

Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations

 ,
,  , , ,
, , ,  ,
,  and
and
Abstract
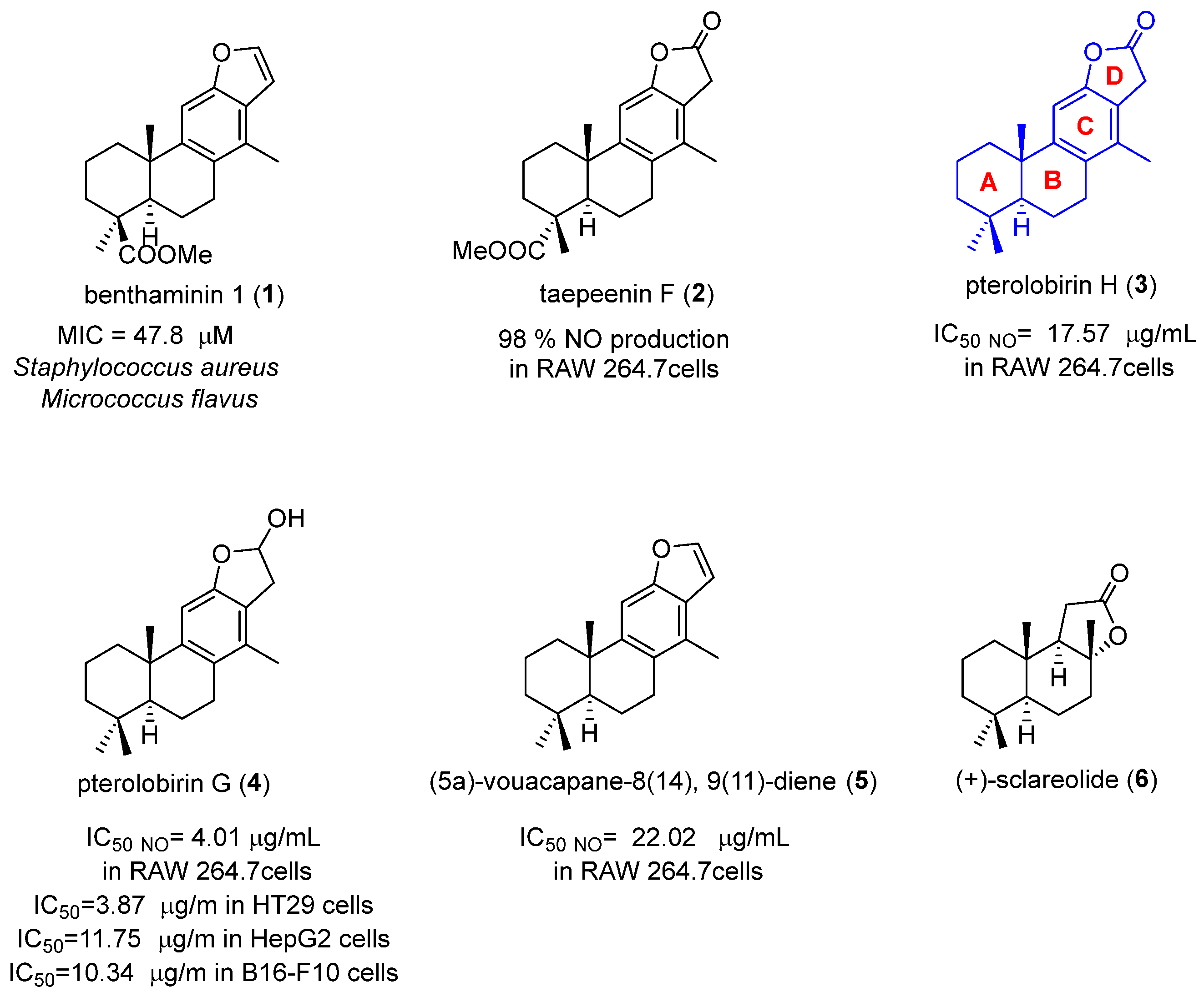
:1. Introduction
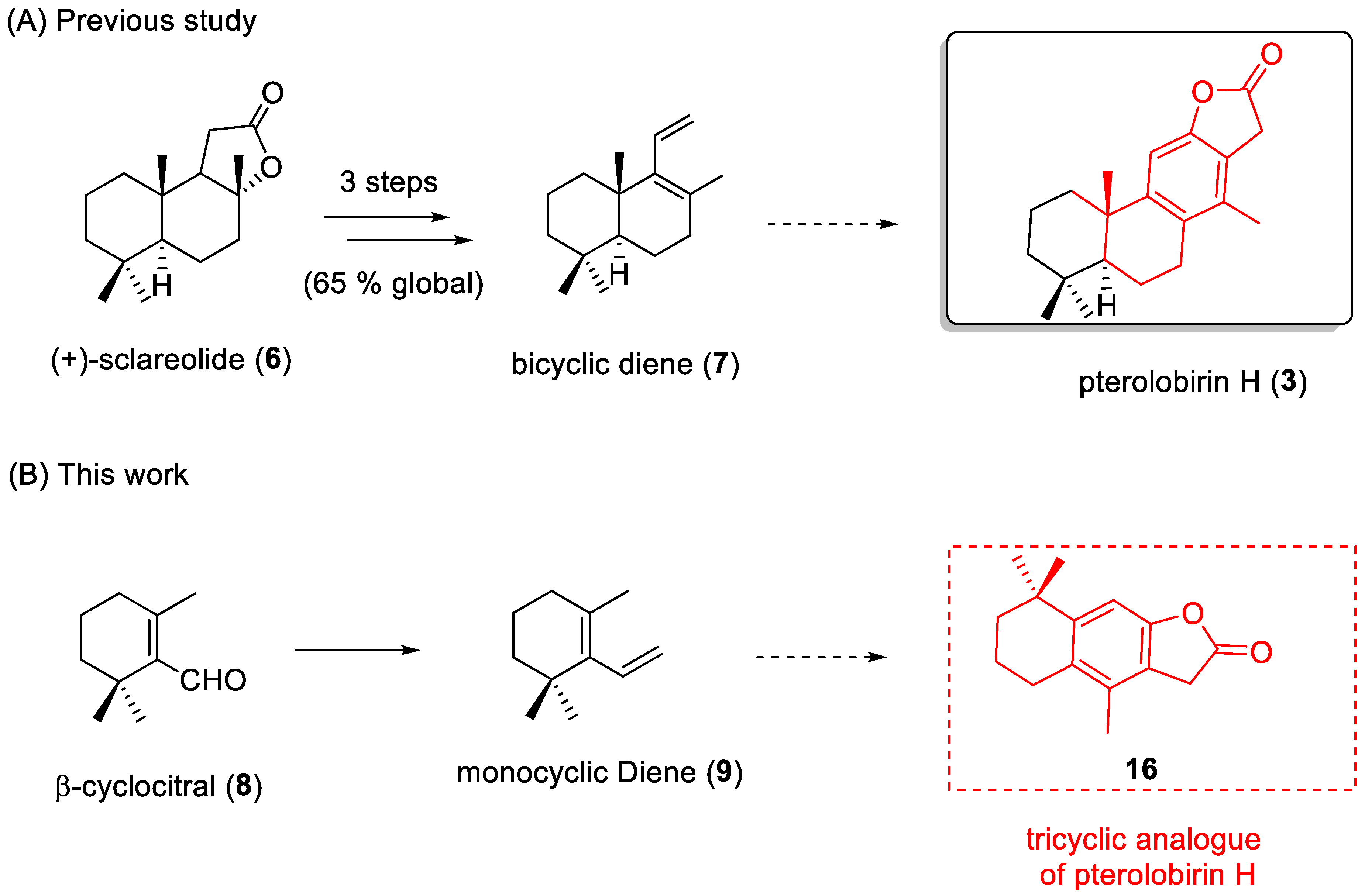
