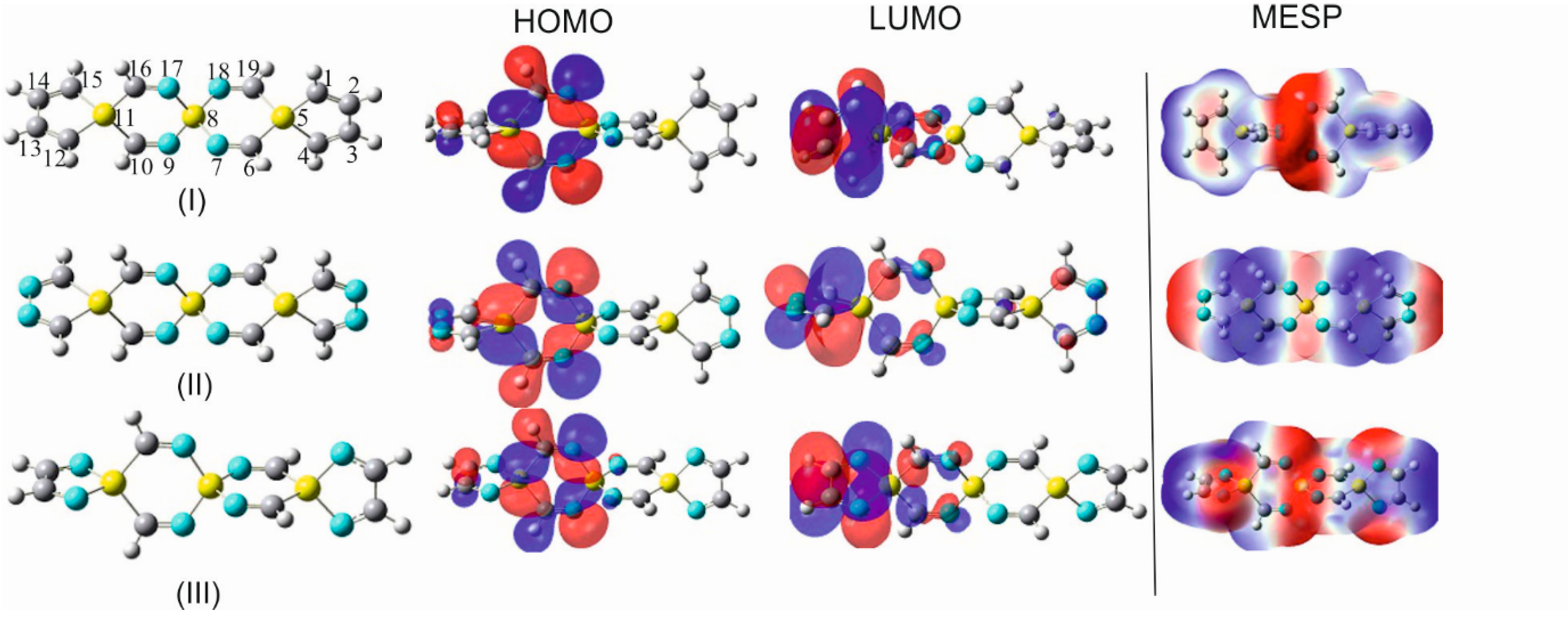
6.1. NPA versus MPA Charge Population Analyses
An additional tool that is, in many respects, helpful in the analysis and rationalization of structural results and substituent effects is the charge distribution analysis.
In the literature, there is a variety of approaches aiming to derive more reasonable definitions and reliable values for atomic charges [
60,
61].
Among these methods are MPA (Mulliken population analysis), NPA (natural population analysis), AIM (atoms in molecules), atomic polar tensor (APT) [
62], and the Merz–Kollman (MK) scheme [
63], in which semiempirical methods were applied to obtain atomic point charges from the electrostatic potential (ESP). In this work, the focus will be exclusively on the two most popular approaches: the natural population analysis (NPA) [
64] and the Mulliken population analysis [
65].
