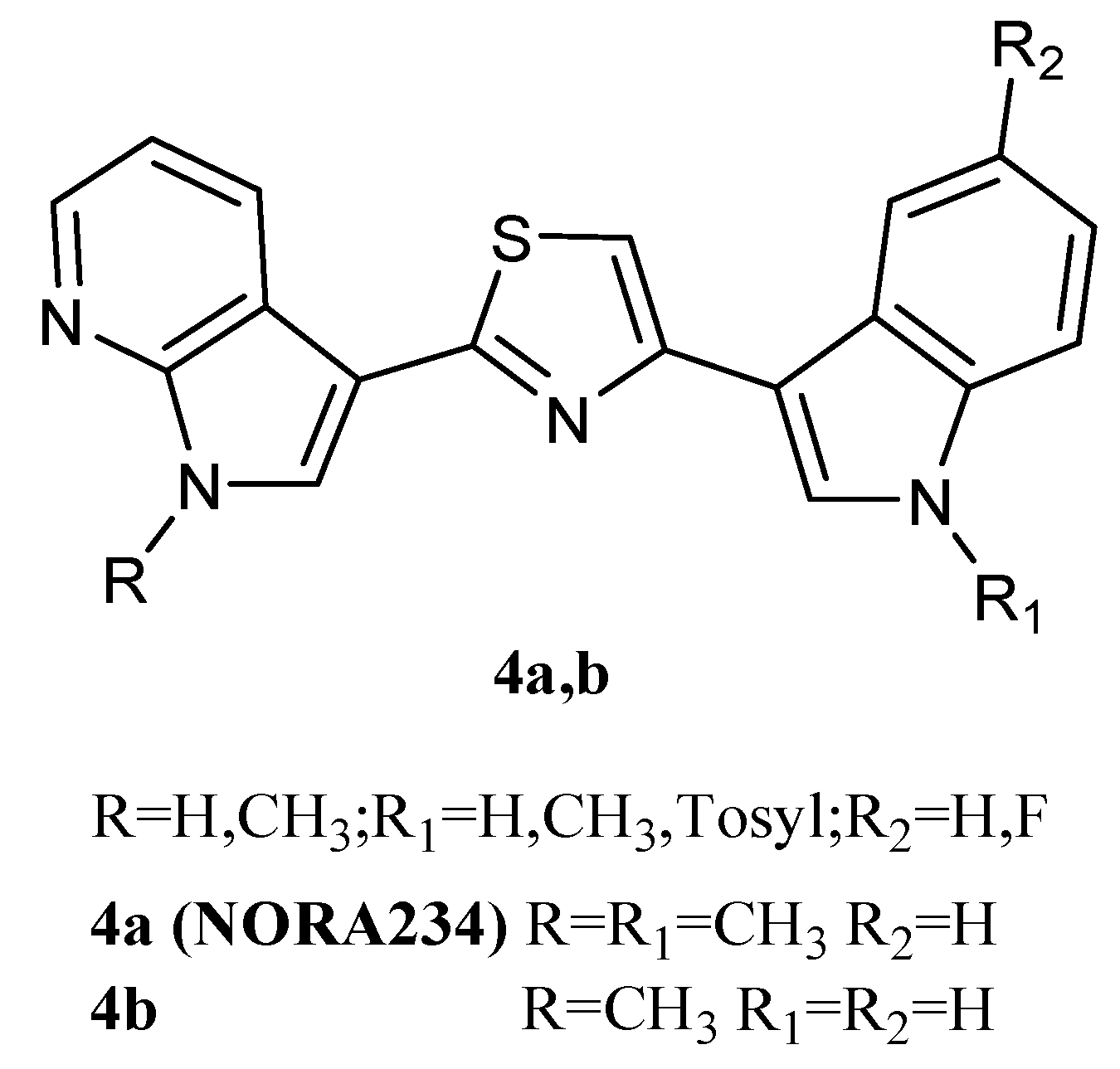
Thiazole derivatives
4a,
b (
Figure 6), in which indole and 7-azaindole units were switched compared to derivatives
2, were also reported. The derivatives
4a and
4b showed good antiproliferative activity against the NCI full panel (about 60 human tumour cell lines) with GI
50 values ranging from low micromolar to nanomolar levels (0.03–13.0 and 0.04–14.2 μM, respectively) (
Table 11). They also exhibited potent cytotoxicity on HepG2 hepatocarcinoma cells, a cell line not included in the NCI panel. Both compounds inhibited the HepG2 cells growth in a dose-dependent manner, with GI
50 values of 1.69 and 0.21 µM for
4a and
4b, respectively. Under the same conditions, both compounds did not affect the viability of normal immortalised human liver cells Chang, proving to be selective towards tumour cells. The mechanism of action of these derivatives was pro-apoptotic, being associated with the externalisation of plasma membrane phosphatidylserine and mitochondrial dysfunction. Both compounds
4a and
4b caused a significant dose-dependent decrease in the percentage of cells in the G0/G1 and S phases, coupled with an increase in cells in the G2/M phase, and the appearance of a subG1-cell population [
20].