2. Results and Discussion
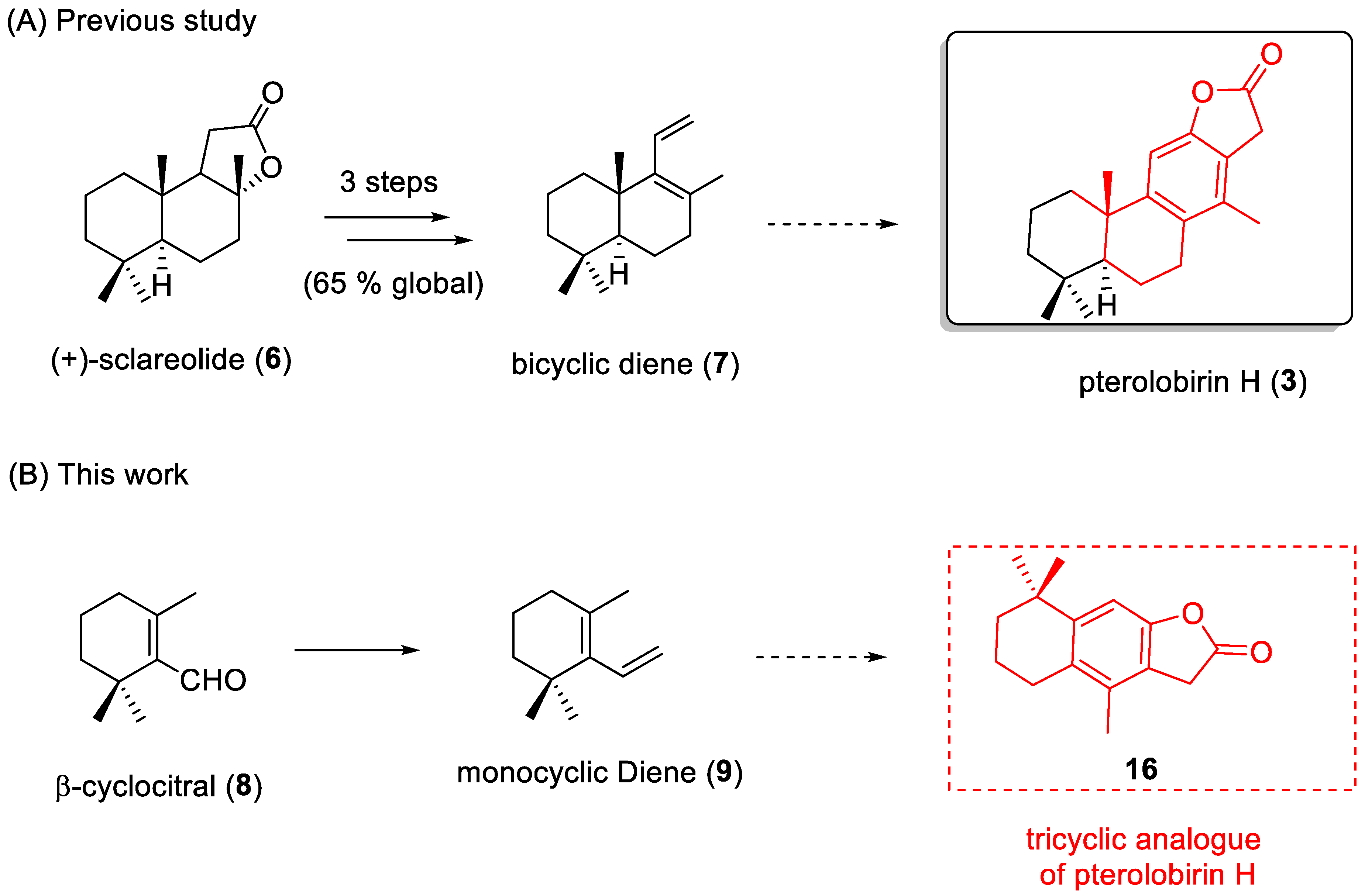
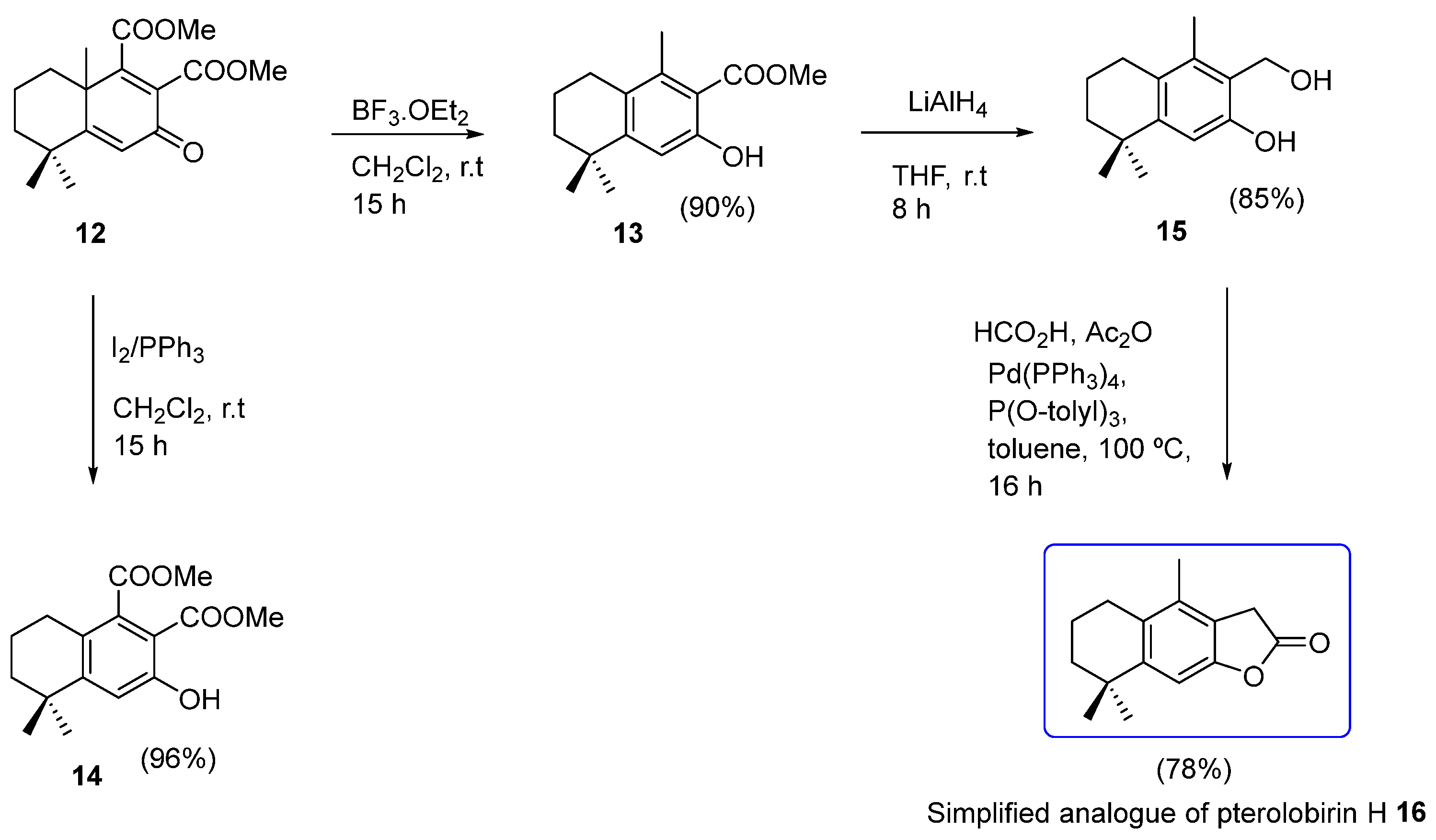