Table 7 shows the NPA charges in some selected SCIs and their tetracyano derivatives. Particularly interesting is the behavior of the distribution of charges between atoms participating in bonds involving silicon or germanium spiro centers (Si
6,12 in (
V) and (
VI) and Si
6/Ge
6 in all remaining SCIs. As is apparent from this table, the largest positive charges are located on Si
6,12 (
qSi6,12 = 2.26 e
−), where the silicon atoms are surrounded by four nitrogen atoms. This is obviously because the electronegativity of nitrogen is considerably higher (χ
N = 3.41) [
27] than that silicon (χ
Si = 1.91 [
27]. In (
III) and (
IV) where the spiro center Si
6 and similarly Si
9 in (
V) and (
VI) are surrounded by four carbon atoms (C-Si-C centers), the positive charges on those silicon spiro centers drop down to 1.46 e
−, which can be anticipated because the electronegativity of the carbon atom (χ
C = 2.471) [
27] is clearly smaller than nitrogen. For the same reasons and based on the indicated difference between the electronegativities of N and C on one hand, and Si on the other hand, the positive charges on the terminal silicon atoms (Si
3,9 in (
I) and Si
3,15 in (
V) amount to 1.10 e
−, whereas in (
III), where the terminal silicon atoms are adjacent to nitrogen atoms, the positive charge on Si
3,9 is 0.44 e
− larger than in (
I) and (
V). A comparison of the NPA atomic charges in TISS
NU-19 and its germanium counterpart TIGS
NU-19 shows that Ge
3,9 and Ge
6 are slightly less positively charged (by 0.13 e
− and 0.14 e
−, respectively) than the corresponding Si
3,9 and Si
6, which is explicable by the slightly higher electronegativity of the germanium atom (χ
Ge = 2.01) [
27].
It is also evident in
Table 7 that the terminal geminal substitution of the addressed SCIs with a strong electron-accepting group, like the C≡N group, leads to an anticipated depletion of charges on the anchor silicon atoms and, thus, an increase in the positivity of these atoms. Nonetheless, the influence of the C≡N substituent on the charge distribution is predominantly a local effect and affects only marginally the charge population on the remaining atoms in the spirocyclic framework.
To scrutinize the performance of the Mulliken charge and its comparability with the natural charges, the Mulliken atomic charges for TISS
NU-19, OITSSHC-33, and their tetracyano derivatives were calculated, and the predicted results are summarized in
Table 8.
In this table, it is apparent that the atomic charges deviate considerably from the corresponding NPA charges in
Table 7. The most striking inconsistency is demonstrated by the negative charges on Si
3,9 and H(-C) and the positive charges on C
2,4. This charge inversion reveals that the Mulliken scheme clearly fails to produce the correct charges on most of the participating atoms within the selected SCIs.
This deficiency and all other flaws of the Mulliken concept have been repeatedly discussed in numerous papers expressing fundamental criticism regarding the occasionally quite poor performance of this charge population concept [
64]. The main criticism is directed toward the accentuated dependency of the Mulliken population analysis on the quality of the applied basis set, particularly its augmentation with diffuse functions. Moreover, in many examples, it was shown that the Mulliken assignment of negative and positive charges to atoms is sometimes interchanged and contradicts the requirement of their electronegativities. The major reasons for these drawbacks originate from its initial formalism, which arbitrarily suggests an equal partitioning of electron density between participating atoms, and the assumption that diffuse functions assigned to one atom in a chemical bond reside only on this atom.
To gain more insight into the response of the charge distribution to the attachment of substituents at various positions of an SCI skeleton, the NPA, as well as the Mulliken charge populations within the fluorinated derivatives of OITSSHC-33, were calculated, and the obtained results are shown in
Tables S12 and S13.
A brief comparison between the NPA charge distributions within OITSSHC-33 in
Table 7 and its fluorinated derivatives in
Table S12 unveils that (1) the charges on the nitrogen atoms in the imino groups are only slightly affected by the addition of fluorine substituents on the neighboring carbon atoms. (2) In accordance with the expectation, the electron depletion on the anchor atoms where the fluorine atoms are attached to is considerable. (3) Strikingly, the positively charged silicon spiro centers, Si
6,12 and Si
9, remain almost unaffected by the addition of fluorine substituents at various positions in the spiro scaffold, in contrast to the corresponding Mulliken atomic charges that are listed in
Table S13. This table shows fluctuating and, in some cases, irrational values (among them, negative charges on silicon atoms).
