
Discovery of a Novel Ubenimex Derivative as a First-in-Class Dual CD13/Proteasome Inhibitor for the Treatment of Cancer

 ,
,  and
and
Abstract
:
1. Introduction
2. Results
2.1. Chemistry
2.2. Biological Evaluation of Compound BC-05
2.2.1. BC-05 Is a CD13 and 20S Proteasome Dual Inhibitor
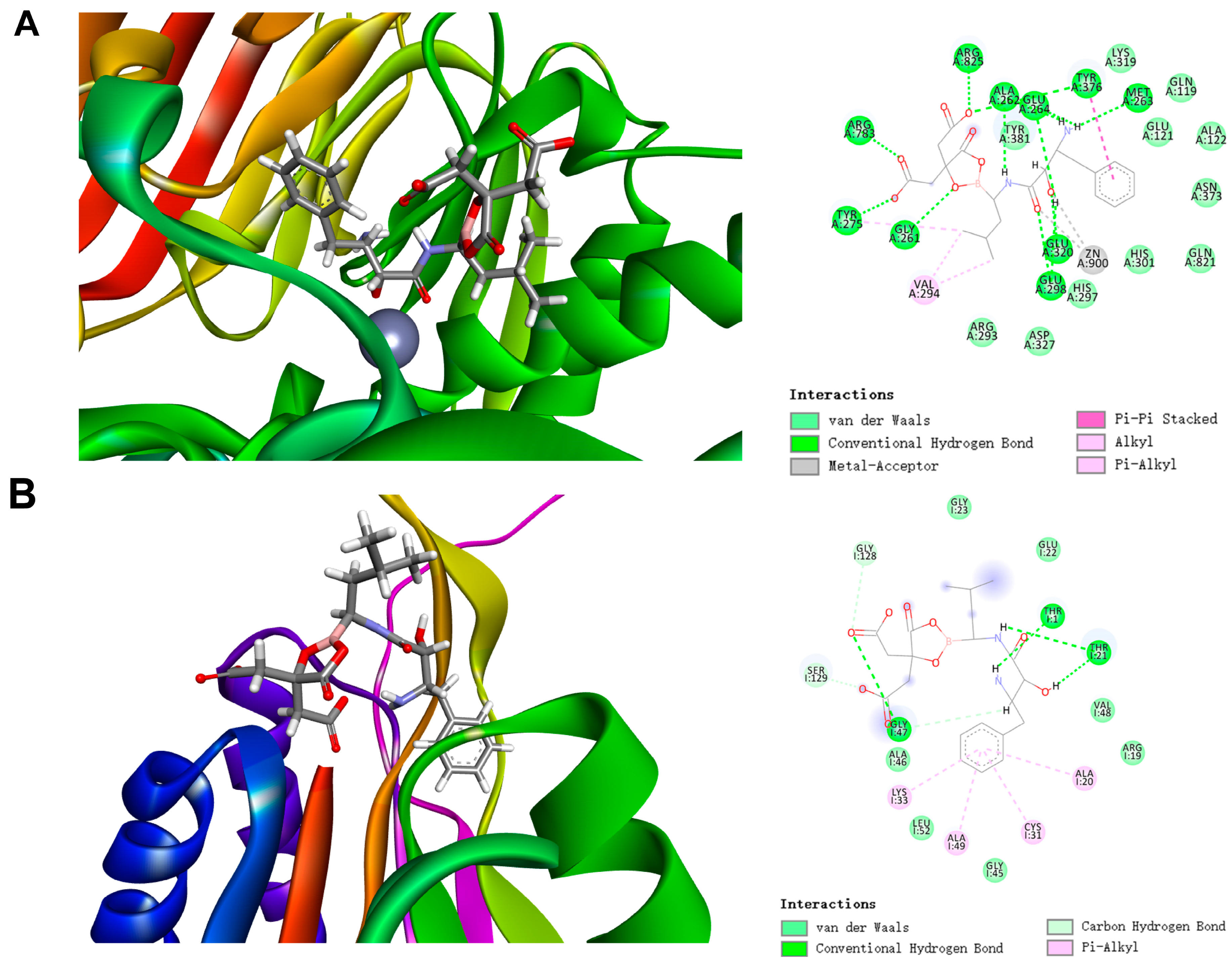
2.2.2. Docking Study of BC-05 with CD13 and 20S Proteasome
2.2.3. BC-05 Displayed Potent Anti-Proliferative Activity toward Cancer Cell Lines
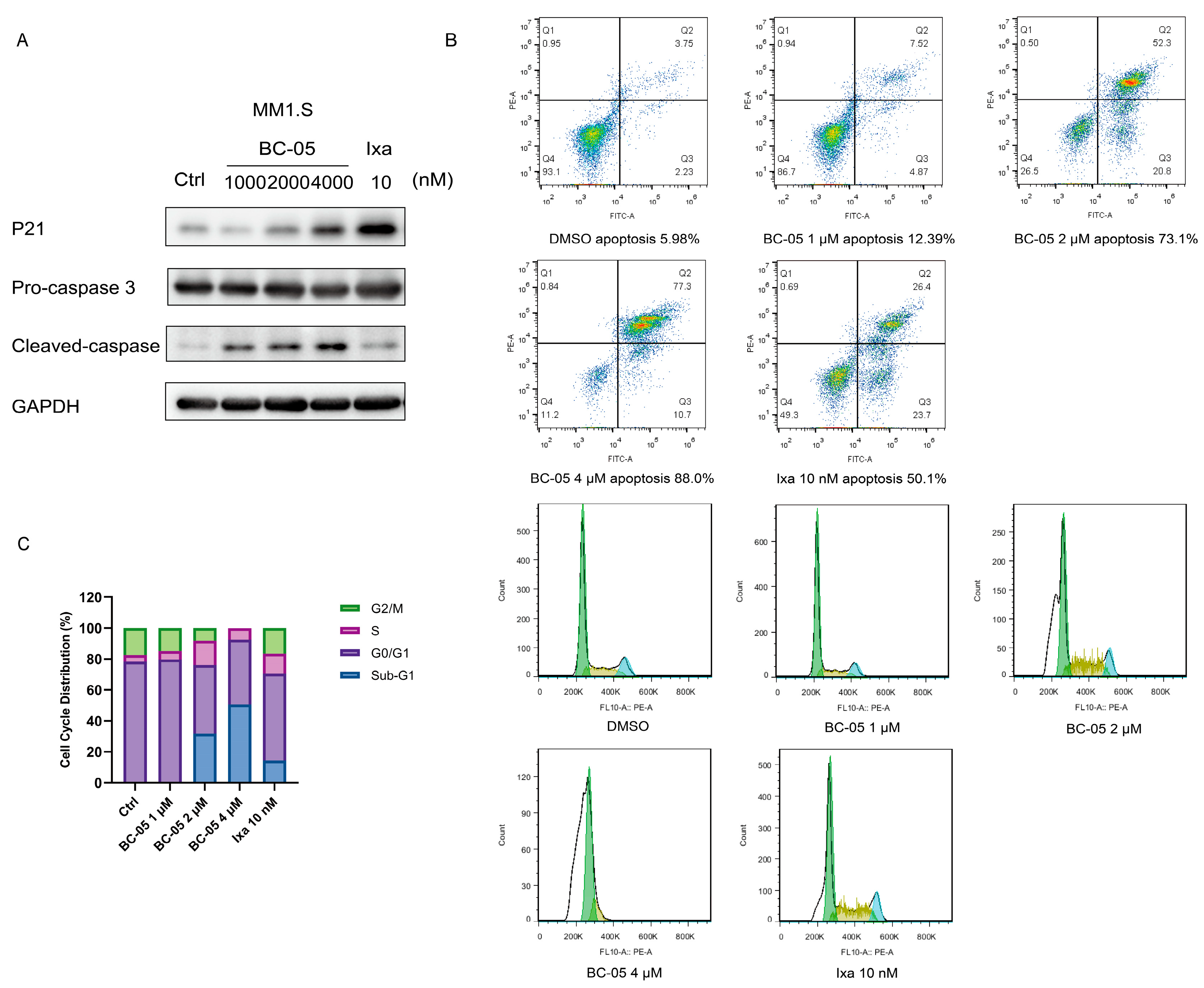
2.2.4. BC-05 Induced Cancer Cell Apoptosis and Cell Cycle Arrest
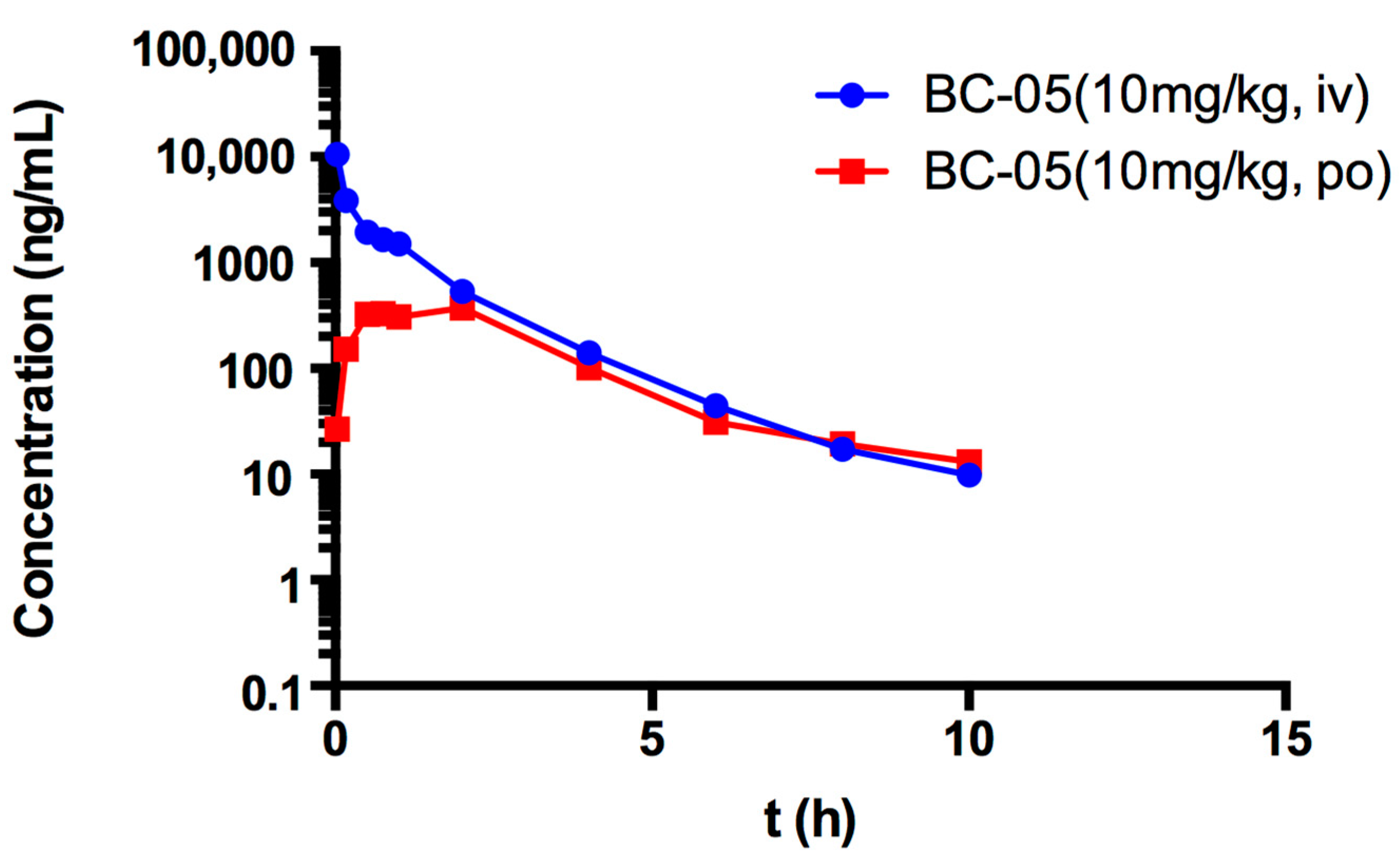
2.2.5. In Vivo Pharmacokinetic Study of BC-05
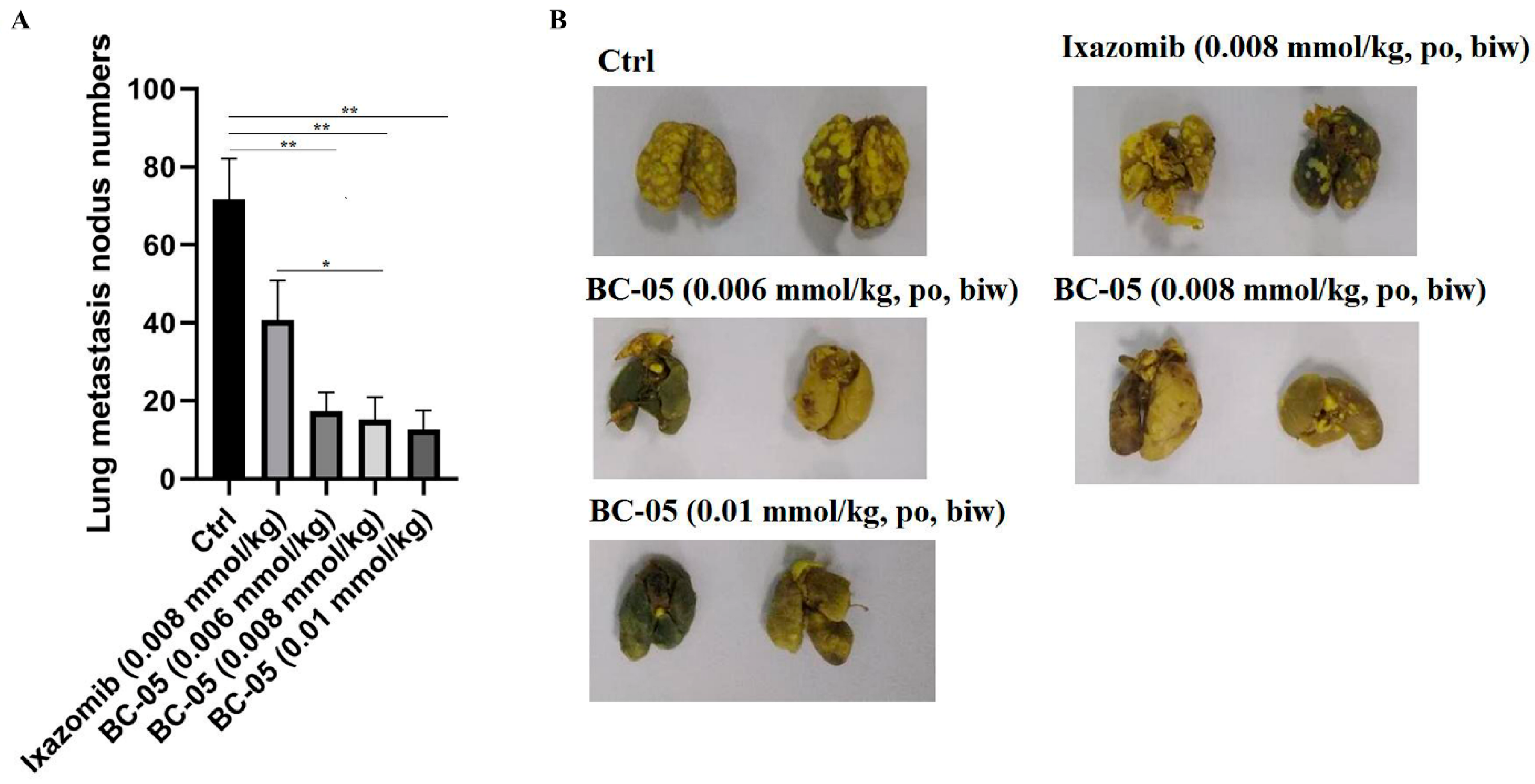
2.2.6. BC-05 Exhibits Remarkably Anti-Metastasis Efficacy In Vivo
2.2.7. BC-05 Exhibits Immunostimulating Potency In Vivo
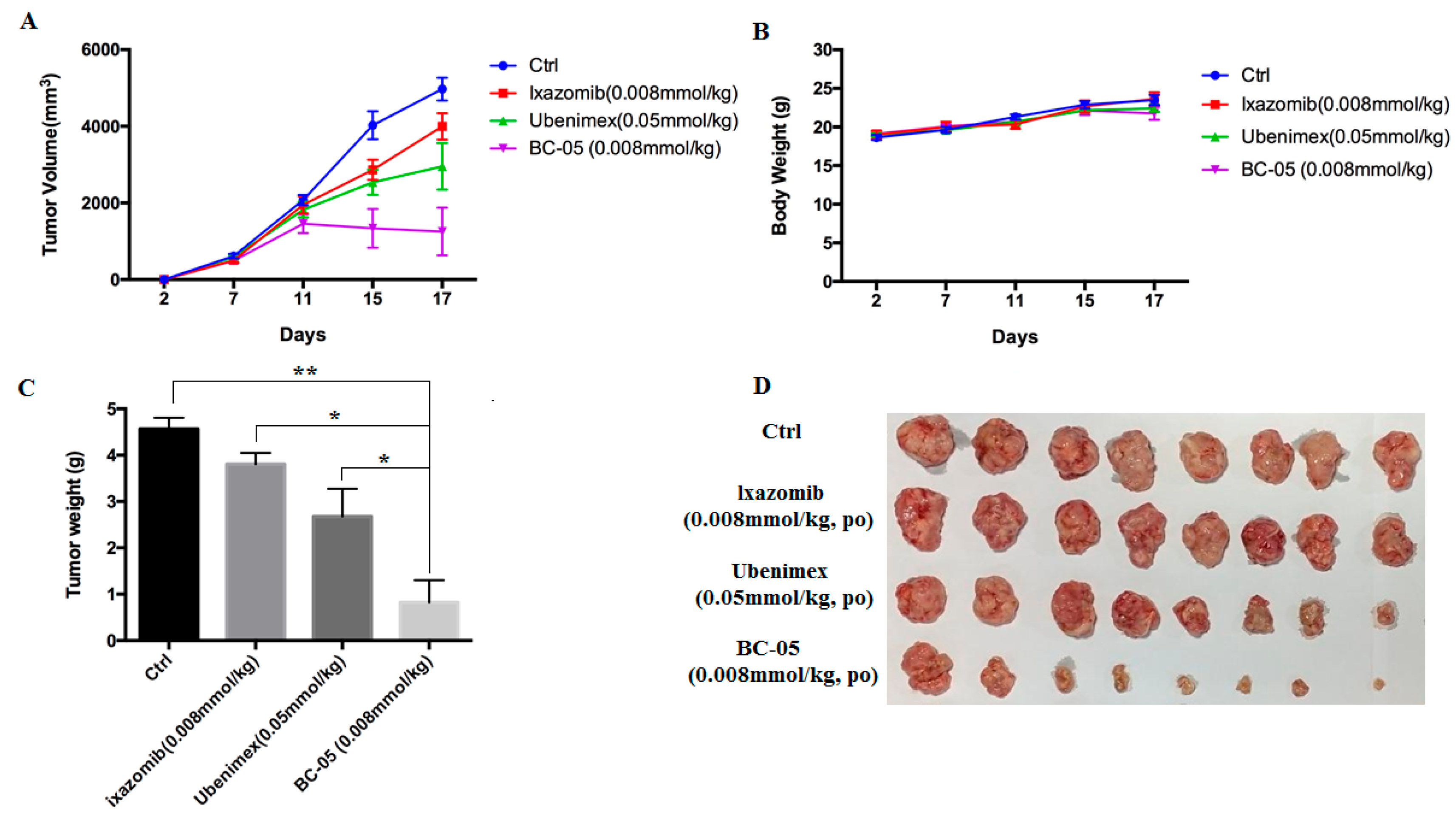
2.2.8. BC-05 Exhibits Superior Anti-Tumor Activity In Vivo
2.2.9. Acute Toxicity Assessment of BC-05 in Healthy Mice
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. CD13 Inhibition Assay [20]
4.3. 20S Proteasome Inhibition Assay
4.4. Molecular Docking
4.5. Cell Proliferation Analysis
4.6. Cell Cycle and Apoptosis Study
4.7. Western Blot Analysis
4.8. In Vivo Pharmacokinetics
4.9. In Vivo H22 Pulmonary Metastasis Model
4.10. In Vivo P3x63Ag8.653 Tumor Transplant Models
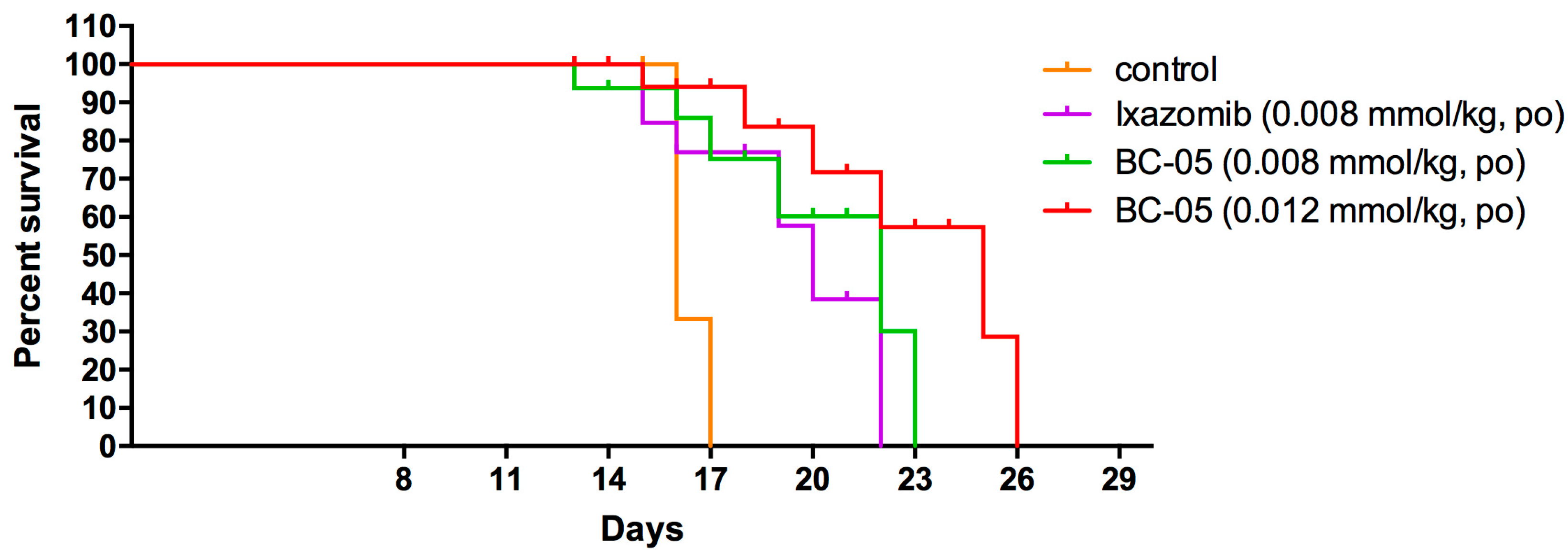
4.11. Survival Time in Mice Bearing B16F10 Cells
4.12. Macrophage Phagocytosis by Carbon Clearance Test
4.13. Acute Toxicity Experiment
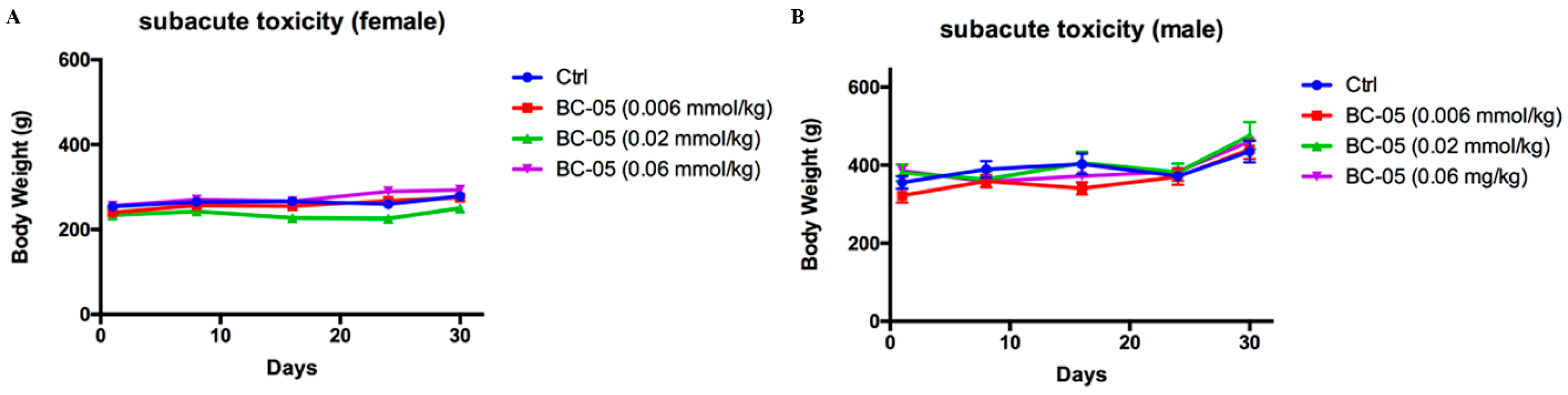
4.14. Subacute Toxicity Experiment