2.1. Chemistry
2.2. Anti-inflammatory Activity
2.2.1. Cell Viability Assay on RAW 264.7 Cell Line
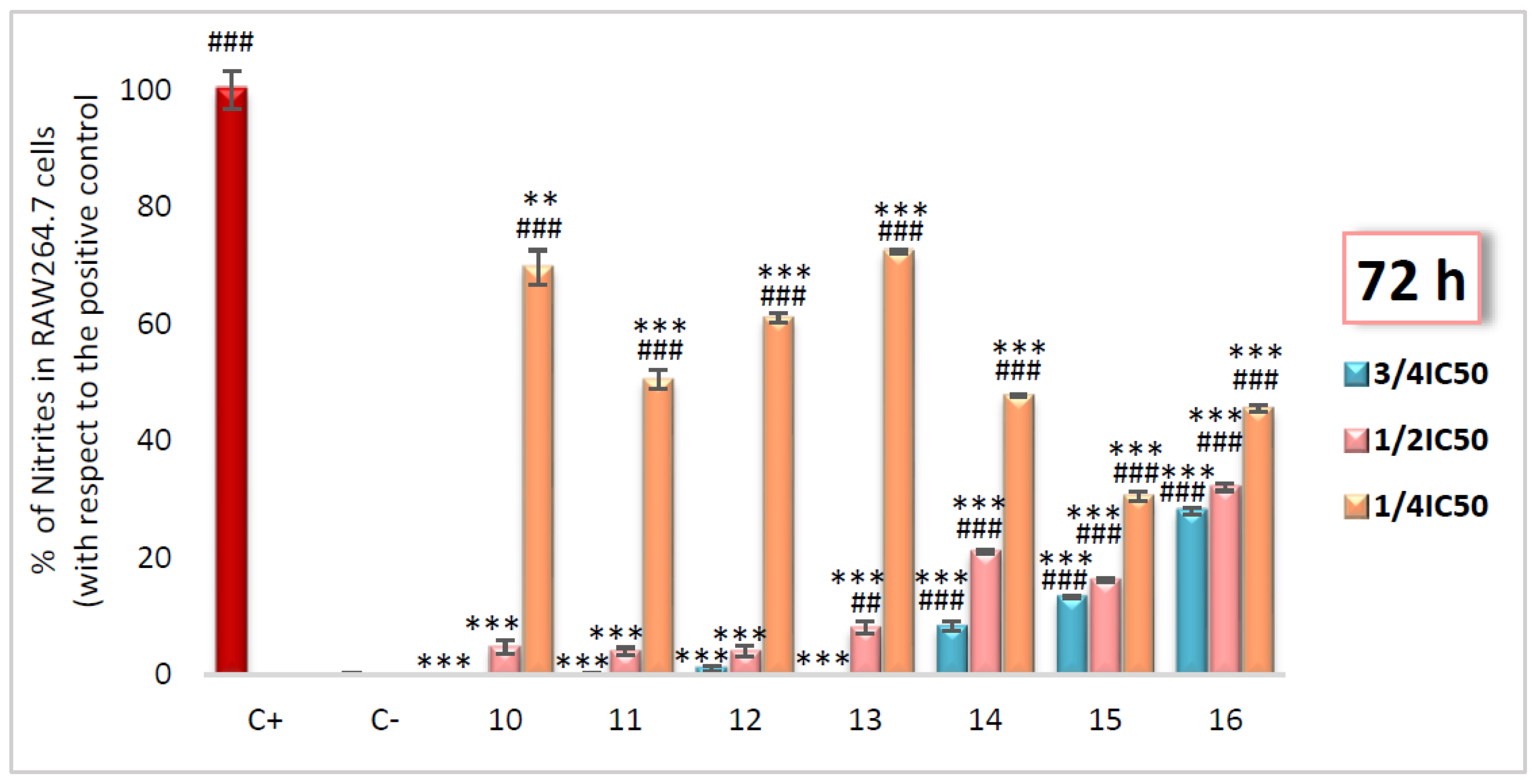
2.2.2. Inhibition of NO Production Assay