To demonstrate the notorious dependency of the Mulliken charge distribution and the large independency of the NPA charges on the quality of the applied computational method and basis set, both population analyses were conducted on a variety of substituted and non-substituted SCIs by employing the B3LYP and B2PLYP functions and Dunning’s aug-cc-pVDZ basis set (in some cases the non-augmented version of this basis set was also employed). A comparison of the collected results, which are shown in
Tables S14–S16, with those that were obtained from the B2PLYP/aug-cc-pVDZ computational method (
Table 7 and
Table 8) confirms the apparent dependency of the Mulliken atomic charges on the level of the utilized method and basis set. For instance, the double-hybrid functional scheme in combination with the basis set aug-cc-pVDZ predicts the following Mulliken atomic charges: 0.284, −0.204, 2.493, 0.356, and 0.915 electrons on the atoms C
2, Si
3, Si
6, C
8, and Si
9 (
Table 8) in OITSSHC-33 using the B3LYP/cc-pVDZ method. However, it also provides the values −0.024, 0.322, 0.614, −0.044, and 0.449 electrons, whereas the B3LYP/aug-cc-pVDZ level of theory suggests 0.390, −0.700, 1.898, 0.421, and 0.771 electrons (
Table S14) for the same order of atoms. From these apparent variations (signs and magnitudes) of the Mulliken charges, it is evident how strongly this charge population procedure is dependent on the chosen level of theory.
Another example showing this inherent methodical deficiency of the Mulliken population analysis (at variance with the NPA charge population scheme) is the investigation of the response of both addressed charge population analyses to the choice of the employed computational routine, which is illustrated by the fluoro-derivatives of OITSSHC-33 (Fx-OITSSHC-33). The predicted results by the B3LYP/aug-cc-pVDZ method are presented in
Tables S15 and S16 with brief comments and a comparison of these data with those that were produced by the B2PLYP/aug-cc-pVDZ method (
Tables S12 and S13).
To obtain additional knowledge about the behavior of the charge population in a spiro system, an NPA analysis was carried out on the cyclic sila-imines TSISS
NU-19 and OSITSSHC-33 (in analogy to TISS
NU-19 and OITSSHC-33) by applying the B3PLYP and B2PLYP functionals in combination with the basis set aug-cc-pVDZ. A cursory comparison between the natural charges in
Table S17 discloses that the positive charges on the terminal silicon atoms Si
3,15 in OSITSSHC-33 are almost negligible (0.04 e
−), and the central silicon Si
9 is negatively charged (−0.667 e
−). Perhaps the unique position of this silicon atom as a spiro center surrounded by four silicon atoms that are attached to nitrogen atoms is responsible for this peculiarity. Attracting electrons from silicon by nitrogen in the N=Si bond results in a lower effective electronegativity of this silicon atom in comparison to the electronegativity of the silicon spiro center. This striking finding prompted me to perform an NPA charge distribution analysis on two silaspiro compounds, in which the silicon spiro center is surrounded by four silicon atoms, i.e., 1,5,7,11-tetrasilaimino-3,9-disila-6-silaspiro [5.5]undecane (TSIDSS
SiU-19) and 1,4,7,10-tetradisilene-3,9-disila-6-silaspiro [5.5]undecane (TDSEDSS
SiU-23), which consist of two unsaturated six-membered rings (two Si = Si double bonds in each) fused by a silicon spiro center that exclusively contain silicon atoms. The applied B2PLYP/cc-pVDZ and B3LYP/aug-cc-pVDZ provided NPA charges of −0.216 e
− and −0.236 e
−, respectively, on the silicon spiro center Si
6. In addition, the NPA charges on the spiro center Si
6 in the tetrasilaimino analog (TSIDSS
SiU-19) amount to −0.661 e
− and −0.665 e
−, as predicted by B2PLYP/cc-pVDZ and B3LYP/aug-cc-pVDZ, respectively. Another example in this regard is 1,5,7,11-tetrasilaethylene-3,9-disila-6-silaspiro [5.5]undecane (TSESS
SiU-23), where the spiro silicon atom Si
6 is negatively charged with −0.445 electrons. Furthermore, an NPA analysis was carried out on the acyclic tetradisilene-silane (Si (-SiH=SiH
2)
4) and tetradisilane-silane (Si (SiH
2-SiH
3)
4), and it turned out that in these cases, negative charges are concentrated on the central Si atom (−0.229 e
− in the former compound and −0.238 e
− in the latter). Interestingly, the NPA charge distribution in the germanium counterparts to all the above-cited compounds has exhibited the same pattern, i.e., the central germanium atom is always negatively charged when it is surrounded by Ge atoms. This remarkable behavior of the NPA charge distribution (the silicon/germanium spiro center is positively charged when the surrounding bonded atoms are doubly bonded nitrogen or carbon but negatively charged when the Si/Ge spiro center is surrounded by Si(Ge)=X double bonds (X=C, Si, Ge)) or Si-Si (Ge-Ge) single bonds will be the subject of a future investigation.