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bauvois, B.; Dauzonne, D. Aminopeptidase-n/cd13 (ec 3.4.11.2) inhibitors: Chemistry, biological evaluations, and therapeutic prospects. Med. Res. Rev. 2006, 26, 88–130. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase n. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Design of aminopeptidase n inhibitors as anti-cancer agents. J. Med. Chem. 2018, 61, 6468–6490. [Google Scholar] [CrossRef] [PubMed]
- Mina-Osorio, P. The moonlighting enzyme cd13: Old and new functions to target. Trends Mol. Med. 2008, 14, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Amin, M.A.; Fox, D.A. Cd13/aminopeptidase n is a potential therapeutic target for inflammatory disorders. J. Immunol. 2020, 204, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Zhai, M.; Yang, Z.; Zhang, C.; Li, J.; Jia, J.; Zhou, L.; Lu, R.; Yao, Z.; Fu, Z. Apn-mediated phosphorylation of bckdk promotes hepatocellular carcinoma metastasis and proliferation via the erk signaling pathway. Cell Death Dis. 2020, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, M.; Lo, R.; Ivic, I.; Aguilera, B.; Qendro, V.; Devarakonda, C.; Shapiro, L.H. Cd13 tethers the iqgap1-arf6-efa6 complex to the plasma membrane to promote arf6 activation, β1 integrin recycling, and cell migration. Sci. Signal. 2019, 12, eaav5938. [Google Scholar] [CrossRef] [PubMed]
- Dondossola, E.; Rangel, R.; Guzman-Rojas, L.; Barbu, E.M.; Hosoya, H.; St. John, L.S.; Molldrem, J.J.; Corti, A.; Sidman, R.L.; Arap, W.; et al. Cd13-positive bone marrow-derived myeloid cells promote angiogenesis, tumor growth, and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 20717–20722. [Google Scholar] [CrossRef]