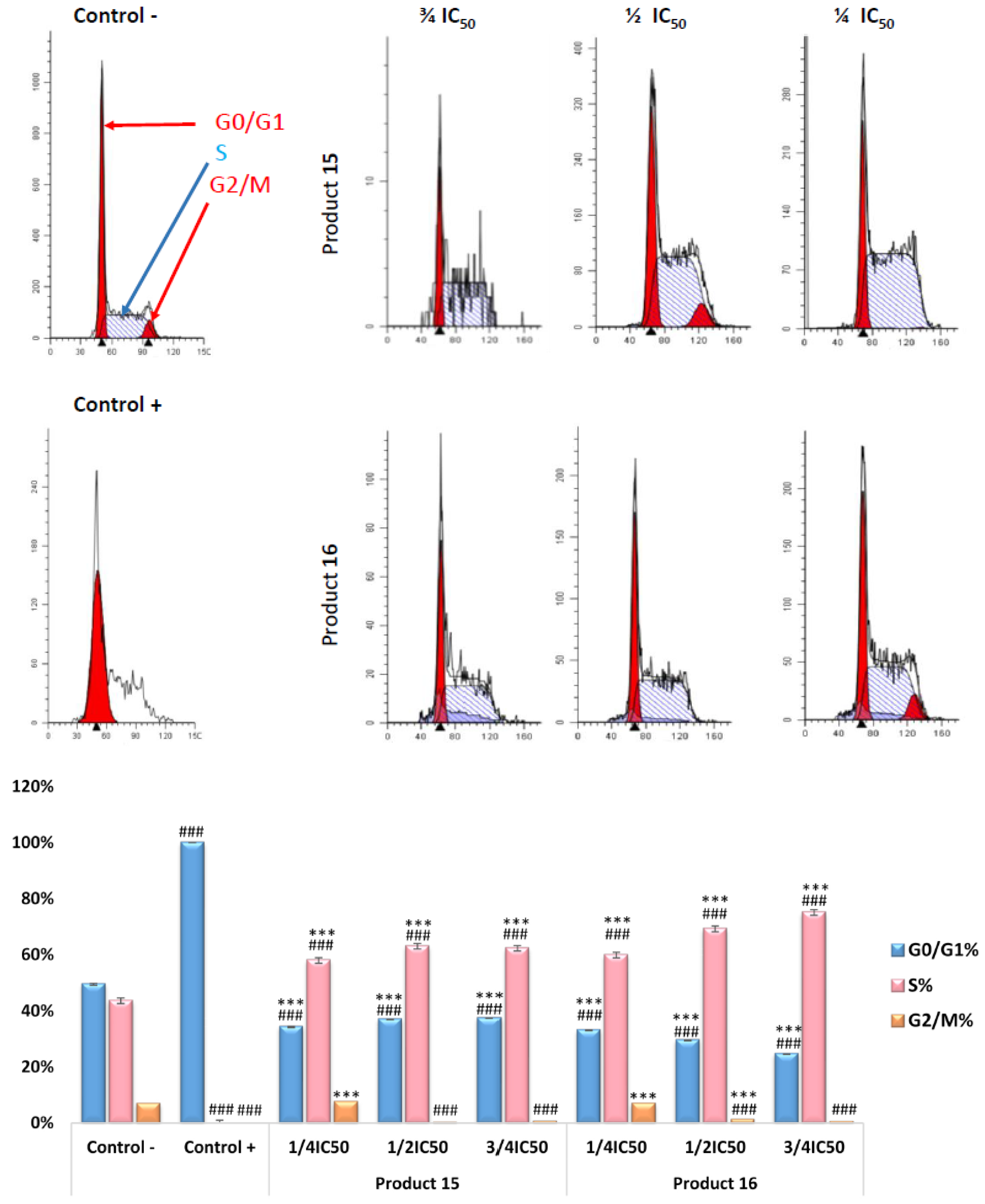
2.2.3. RAW 264.7 Cell-Cycle Assay by Flow Cytometry
2.3. Anticancer Activity