6.2. Bond–Antibond (Lewis Non-Lewis) Interactions
For the evaluation of the criteria for the stabilizing bond–antibond NBO interactions, the NBO computational routine [
66], which is incorporated in the Gaussian 16 program, has been utilized.
Table 9 summarizes the values of the donor–acceptor delocalization energy, which are E(2) for sila-/germa-spirocyclic imines TISS
NU-19 and TIGS
NU-19 and their tetracyano derivatives, and
Table 10 presents some relevant E(2)
values for OITSSHC-33 and its fluorine derivatives, as predicted by the second order perturbation theory (SOPT) in the NBO basis [
67].
These energy values depict the extent of charge delocalization from Lewis- to non-Lewis (bond–antibond) NBOs and, thus, indicate the strength of bond–antibond conjugative or hyperconjugative interactions between NBOs.
Scrutiny of the values comparing the second-order perturbation energies (SOPE) of some important intra-molecular donor–acceptor charge delocalization in
Table 9 and
Table 10 leads to the following intrinsic conclusions (only values larger than 2.0 kcal∙mol
−1 are listed in these tables, and donor/acceptor interactions involving C-H bonds have not been considered):
(i) Perhaps the most decisive Lewis to non-Lewis charge transfer is the
(2)N1=C2 → σ*
Si6-N7= donor–acceptor interaction in TISS
NU-19 and TC-TISS
NU-23 (and similarly in the germa counterparts) with a stabilization energy of 3.3 kcal mol
−1 and 3.2 kcal mol
−1, respectively. Such kind of NBO interaction can be considered clear evidence for the cross-hyperconjugative interaction in this class of sila spirocyclic imines. This essential assessment will be affirmed later in this work by discussing the spiro-aromaticity of SCIs. For the sake of brevity and because the E(2) values for the analog germanium compounds in
Table 9 parallel those that have been presented above, a consideration of the E(2) values for the latter compounds will be renounced. It should only be noted that except for some values, the SOPT analysis provides mostly larger delocalization energies for the germanium compounds than the silicon counterparts.
(ii) The considerably high stabilization energy for the delocalization from the donor orbitals of the lone pairs on the imino nitrogen N1 into the acceptor NBOs σ*Si3-C2= and σ*Si6-N5= undoubtedly play a prominent role by stabilizing the rigidity of the spiro skeleton.
(iii) The NBO analysis furnishes pertinent evidence for charge transfer and redistribution of the entire electronic structure of a specific molecular entity. In this respect, structural and electron density alterations provoked by the introduction of substituents are clearly detectable by applying the perturbation theory energy analysis on the NBO basis. The validity of such valuation is illustrated by the NBO analysis of the cyano- and fluorine-derivatives of some SCIs (
Table 9 and
Table 10). The addition of such substituents to SCIs engenders perceptible structural changes and has a noticeable effect on some crucial properties of this class of compounds like increased polarity, intra-, and inter-molecular charge transfer dynamics, electronic chemical potential, and global hardness/softness, as will be shown below. The SOPT analysis of TCTISS
NU-23 (
Table 9) has shown that the interaction between the nitrogen lone pair in the cyano group and the localized Si-C≡ antibond, (LP)
N≡17 → σ*
Si3-C≡16), possesses an appreciable delocalization energy of 7.9 kcal mol
−1. From this analysis, it also emerges that charge transfer occurs from the Lewis-type donor NBO σ
C16≡N17 to the non-Lewis acceptor NBO σ*
Si3-C≡16 (E(2) = 2.6 kcal mol
−1), and a donation of electrons from the occupied
(2)C16≡N17 bonding NBO to the antibonding acceptor σ*
Si3-C≡18 orbital results in a stabilization energy of 2.4 kcal mol
−1. These two orbital interactions manifest the hyperconjugative interaction within the N≡C-Si-C≡N fragment (electrons are commuting between the terminal nitrogen atoms), which rationalizes the abovementioned shortening of the Si
3-C≡ bond and, at the same time, they confirm the specific bonding character of the cyano group as a simultaneous strong 6 and π acceptor. To add more evidence to the justification of this kind of internal orbital interaction between the geminal cyano groups, a combined structural and NBO analysis on 3,9-dicyano-TISS
NU-21 and 3,9-dicyano-TIGS
NU-21 (only one cyano group on each side) has been performed. Interestingly, the elimination of the geminal arrangement of the cyano groups has led to the following consequences. (1) The Si
3-C≡ bond lengthens by 0.01 Å in comparison to the Si
3-C≡ bond in TCTISS
NU-23. (2) The C≡N bond length remains unaffected (1.174 Å). (3) The endo cyclic bond angle =C
2-Si
3-C
4= decreases by 1.6°. Similarly, the Ge
3-C≡ bond length in 3,9-dicyano-TIGS
NU-21 increases by 0.01 Å in comparison to the corresponding bond in TCTIGS
NU-23, and the endo cyclic bond angle =C
2-Ge
3-C
4= narrows by 3.2° due to the absence of the electronic coupling, which was prevailing in the case of the geminal substitution.