- Tsou, P.S.; Lu, C.; Gurrea-Rubio, M.; Muraoka, S.; Campbell, P.L.; Wu, Q.; Model, E.N.; Lind, M.E.; Vichaikul, S.; Mattichak, M.N.; et al. Soluble cd13 induces inflammatory arthritis by activating the bradykinin receptor b1. J. Clin. Investig. 2022, 132, e151827. [Google Scholar] [CrossRef]
- Domínguez, J.M.; Pérez-Chacón, G.; Guillén, M.J.; Muñoz-Alonso, M.J.; Somovilla-Crespo, B.; Cibrián, D.; Acosta-Iborra, B.; Adrados, M.; Muñoz-Calleja, C.; Cuevas, C.; et al. Cd13 as a new tumor target for antibody-drug conjugates: Validation with the conjugate mi130110. J. Hematol. Oncol. 2020, 13, 32. [Google Scholar] [CrossRef]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. Cd13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef]
- Guzman-Rojas, L.; Rangel, R.; Salameh, A.; Edwards, J.K.; Dondossola, E.; Kim, Y.G.; Saghatelian, A.; Giordano, R.J.; Kolonin, M.G.; Staquicini, F.I.; et al. Cooperative effects of aminopeptidase n (cd13) expressed by nonmalignant and cancer cells within the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2012, 109, 1637–1642. [Google Scholar] [CrossRef]
- Guan, L.; Zhang, Z.; Gao, T.; Fu, S.; Mu, W.; Liang, S.; Liu, Y.; Chu, Q.; Fang, Y.; Liu, Y.; et al. Depleting tumor infiltrating b cells to boost antitumor immunity with tumor immune-microenvironment reshaped hybrid nanocage. ACS Nano 2022, 16, 4263–4277. [Google Scholar] [CrossRef]
- Shim, H.; Ha, J.H.; Lee, H.; Sohn, J.Y.; Kim, H.J.; Eom, H.S.; Kong, S.Y. Expression of myeloid antigen in neoplastic plasma cells is related to adverse prognosis in patients with multiple myeloma. BioMed Res. Int. 2014, 2014, 893243. [Google Scholar] [CrossRef] [PubMed]
- Saida, S.; Watanabe, K.; Kato, I.; Fujino, H.; Umeda, K.; Okamoto, S.; Uemoto, S.; Hishiki, T.; Yoshida, H.; Tanaka, S.; et al. Prognostic significance of aminopeptidase-n (cd13) in hepatoblastoma. Pediatr. Int. 2015, 57, 558–566. [Google Scholar] [CrossRef]