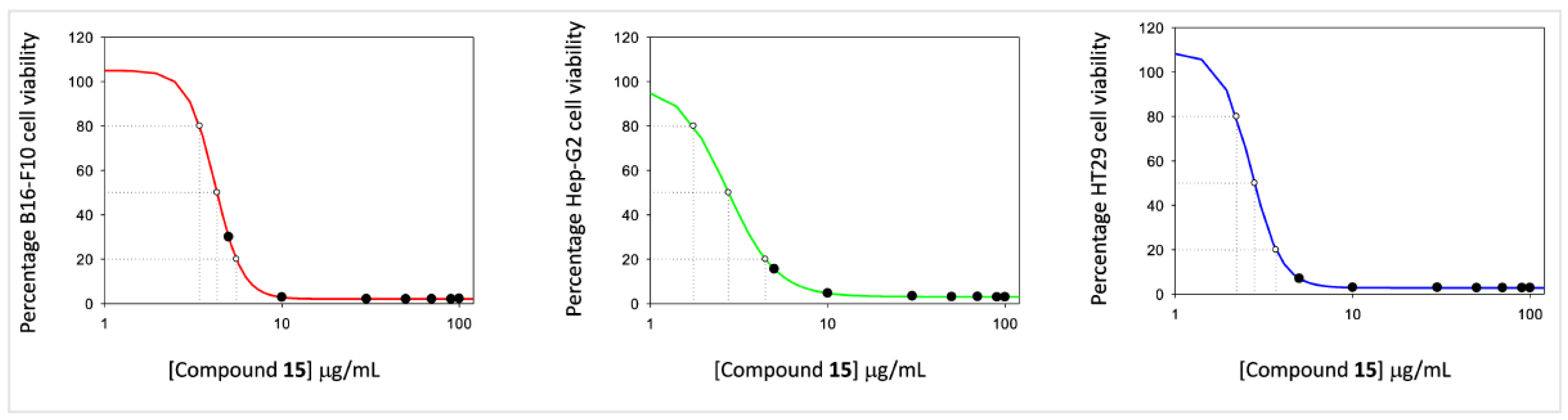
2.3.1. Cell Viability Assay
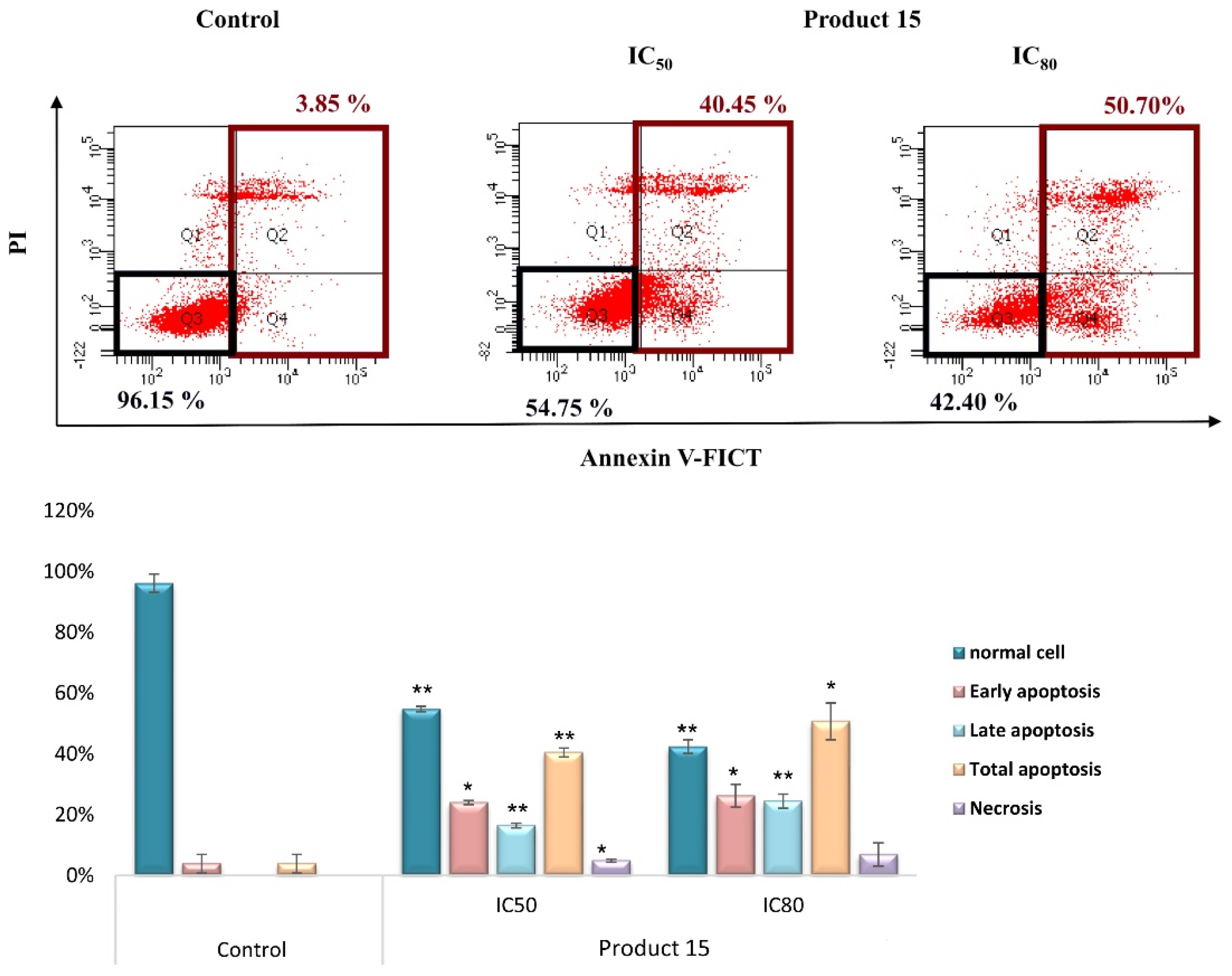