(iv) In
Table 9, it is also apparent that the most energetically favorable Lewis to non-Lewis charge delocalization occurs by the donation of the electron lone pairs on the nitrogen atoms in the imino and cyano groups. In this respect,
Table 9 indicates that the largest charge delocalization energy of 12.2 kcal mol
−1 in TISS
NU-19 andTCTISS
NU-23 (and correspondingly, 13.1 and 13.4 kcal mol
−1 in TIGS
NU-19 and TCTIGS
NU-23) is associated with the (LP)
N1 → σ*
Si3-C2= (and (LP)
N1 → σ*
Ge-C2=) donor–acceptor orbital interaction, leading to a strengthening of the Si
3-C
2= (and Ge
3-C
2=) bond. The markedly large value of the stabilization energy of 8.6 kcal mol
−1 for the (LP)
N≡17 → σ*
Si3-C≡16 (and obviously for the (LP)
N≡19 → σ*
Si3-C≡18) charge delocalization contributes to an additional strengthening of the Si
3-C≡ bond and simultaneously to a slight weakening of the C≡N bond.
Among the essential information provided by the NBO analysis is the occupancy of bond orbitals and their departure from the idealized Lewis structure.
Table S18 reveals that all 6- and π-bonding orbitals in the C=N and C≡N bonds are obviously polarized toward the higher electronegative nitrogen atom. For instance, the 6
N1-C2 NBO consists of 60.7% electrons localized on a nitrogen natural atomic hybrid and 39.3% on a carbon natural hybrid (this partitioning of the NBO is in TISS
NU-19 and TCTISS
NU-23, which are almost alike). The accentuated polarity of the =N-Si bond is demonstrated by the large polarization coefficient ξ(N
1) of 0.898 (80.6% of the NBO) on the nitrogen atom and ξ(Si) = 0.44 (19.4% of the NBO) on silicon. As is also shown in
Table S18, the Si-C≡ bond in TCTISS
NU-23 is less polar than the =N-Si bond. There is a distinct decrease in the nitrogen (all nitrogen atoms surrounding the silicon spiro center) and lone pair NBO occupancy down to 1.88 and 1.87 electrons in TISS
NU-19 and TCTISS
NU-23, respectively. This reduction in the lone pair occupancy on each of the nitrogen atoms, N
1, N
5, N
7, and N
11, around the spiro silicon atom Si
6 is due to the donation of electrons mainly into the non-Lewis antibond orbitals σ*
Si6-N15,7,11 (0.095 e
− on each). Moreover, the loss of occupancy on the cyano nitrogen (1.97 electrons) is caused by the donation of charges into the localized antibonding NBOs σ*
Si3-C≡16,18 (0.05 e
− on each).