- Ikeda, N.; Nakajima, Y.; Tokuhara, T.; Hattori, N.; Sho, M.; Kanehiro, H.; Miyake, M. Clinical significance of aminopeptidase n/cd13 expression in human pancreatic carcinoma. Clin. Cancer Res. 2003, 9, 1503–1508. [Google Scholar]
- Ota, K.; Kurita, S.; Yamada, K.; Masaoka, T.; Uzuka, Y.; Ogawa, N. Immunotherapy with bestatin for acute nonlymphocytic leukemia in adults. Cancer Immunol. Immunother. 1986, 23, 5–10. [Google Scholar] [CrossRef]
- Ota, K.; Uzuka, Y. Clinical trials of bestatin for leukemia and solid tumors. Biotherapy 1992, 4, 205–214. [Google Scholar] [CrossRef]
- Aozuka, Y.; Koizumi, K.; Saitoh, Y.; Ueda, Y.; Sakurai, H.; Saiki, I. Anti-tumor angiogenesis effect of aminopeptidase inhibitor bestatin against b16-bl6 melanoma cells orthotopically implanted into syngeneic mice. Cancer Lett. 2004, 216, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Cao, J.; Jia, Y.; Zhang, X.; Fang, H.; Xu, W. Development of synthetic aminopeptidase n/cd13 inhibitors to overcome cancer metastasis and angiogenesis. ACS Med. Chem. Lett. 2012, 3, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Wada, H.; Eguchi, H.; Ogawa, H.; Yamada, D.; Noda, T.; Asaoka, T.; Kawamoto, K.; Gotoh, K.; Umeshita, K.; et al. A cd13 inhibitor, ubenimex, synergistically enhances the effects of anticancer drugs in hepatocellular carcinoma. Int. J. Oncol. 2016, 49, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Ma, Y.; Xing, X.; Ge, B.; Li, Y.; Xu, X.; Song, J.; Xiao, M.; Gao, F.; Jiang, W.; et al. Suppression of cd13 enhances the cytotoxic effect of chemotherapeutic drugs in hepatocellular carcinoma cells. Front. Pharmacol. 