In addition, the compound
4a, called Nortopsentin 234 (NORA234) (
Figure 6) was found to lead to an initial reduction in the proliferative and clonogenic potential of advanced colorectal cancer sphere cells (CR-CSphCs), followed by an adaptive response selecting the CR-CSphC-resistant compartment. Cells saved from the treatment with NORA234 expressed high levels of CD44v6, combined with constitutive activation of the Wnt pathway. In CR-CSphC-based organoids, NORA234 caused genotoxic stress with concomitant G2/M cell cycle arrest and activation of CHK1, driving the DNA damage repair of CR-CSphCs, regardless of the mutational background, microsatellite stability, and consensus molecular subtype. The synergic combination of NORA234 and a CHK1 inhibitor (rabusertib) targeted synthetic lethal inducing death in both CD44v6-negative and CD44v6-positive CRC stem cell fractions, apart from Wnt pathway activity. These data could provide a rationale for developing an effective strategy for the treatment of colorectal cancer (CRC) [
61].
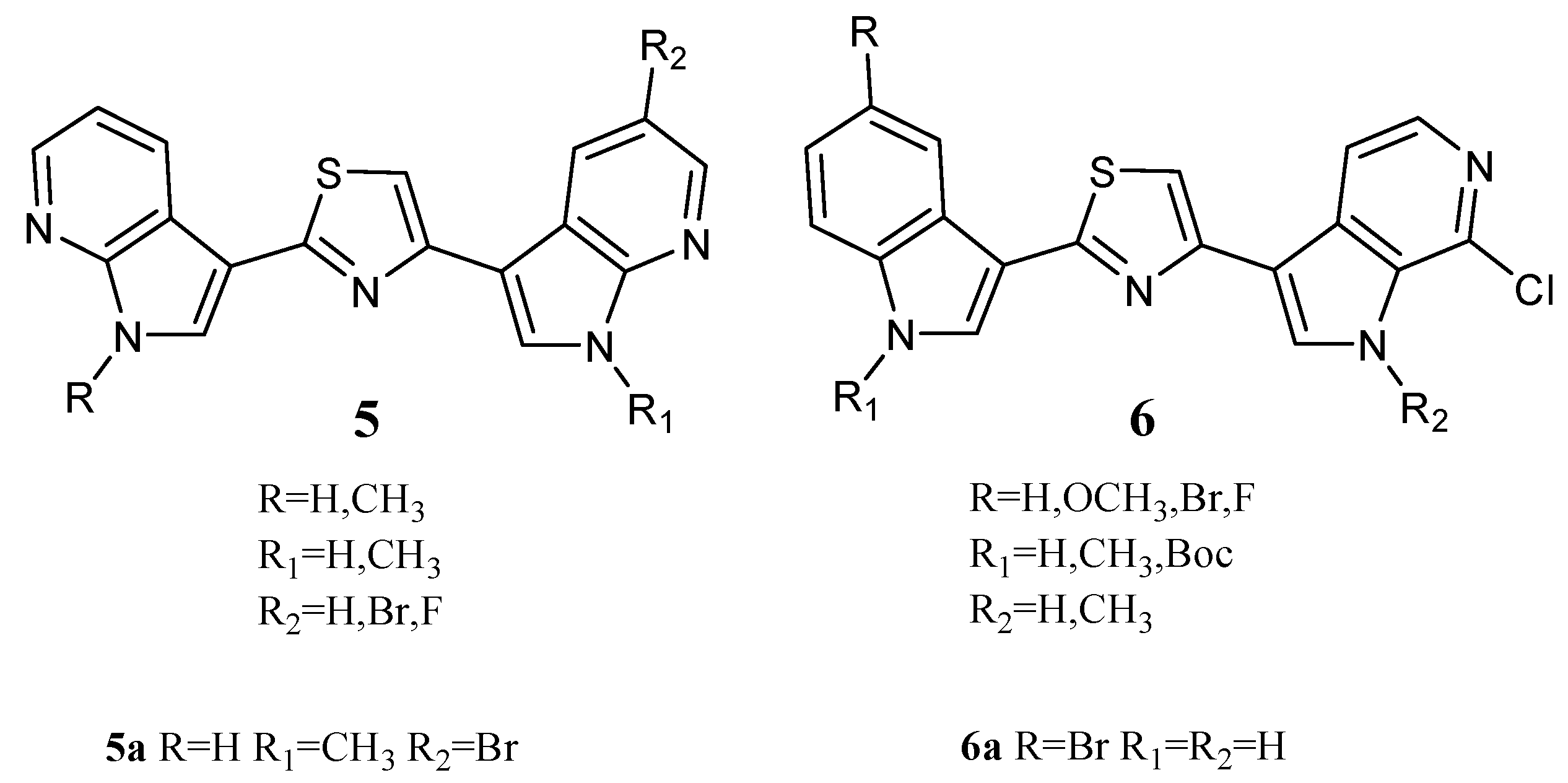
Two series of Nortopsentin thiazolyl analogues
5 and
6 (
Figure 7) were also synthesized. Compared to derivatives
2, in derivatives
5, both indole units were replaced by 7-azaindole moieties, while in derivatives
6, one indole unit was replaced by a 6-azaindole unit. In these series, compounds
5a and
6a showed cytotoxic activity against a broad spectrum of human cancer cell lines included in the NCI panel, having GI
50 values of 0.81–27.7 and 0.93–4.70 µM, respectively (
Table 11). The indolyl-thiazolyl-pyrrolo[2,3-
c]pyridine derivative
6a resulted in more active than thiazolyl-bis-pyrrolo[2,3-
b]pyridines derivative
5a in terms of GI
50. Interestingly, the compounds did not significantly compromise the vitality of intestinal normal-like differentiated Caco-2 cells, providing tumour cells as the main target of their cytotoxicity. Investigation of the mechanisms behind the antiproliferative activity in HCT-116 colon cancer cells showed that derivative
5a caused a dose-dependent increase in the apoptotic cell population, engaging the mitochondria-mediated pathway and causing cell cycle arrest at the G2/M phase. On the other hand, derivative
6a, at a concentration lower than its GI
50, exhibited antiproliferative effects with a great accumulation of autophagic vacuoles without apparent signs of apoptosis. The arrest of the cell cycle at G1 phase proved the autophagic fate of the cells. Evidence indicates that autophagic cell death can be induced as an alternative to apoptosis with therapeutic finality in cancer cells that are resistant to apoptosis. It follows that thiazole compound
6a could be considered a lead compound for Nortopsentin derivatives with autophagic activity [
20].