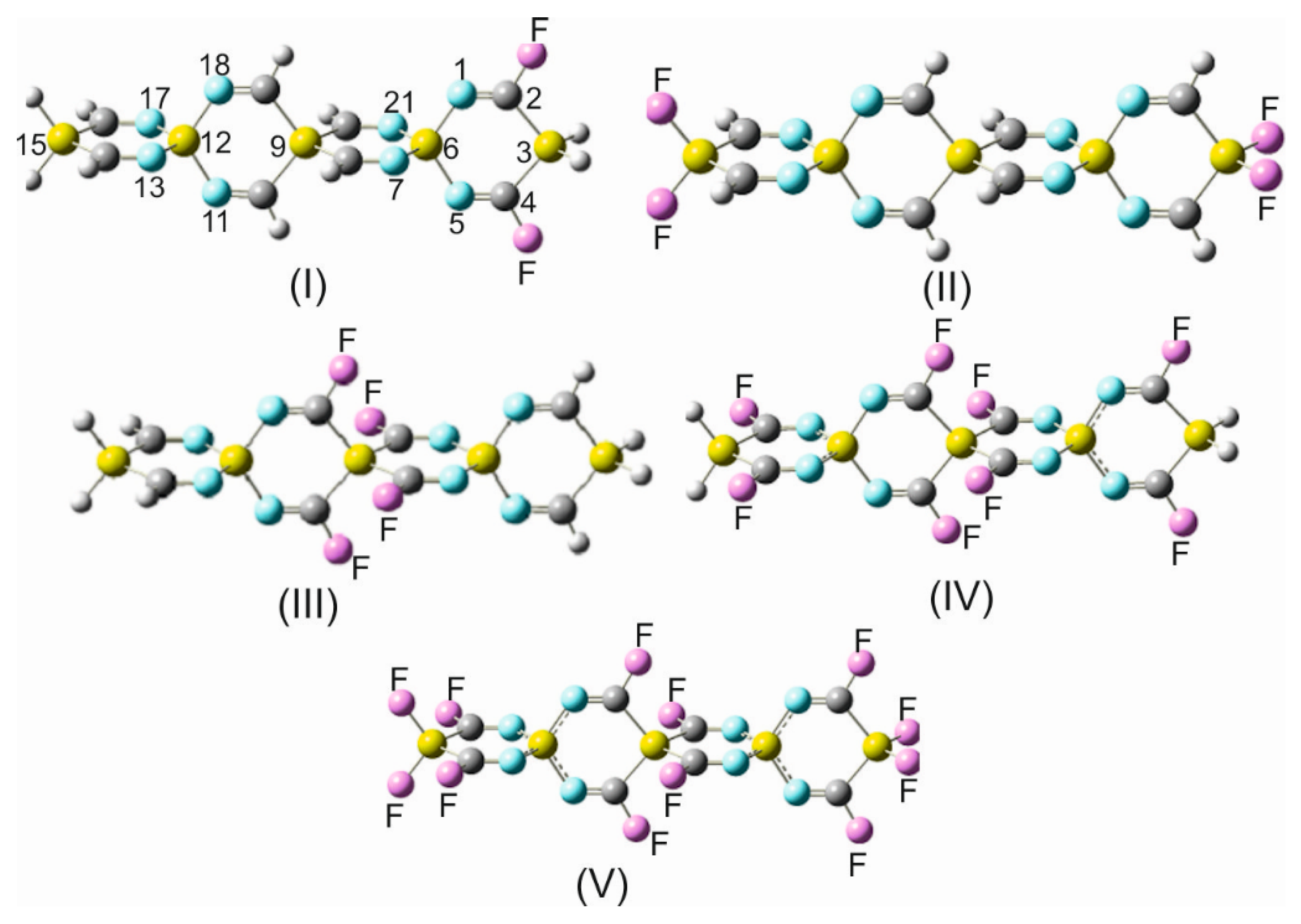
Before analyzing the consequences of the fluorine substitution on the donor–acceptor interactions, in OITSSHC-33, a few remarks should be addressed. As was alluded to in the introduction, the replacement of a carbon by an isostere, like silicon, in pharmacologically active agents usually has a marked impact on its pharmacodynamics and pharmacokinetics. Tacke and his research group [
5] have thoroughly investigated this interesting isosteric substitution and its relevance for developing new classes of drug-active ingredients with higher efficiency than their carbon counterparts. Moreover, in numerous papers, it was shown that the C-Si-N bonding arrangement possesses a distinctive biological activity and chemical reactivity [
68,
69]. Provided that the new category of sila-/germa-spirocyclic imines possess the predicted pharmacological activity, the attachment of a unique substituent, like fluorine, with its small size and high electronegativity to these compounds would add some novel physicochemical properties comprising increased lipophilicity, charge mobility, and affinity for interacting with adjacent molecular entities. The main rationale for a successive fluorine addition (
Table 10) is to systematically investigate the gradual impact of such systematic substitution on the internal donor–acceptor interactions and charge distribution along the framework of a spirocyclic imine unit. Additionally, it can be anticipated that the incorporation of fluorine substituents in an SCI unit will lead to the rearrangement of the electrophilic and nucleophilic centers, geometrical alterations, and the influence of the charge transport mobility and ability for docking efficacy to other molecular assemblies. The impact of fluorination on charge transfer dynamics and its efficiency across benzonitrile-based self-assembled monolayers has been investigated [
46].