2021, 12, 660377. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Sui, Z.G.; Xu, W.; Quan, X.H.; Sun, J.L.; Li, X.; Ji, H.Y.; Jing, F.B. Ubenimex suppresses pim-3 kinase expression by targeting cd13 to reverse mdr in hcc cells. Oncotarget 2017, 8, 72652–72665. [Google Scholar] [CrossRef] [PubMed]
- Sakuraya, M.; Tamura, J.; Itoh, K.; Kubota, K.; Naruse, T. Aminopeptidase inhibitor ubenimex inhibits the growth of leukaemic cell lines and myeloma cells through its cytotoxicity. J. Int. Med. Res. 2000, 28, 214–221. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Hou, J.; Huang, Y.; Wang, X.; Jia, Y.; Wang, Q.; Xu, W.; Zhang, J.; Zhang, Y. Synthesis and biological characterization of ubenimex-fluorouracil conjugates for anti-cancer therapy. Eur. J. Med. Chem. 2018, 143, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, V.; Mussetti, A.; Salas, M.Q.; Martinelli, G.; Cerchione, C. Old and new generation proteasome inhibitors in multiple myeloma. Panminerva Med. 2020, 62, 193–206. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor mln9708 against multiple myeloma cells. Clin. Cancer Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef]
- Gupta, N.; Hanley, M.J.; Venkatakrishnan, K.; Perez, R.; Norris, R.E.; Nemunaitis, J.; Yang, H.; Qian, M.G.; Falchook, G.; Labotka, R.; et al. Pharmacokinetics of ixazomib, an oral proteasome inhibitor, in solid tumour patients with moderate or severe hepatic impairment. Br. J. Clin. Pharmacol. 2016, 82, 728–738. [Google Scholar] [CrossRef]