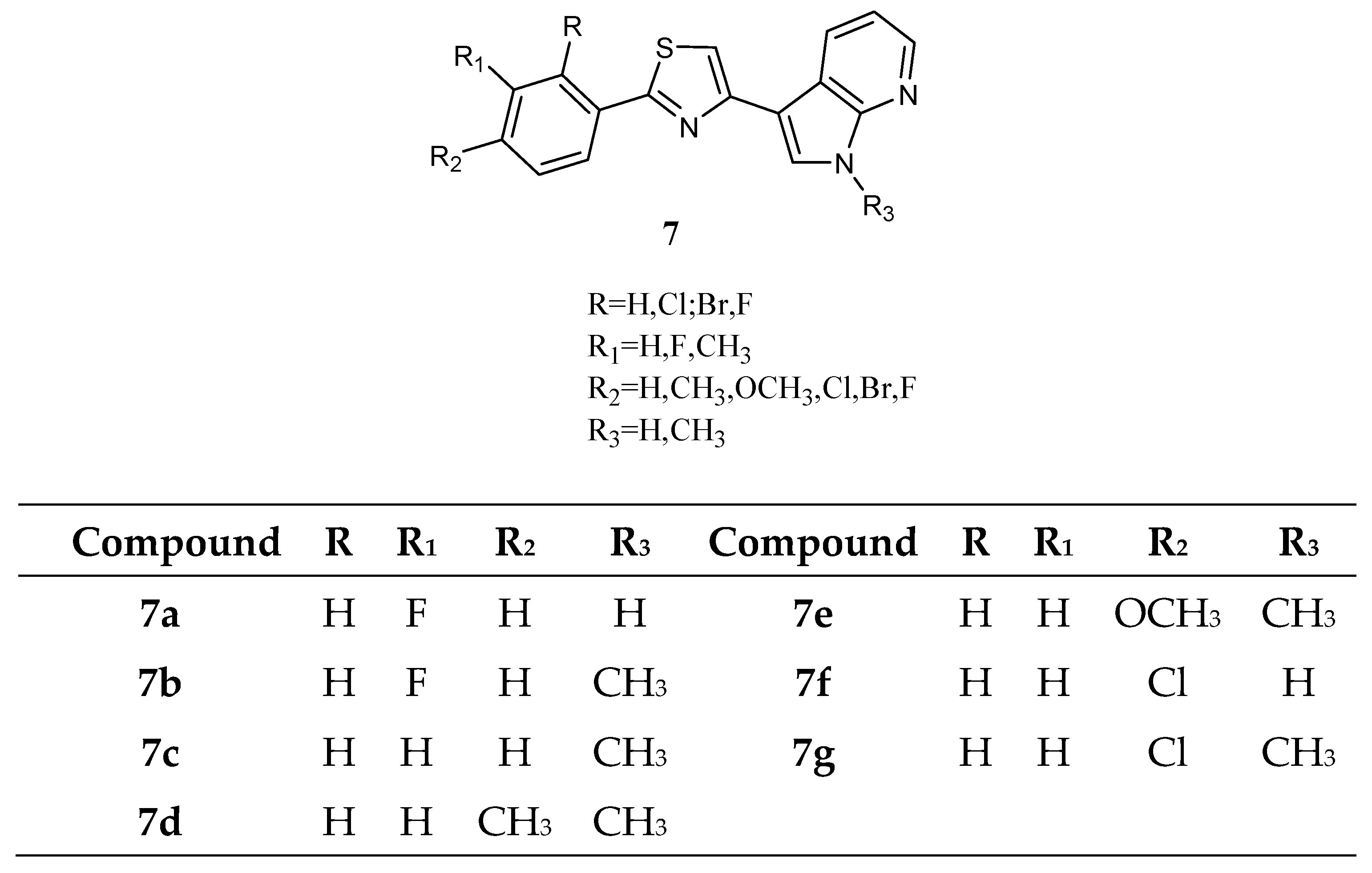
Among thiazole analogues, derivatives
7 (
Figure 8), in which one indole ring was replaced by a phenyl while the other one was replaced by a 7-azaindole, were also reported.