To gain more a pertinent rationalization of the fluorine substituent effect on the spirocyclic imines, a full NBO analysis was conducted, and the emerging delocalization energy values as estimated by the second-order perturbation theory (SOPT) are listed in
Table 10. It is worth indicating that further representatives of the sila-/germa-spirocyclic imine series have been also fluorinated and analyzed by means of the NBO routine but for the sake of brevity, they have not been included in this paper. Rather, the focus has been placed on discussing the effect of the multiple fluorine substitution in SCIs by choosing OITSSHC-33 as an example for all other representatives of this class of compounds. An inspection of
Table 10 reveals that the perturbative stabilization energy E(2) for a cross-hyperconjugative interaction symbolized by the
(2)N1-C2 → σ*
Si6-N7,11 donor–acceptor interaction amounts to 3.1 kcal mol
−1 in OITSSHC-33 and is slightly higher in the fluorinated derivatives, indicating a tendency for the enhancement of this kind of interaction along with the increase in the attached fluorine atoms. Furthermore, in accordance with the expectation of the strong polar Si-N= bonds with a high charge concentration on the nitrogen atoms, a donation of electrons into the unoccupied non-Lewis NBOs of the adjacent =C-F antibonds was made. Remarkably, however, are the particularly large delocalization energy values of 14.6 kcal mol
−1 (and a value for the off-diagonal element in the NBO Fock matrix, F
i,j of 0.098 a. u.) in (
III) and 14.0 kcal mol
−1 in (
IIV) for the σ
Si6-N1,5 → σ*
C2,4-F donor–acceptor NBO interaction. Similarly, appreciable stabilization energy values of 15.9, 15.4, and 15.1 kcal·mol
−1 (and F
i,j,,= 0.100 a.u. for each) are affiliated with the donor–acceptor NBOs σ
Si6-N7 → σ*
C8-F in (
II),
(III), and (
IV), respectively (
Table 10). The reason for such a large E(2) value is the accentuated electron withdrawal by the fluorine atoms. Probably, the most decisive valence shell Lewis and non-Lewis charge delocalizations are those in which the lone pairs on both imine nitrogen and fluorine atoms are participating. For instance, the significant deviation from the idealized Lewis occupancy is related to the valence lone pair NBO (LP)
N7 (and equally the valence lone pairs on the imino nitrogen atoms N
11,18,21), which is occupied by 1.879 electrons in OITSSHC-33 and 1.832 e
− in the fluorine derivatives F4-, F8-, and F12- (
Figure 4). The depletion of occupancy is due to charge distribution into the acceptor NBOs 6*Si
6-N
7,21 (each occupied by 0.103 e
−), 6*C
8,20-F (each occupied by 0.128 e
−), and 6*C
8-Si
9 (0.094 e
−). One further interesting charge delocalization occurs in F12-OITSSHC-33, where the occupancy of the valence (LP
3)
F23 NBO decreases to 1.945 e
−, losing charges to the acceptor non-Lewis NBOs 6*Si
3-C
2,4 and 6*Si
3-F
23. The latter donor–acceptor interactions (LP
3)
F23 → 6*Si
3-F
24 (and obviously (LP
3)
F24 → 6*Si
3-
F23) indicate that the geminal terminal Si-F bonds are stabilized by means of a negative hyperconjugation effect with an appreciable stabilization energy E(2) of 11.8 kcal mol
−1 (
Table 10). This table also shows that in F12-OITSSHC-33, only moderate energy of 3.9 kcal·mol
−1 is required for the delocalization of charges from the σ
C2,4-Si3 donor NBO to the acceptor antibonding σ*
Si3-F NBO.
These peculiar details, which have been derived from the NBO and SOPT analyses, confirm once more the relevance of the Si-N bond for modifying the fundamental physicochemical properties of molecular assemblies by incorporating such a vital bond. As can be anticipated, the successive addition of fluorine substituents to SSCIs forming either a C-F or Si-F bond as in OITSSHC-33 most likely will contribute to a drastic alteration or accentuation of most of the physicochemical properties that have been repeatedly addressed above. Furthermore, the partial or total fluorination of a sila-/germa-spirocyclic imine framework would promote their ablation capability on surfaces of functional carriers and manipulate the interfacial interactions of such functionalized surfaces.