- Liu, T.; Wan, Y.; Xiao, Y.; Xia, C.; Duan, G. Dual-target inhibitors based on hdacs: Novel antitumor agents for cancer therapy. J. Med. Chem. 2020, 63, 8977–9002. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, X.; Hou, J.; Huang, Y.; Jia, Y.; Zou, M.; Zhang, J.; Wang, X.; Xu, W.; Zhang, Y. Discovery of bc-01, a novel mutual prodrug (hybrid drug) of ubenimex and fluorouracil as anticancer agent. Eur. J. Med. Chem. 2016, 121, 649–657. [Google Scholar] [CrossRef]
- Jiang, Y.; Hou, J.; Li, X.; Huang, Y.; Wang, X.; Wu, J.; Zhang, J.; Xu, W.; Zhang, Y. Discovery of a novel chimeric ubenimex-gemcitabine with potent oral antitumor activity. Bioorg. Med. Chem. 2016, 24, 5787–5795. [Google Scholar] [CrossRef]
- Yue, K.; Hou, X.; Jia, G.; Zhang, L.; Zhang, J.; Tan, L.; Wang, X.; Zhang, Z.; Li, P.; Xu, W.; et al. Design, synthesis and biological evaluation of hybrid of ubenimex-fluorouracil for hepatocellular carcinoma therapy. Bioorg. Chem. 2021, 116, 105343. [Google Scholar] [CrossRef]
- Tsukagoshi, S. A new antitumor drug with immunomodulating activity, ubenimex (bestatin). Gan Kagaku Ryoho 1987, 14, 2385–2391. [Google Scholar]
- Tsukamoto, H.; Shibata, K.; Kajiyama, H.; Terauchi, M.; Nawa, A.; Kikkawa, F. Aminopeptidase n (apn)/cd13 inhibitor, ubenimex, enhances radiation sensitivity in human cervical cancer. BMC Cancer 2008, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, M.; Masuda, T.; Mizutani, S.; Takeuchi, T.; Umezawa, H. Antitumor cells found in tumor-bearing mice given ubenimex. J. Antibiot. 1987, 40, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Cooper, B.M.; Iegre, J.; Donovan, D.H.O.; Halvarsson, M.; Spring, D.R. Peptides as a platform for targeted therapeutics for cancer: Peptide-drug conjugates (pdcs). Chem. Soc. Rev. 2021, 50, 1480–1494. [Google Scholar] [CrossRef]