Derivatives
7b,
c,
d, and
g were active against all cancer cell lines tested by NCI, while derivatives
7a,
e, and
f were cytotoxic against a good percentage of the tested cell lines (50%, 70% and 15% respectively), having GI
50 values in the micromolar to sub-micromolar/nanomolar range (
Table 11). The most active compounds were the
N-methyl derivatives, and four of them,
7b,
7c,
7d, and
7g, were further tested against pancreatic carcinoma (MiaPaCa-2) and malignant peritoneal mesothelioma (STO) showing IC
50 values in the range 4.3–41.6 µM and 0.41–17.2 µM, respectively (
Table 12). Kinase activity assays (CDK1/cyclin, CDK5/p25, or GSK3β) were also performed to explain the mechanism of action of this series of compounds. Only the compounds with the highest antiproliferative activity (
7c and
7d) exhibited affinity for CDK1, with IC
50 values of 0.41 and 0.85 µM, respectively. Such values were like those of roscovitine and purvanalol A, used as reference drugs (
Table 13). Moreover, exposure of asynchronously growing STO cells to both compounds affected cell-cycle phase distribution, leading to a concentration-dependent accumulation of cells in the G2/M phase with a concomitant increase in the sub-G1 apoptotic cell population [
62].
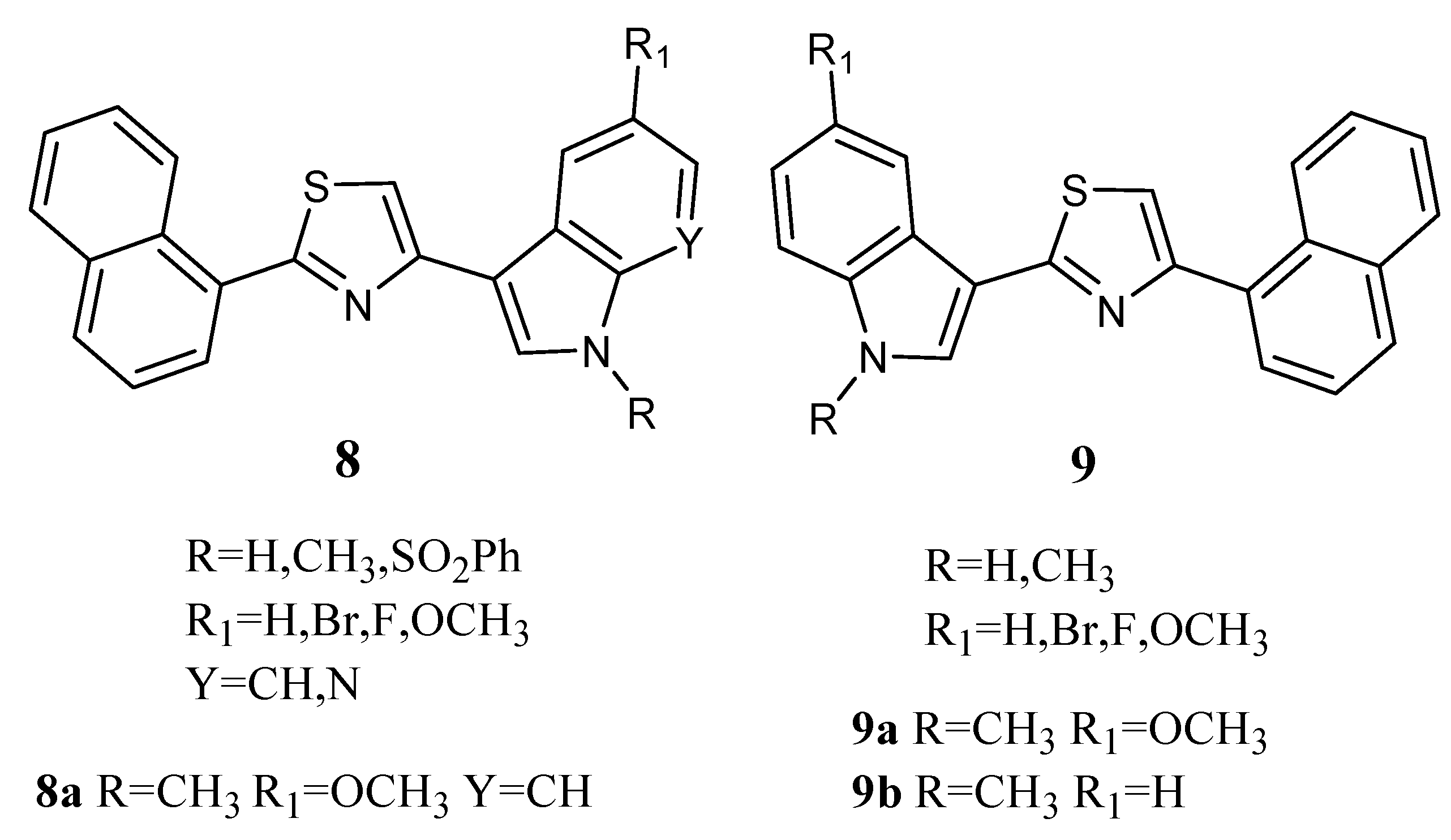
Thiazole derivatives
8 and
9 (
Figure 9) combine an indole unit with a naphthalyl portion. Derivatives
8a,
9a, and
9b in particular displayed good antiproliferative activity against the MCF-7 cell line with GI
50 values in the micromolar range (2.13, 3.26 and 5.14 µM, respectively,
Table 14). Their mechanism of action was found to be pro-apoptotic, inducing early apoptosis in MCF-7 cells after 24 h of treatment without necrotic effects. They also caused a decrease in the percentage of cells in the G0/G1 and S phases, with a concomitant percentage increase in cells in the G2/M phase [
63].