To examine the response of the charge distribution in spirocyclic silaimines (SCSIs) to fluorination, an NBO analysis was conducted on various fluorine derivatives of TSISS
NU-19 (
Figure S7). A review of the most prominent Lewis-occupied NBOs and non-Lewis-localized NBOs in F8-TSISS
NU-19 (
Table S19) leads to the following brief conclusions. The charge delocalization π
N1=Si2 → 6*
Si6-N7 is stabilized by 5.0 kcal mol
−1, confirming the role of the cross-hyperconjugation in this class of SCSIs. Mainly by virtue of the strong electron withdrawal by the attached fluorine atoms, the occupancy of the Si
2-Si
3 bonds drops down to 1.856 e
− (at the B3LYP-aug-cc-pVDZ level of theory), as shown in
Table S19. This perceptible departure from the idealized Lewis occupancy is provoked by the donation of charges into the acceptor antibond orbitals 6*
Si2-F and 6*
N1-Si2 that are associated with delocalization energies of E(2) = 10.5 kcal mol
−1 and 9.9 kcal mol
−1, respectively.
In accordance with the expectation, the NBO routine has also shown that the lone pairs on nitrogen and fluorine donate electrons into various non-Lewis antibond NBOs, like, for instance (LP)N
1 → 6*
N5-Si6, with a delocalization energy of (E(2) = 10.2 kcal·mol
−1 and (LP
3)F
15 → 6*
Si8-N7 (E(2) = 14.3 kcal·mol
−1), leading to a reduction in the occupancy of the (LP)N
1 NBO down to 1.843 e
− and the occupancy of the (LP
3)F
15 to 1.935 e
− (
Table S19). Of particular interest is the donation of charges from the occupied (LP
3)F
17 NBO into the non-Lewis NBO 6*
Si3-F18 (and similarly the delocalization of charges from (LP
3) F
18 into 6*
Si3-F17), resulting in a loss of occupancy of the (LP
3)F
17 NBO (1.946 e
− in
Table S19) with a stabilization energy E(2) of 10.3 kcal mol
−1. These mutual donor–acceptor NBO interactions symbolize, once more, the negative hyperconjugative stabilizing effect between the geminally substituted fluorine atoms. For comparison reasons, the NBO occupancy values for the counterpart TISS
NU-19 have been included in
Table S19. A brief comparison of the data in this table reveals that the Si
2-F bond in F8-TSISS
NU-19 is clearly more polar than the C
2-F bond F8-TISS
NU-19, which is apparent from the polarization coefficient values of ξ(Si
2) = 0.364 and ξ(F) = 0.932 in the Si
2-F bond and ξ(C
2) = 0.517 and ξ(F) = 0.856 in the C
2-F bond. Moreover, it is interesting that the terminal Si
3-F bond is equally polar in both F8-TSISS
NU-19 and F8-TISS
NU-19, which is discernible from the equal polarization coefficients of Si
3 on one hand and the fluorine atom in the Si
3-F bond on the other hand.
Finally, it is worth noting that some caution is advised when trying to derive decisive conclusions from comparing perturbative E(2) values originating from quantum chemical calculations carried out by applying different methods and basis sets. In this respect, a full NBO analysis on TISS
NU-19 and TIGS
NU-19 was performed by varying the computational method and using the same basis set to verify the effect of the applied method on the NBO occupancies and the donor–acceptor second-order stabilization energy E(2). Furthermore, the same computational method but in combination with different basis sets (with and without augmentation with diffuse functions) was applied.
Table S20 visualizes the dependency of the E(2) values on the applied method and basis set for some important donor–acceptor interactions in the above-indicated SCIs. In
Table S20, it was found that (1) using the B3LYP method in combination with basis sets aug-cc-pVDZ and cc-pVDZ suggests E(2) values that are by 6–13% larger, as provided by using the former basis sets than those that were obtained by employing the non-augmented basis set cc-pVDZ, demonstrating the effect of the inclusion of diffuse functions and, therefore, the variation of the basis set. (2) A comparison between the E(2) values, as predicted by B3LYP and B2PLYP in combination with the basis set aug-cc-pVDZ, provides an increase in the E(2) energy values by 7–21% when applying the double hybrid functional method, certifying the effect of the applied computational method. (3) In the final step, the wave function HF/6-311G(d, p) was used to inspect the consequence of the variation of both the utilized computational method and the applied basis set. In this case, the alterations of the proposed perturbative delocalization energy E(2) values in comparison to those resulting from the above-mentioned methods were nonuniform (some are smaller and others are larger), as is apparent in
Table S20. Additionally, the calculated E(2) values for TIGS
NU-19 exhibit the same trend as those that were found in TISS
NU-19 by utilizing the same level of theory, but they are generally smaller (
Table S20).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}