- Srinivasarao, M.; Low, P.S. Ligand-targeted drug delivery. Chem. Rev. 2017, 117, 12133–12164. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd. | IC50(μM) a toward CD13 from Porcine Kidney | IC50(μM) a toward Human CD13 | IC50(μM) b toward Chymotrypsin-like (CT-L) |
|---|---|---|---|
| BC-05 | 0.65 ± 0.05 | 0.13 ± 0.01 | 1.39 |
| Ixazomib | >20 | N.D. | 0.0024 |
| Ubenimex | 4.91 ± 0.13 | 2.03 ± 0.18 | >10 |
| Cell Lines | BC-05 | Ixazomib | Ubenimex |
|---|---|---|---|
| U266 | 13.66 ± 1.43 | 0.12 ± 0.01 | >200 |
| RPMI-8266 | 15.12 ± 2.23 | 0.098 ± 0.01 | >200 |
| ARP-1 | 10.23 ± 1.23 | 0.099 ± 0.002 | >200 |
| MM.1S | 1.53 ± 0.24 | 0.007 ± 0.002 | >200 |
| HEL | 13.26 ± 2.81 | 0.11 ± 0.003 | >200 |
| KG1 | 41.55 ± 4.11 | 0.40 ± 0.05 | >200 |
| P3x63Ag8.653 | 17.73 ± 0.75 | 0.08 ± 0.005 | >200 |
| SP2 | 14.67 ± 4.91 | 0.074 ± 0.017 | >200 |
| A2780 | 33.71 ± 0.99 | 0.11 ± 0.01 | >200 |
| HCT116 | 86.66 ± 7.34 | 0.02 ± 0.01 | >200 |
| Hela | 83.11 ± 8.79 | 0.74 ± 0.19 | >200 |
| MDA-MB-231 | 13.30 ± 0.49 | 0.12 ± 0.01 | >200 |
| HT-29 | 12.51 ± 1.21 | 0.06 ± 0.001 | >200 |
| U251 | 47.54 ± 11.89 | 0.92 ± 0.86 | >200 |
| PC3 | 47.41 ± 4.22 | 0.75 ± 0.12 | >200 |
| HEK293 | >200 | 17.10 ± 2.27 | >200 |
| HL-7702 | >200 | 4.04 ± 0.63 | >200 |
| BC-05 | BC-05 | |
|---|---|---|
| Administered dose (mg/kg) | i.v. at 10 mg/kg | oral at 10 mg/kg |
| AUC0–∞ (μg/L∗h) | 5154.34 ± 482.30 | 1307.71 ± 116.74 |
| t1/2 (h) | 1.72 ± 0.67 | 2.35 ± 1.23 |
| CL (L/h/kg) | 1.95 ± 0.18 | 7.69 ± 0.66 |
| Tmax (h) | 0.03 ± 0 | 2.0 ± 0 |
| Cmax (ng/mL) | 10,488.6 ± 1414.99 | 373.2 ± 49.65 |
| F (%) | -- | 24.9 ± 4.32 |
| Groups | Dosage (mmol/kg) | Carbon Clearance Index (K) a | Phagocytosis Index (α) a |
|---|---|---|---|
| Ctrl | 0 | 0.057 ± 0.0061 | 5.36 ± 0.19 |