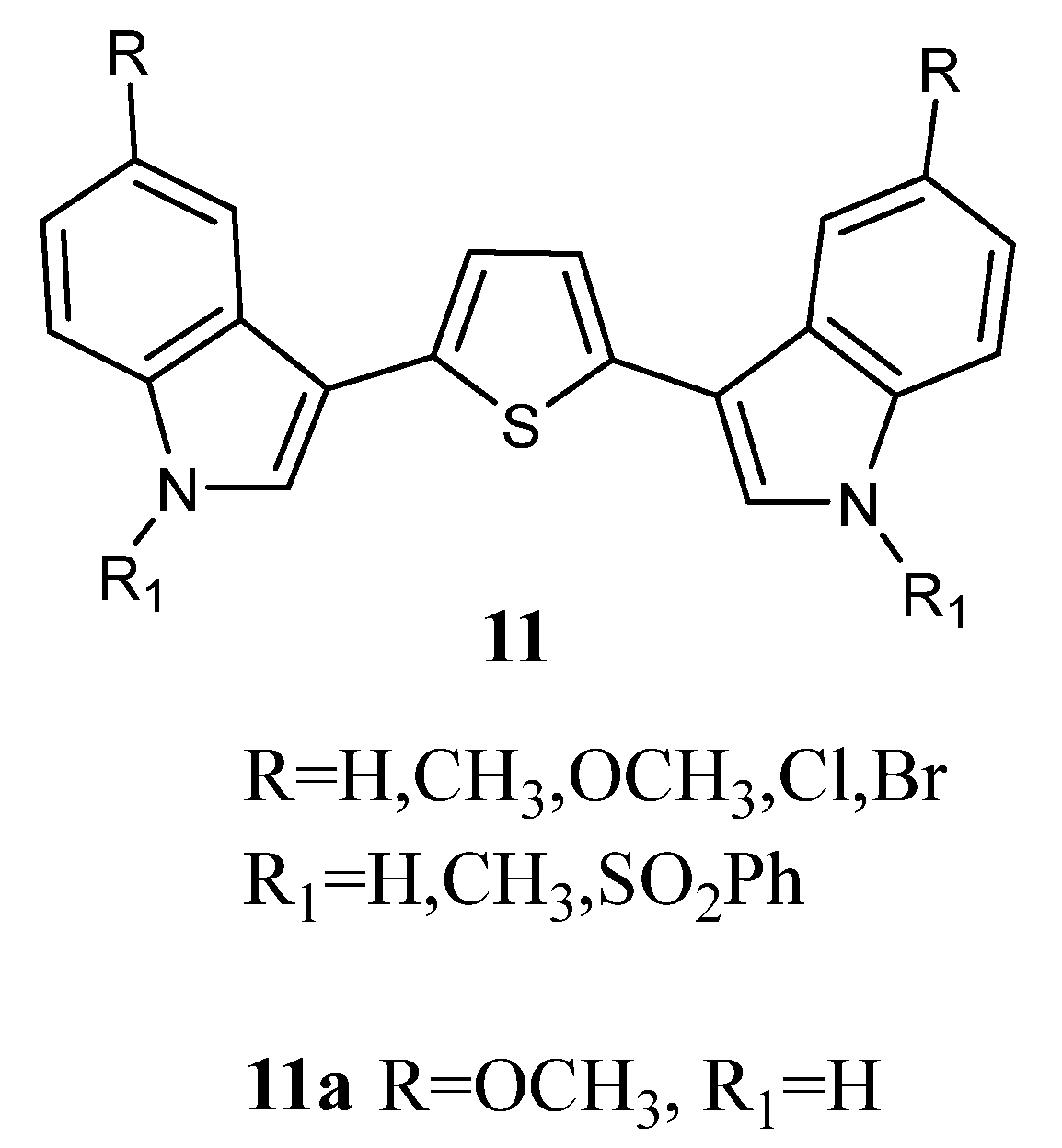
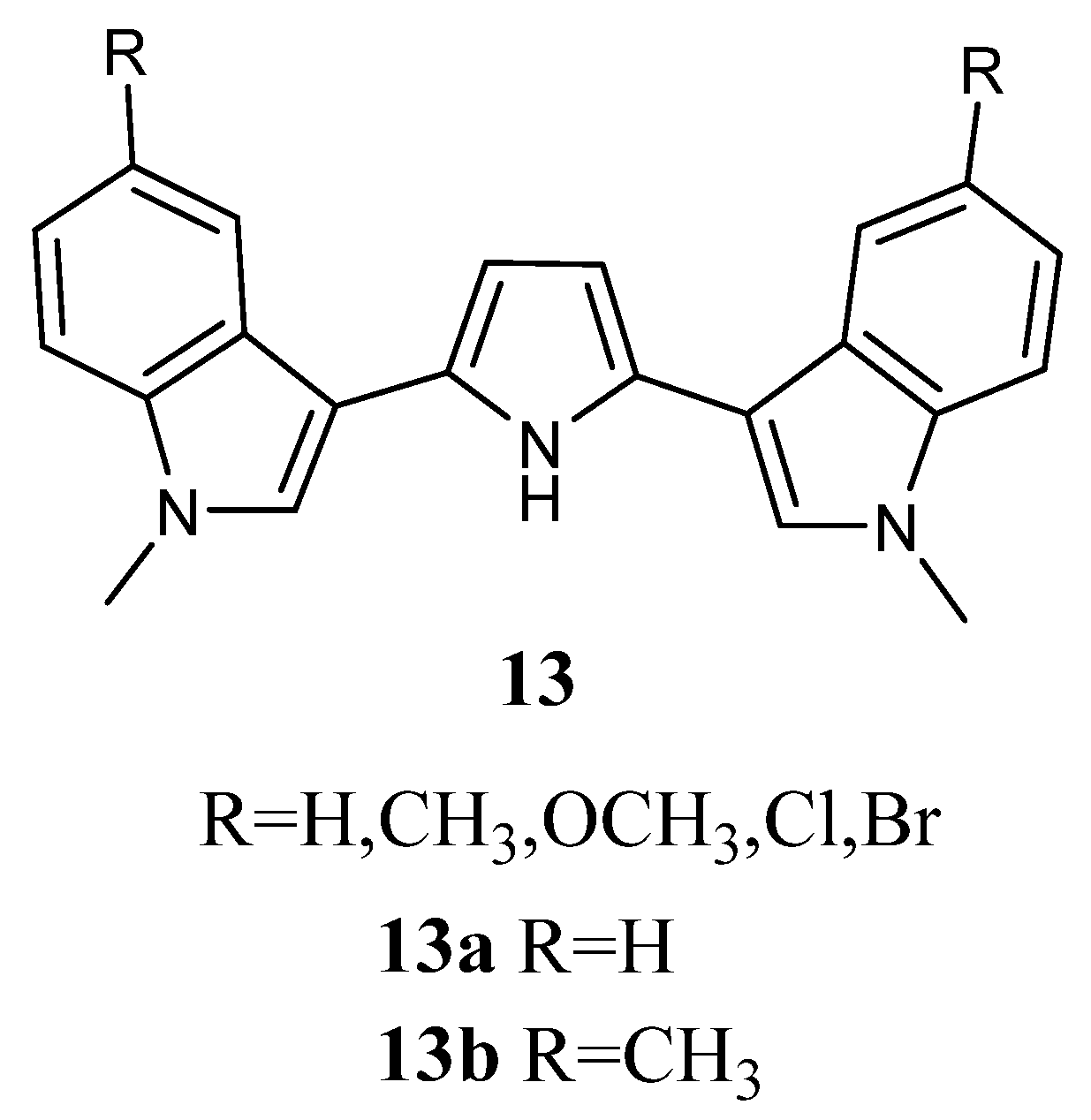
2.1.5. Pyrroles
Bis-indolyl-pyrroles of type
13 (
Figure 13) were investigated
in vitro against human tumour cell lines and by ex-vivo clonogenic assay using human tumour xenografts. Screening in monolayer cultures of 42 human tumour cell lines derived from 15 different solid tumour types (bladder, colon, gastric, head-neck, liver, lung, mammary, melanoma, ovarian, pancreatic, prostate, pleural mesothelioma, renal, sarcoma, and uterus) revealed that the most active derivatives were
13a and
13b. Compounds
13a and
13b affected concentration-dependent inhibition of tumour cell growth with IC
50 values in the range 0.22–6.34 μM and 0.11–2.65 μM, respectively, indicating pronounced cytotoxic potency (
Table 18). Regarding compound
13a, selective activity was observed with submicromolar IC
50 values against some cell lines, such as cell lines of bladder cancer (BXF 1218L, BXF 1352L), gastric cancer cell line (GXA MKN45), head-neck cell line (HNXF CAL27), two melanoma cell lines (MEXF 1341L; MEXF 276L), as well as LXFL 1121L (lung cancer), PAXF PANC-1 (pancreatic cancer), PRXF PC3M (prostate cancer), SXF SAOS-2 (sarcoma), and UXF 1138L (cancer of the uterine body) cell lines. Less sensitive cell lines were found among colon (HCT-116, HT-29), lung (LXFA 289L), ovarian (OVXF 899L), prostate (DU145), and renal cancer (RXF 393NL, RXF 486L). Compound
13b exhibited pronounced activity with submicromolar IC
50 values in 32 cell lines.
The anti-proliferative activity of
13a and
13b was also evaluated in cell suspensions prepared from 44 human tumour xenografts of 13 different tumour types (bladder, colon, gastric, head-neck, lung, mammary, melanoma, ovarian, pancreatic, prostate, pleural mesothelioma, renal, and sarcoma), which were cultured as solid tumours in serial passage on immune-deficient nude mice (
Table 18). The results confirmed the concentration-dependent activity of
13a and
13b on cell lines with IC
50 values in the range of 1.18–54.90 μM and 0.37–40.70 μM, respectively. Selectivity was encountered for
13a against 9 out of the 44 tumours tested, while these sensitive tumours were scattered among various tumour histotypes, like bladder, gastric, head and neck, lung cancer, melanoma, and pleuramesothelioma. Better tumour selectivity was displayed for compound
13b, with 14 out of 44 tumours. Sensitive cancer types were found among the bladder, head and neck, lung, pancreatic, prostate, renal cancer, melanoma, and pleuromesothelioma [
16].