| Ixazomib | 0.008 | 0.045 ± 0.0092 | 5.00 ± 0.34 |
| BC-05 | 0.006 | 0.066 ± 0.0077 | 6.60 ± 0.26 |
| BC-05 | 0.008 | 0.076 ± 0.0016 | 6.61 ± 0.05 |
| Groups | Survival Animals |
|---|---|
| Vehicle | 10/10 |
| BC-05 (0.27 mmol/kg) | 10/10 |
| BC-05 (0.36 mmol/kg) | 6/10 |
| BC-05 (0.49 mmol/kg) | 4/10 |
| BC-05 (0.65 mmol/kg) | 1/10 |
| BC-05 (0.86 mmol/kg) | 0/10 |
| Ixazomib (0.04 mmol/kg) | 0/10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Sun, S.; Liu, J.; Zhang, L.; Guo, D.; Zhang, N.; Zhao, J.; Kong, D.; Xu, T.; Wang, X.; et al. Discovery of a Novel Ubenimex Derivative as a First-in-Class Dual CD13/Proteasome Inhibitor for the Treatment of Cancer. Molecules 2023, 28, 6343. https://doi.org/10.3390/molecules28176343
Zhang J, Sun S, Liu J, Zhang L, Guo D, Zhang N, Zhao J, Kong D, Xu T, Wang X, et al. Discovery of a Novel Ubenimex Derivative as a First-in-Class Dual CD13/Proteasome Inhibitor for the Treatment of Cancer. Molecules. 2023; 28(17):6343. https://doi.org/10.3390/molecules28176343
Chicago/Turabian StyleZhang, Jian, Simin Sun, Jinyu Liu, Liang Zhang, Di Guo, Naixin Zhang, Jun Zhao, Dexin Kong, Tongqiang Xu, Xuejian Wang, and et al. 2023. "Discovery of a Novel Ubenimex Derivative as a First-in-Class Dual CD13/Proteasome Inhibitor for the Treatment of Cancer" Molecules 28, no. 17: 6343. https://doi.org/10.3390/molecules28176343
APA StyleZhang, J., Sun, S., Liu, J., Zhang, L., Guo, D., Zhang, N., Zhao, J., Kong, D., Xu, T., Wang, X., Xu, W., Li, X., & Jiang, Y. (2023). Discovery of a Novel Ubenimex Derivative as a First-in-Class Dual CD13/Proteasome Inhibitor for the Treatment of Cancer. Molecules, 28(17), 6343. https://doi.org/10.3390/molecules28176343