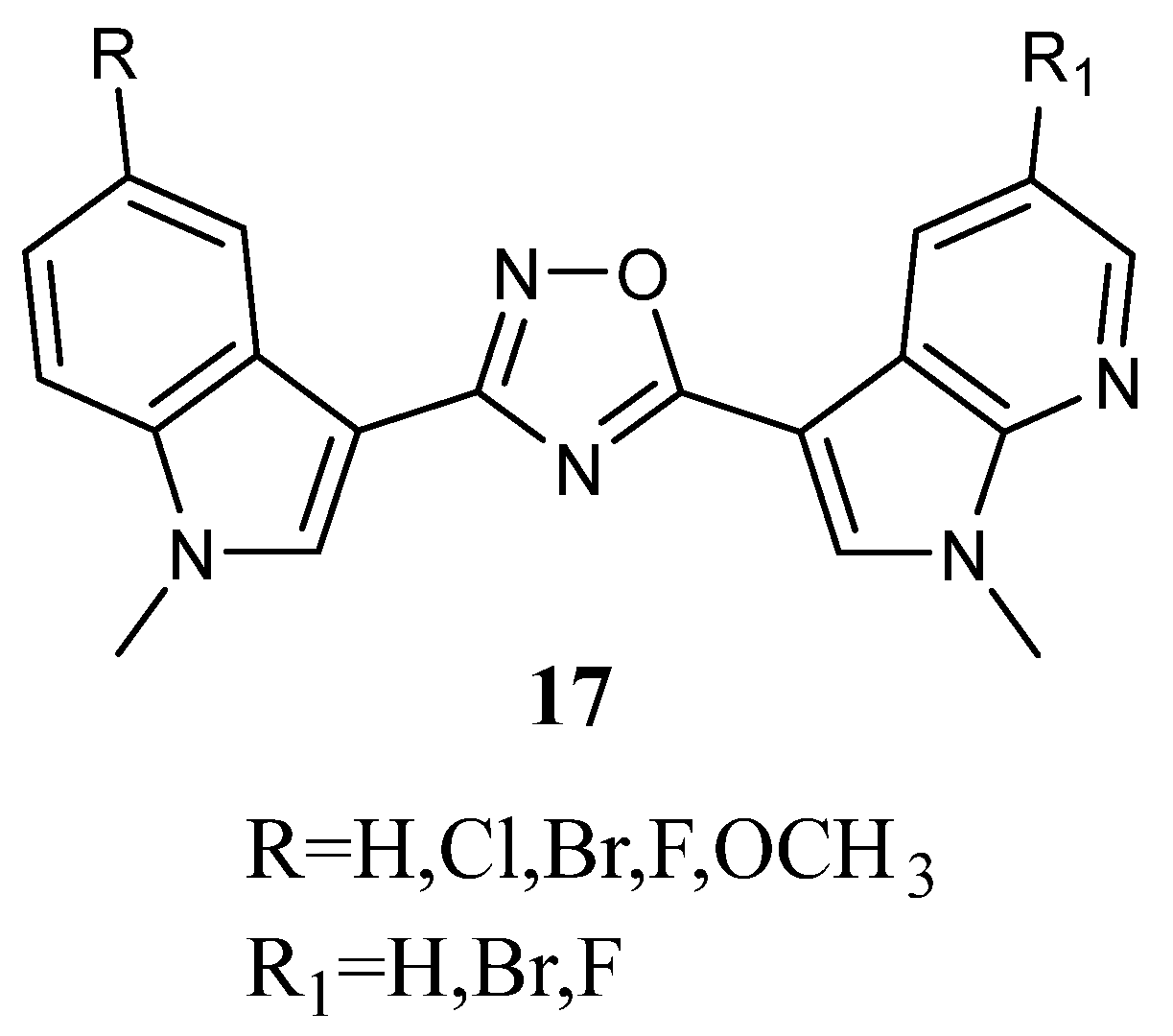
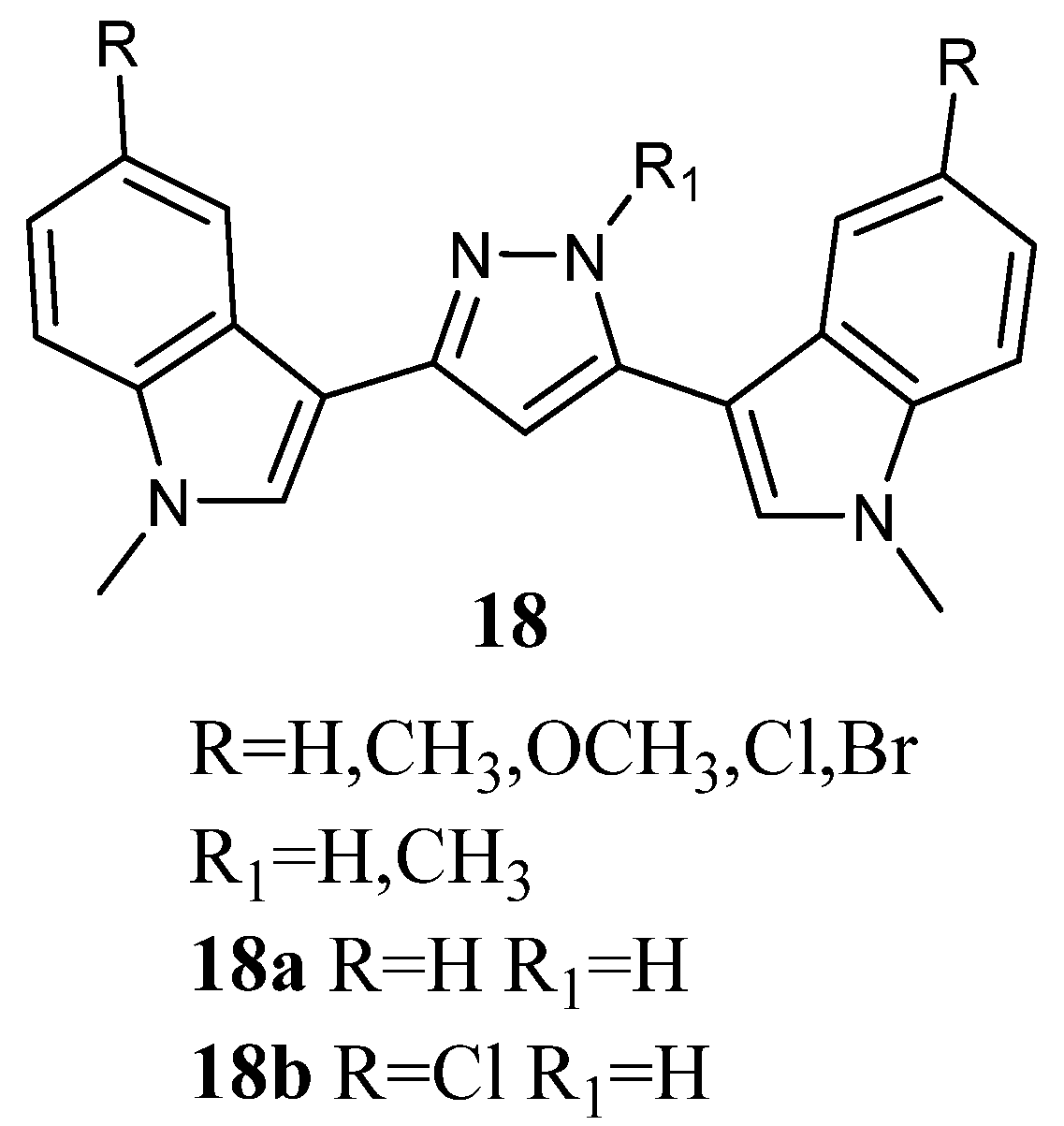
2.1.8. Pyrazoles
Bis-indolyl-pyrazoles
18 (
Figure 18), in which a pyrazole central ring substituted the imidazole ring of Nortopsentin, were screened by the NCI.
Among the investigated compounds, derivatives
18a and
b were the most active, exhibiting antiproliferative activity against most of the human cell lines. The percentage of sensitive cell lines out of the total number of cell lines investigated was 90% and 100%, respectively, while MG_MID was 18.2 and 3.23 µM, respectively. Therefore, compound
18b, bearing a chlorine atom, was more active than the unsubstituted derivative
18a. Derivative
18b was cytotoxic against the totality of cell lines investigated at micromolar concentration, and it proved to be selective for the melanoma subpanel, having all the subpanel cell lines GI
50 values in the range of 1.63–9.64 µM (
Table 23). The most sensitive cell lines were UACC-62, LOX IMVI, and SK-MEL-5 (GI
50 values of 1.63, 1.70, and 1.79 µM, respectively). It also showed selectivity for MOLT-4, SR, and K-562 (GI
50 values of 1.55, 2.36, and 2.78 µM, respectively) of the leukaemia subpanel, HCC-2998 and COLO 205 (GI
50 values of 1.71 and 2.22 µM, respectively) of colon cancer, CAKI-1 (GI
50 value of 1.70 µM) of renal cancer, BT-549 (GI
50 value of 2.03 µM) of breast cancer, and SF-539 (GI
50 value of 1.81 µM) of CNS subpanel. Derivative
18a was particularly effective against the colon subpanel, having GI
50 in the range of 4.58–19.0 µM. The most sensitive colon cell lines were KM12 and HCC-2998 (GI
50 values of 4.58 and 4.74 µM, respectively). Compound
18a showed good selectivity for HOP-92 (GI
50 2.06 µM) and NCIH460 (GI
50 value of 4.48 µM) of the non-small-cell lung cancer subpanel and MCF-7 (GI
50 value of 3.95 µM) of the breast cancer subpanel (
Table 23) [
60]. Experiments aimed at evaluating the ability of derivatives
18a and
b to interact with DNA, revealed that they were unable to form a molecular complex with the macromolecule. In particular, linear flow dichroism spectra obtained by salmon testes DNA, where different concentrations of
18a and
b were used, resulted in nearly overlapping to those recorded without compounds. Furthermore, the ability to interfere with the activity of the nuclear enzyme topoisomerase II, which catalyses the interconversion of different topological forms of DNA, was assayed. As to
18a, the results highlight that the inhibitory capability became detectable at about 50 µM, while in the case of
18b, it happened at 100 µM. Furthermore, both tested compounds presented a scored inhibition, which was considerably weaker than that of
m-amsacrine used at 8 µM concentration. These results proved that DNA cannot be considered the main target of cell death, suggesting that other cellular molecular targets are engaged in the antiproliferative activity of
18a and
b [
63].

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