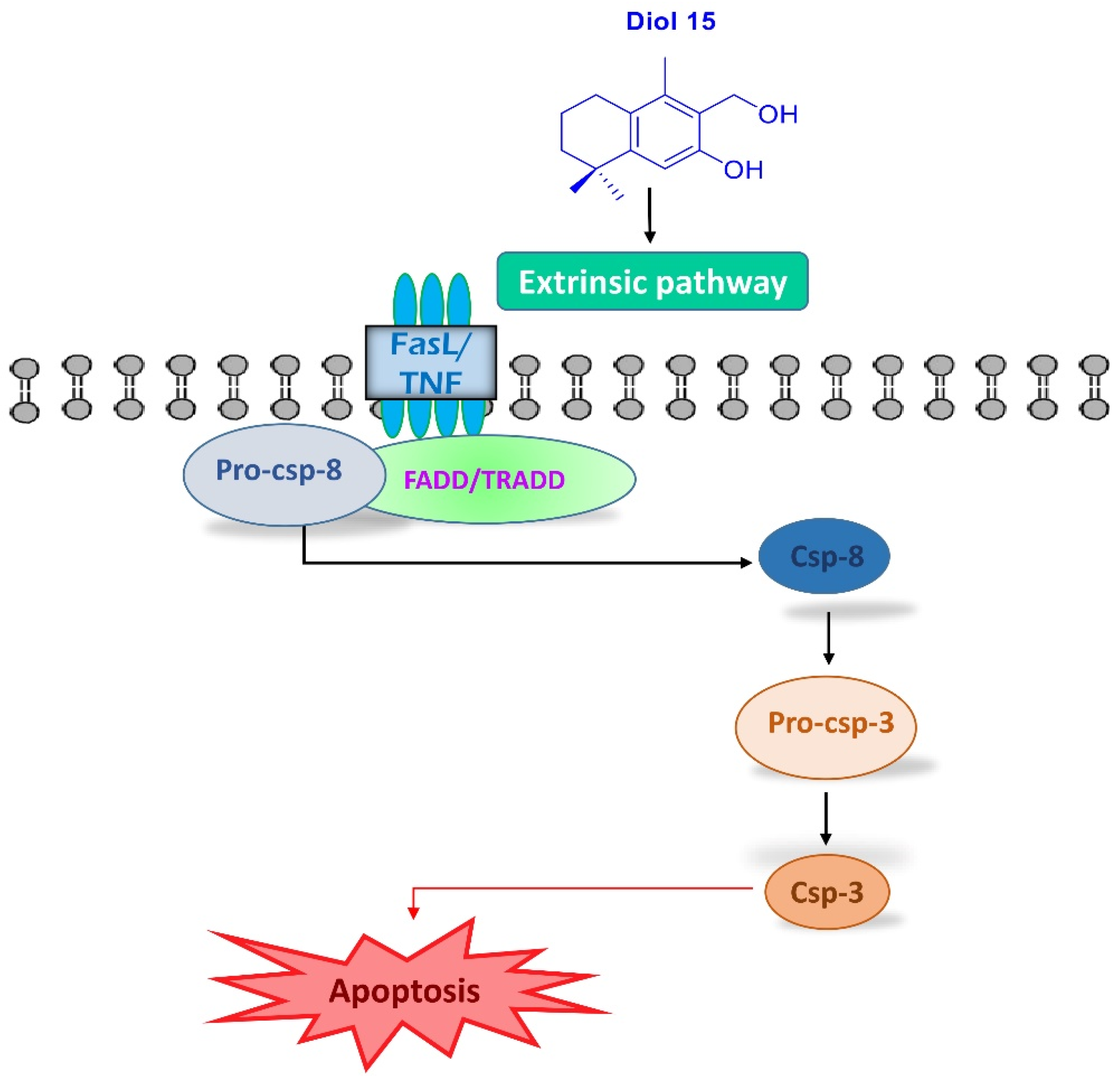
2.3.2. Apoptotic Assay by Flow Cytometry
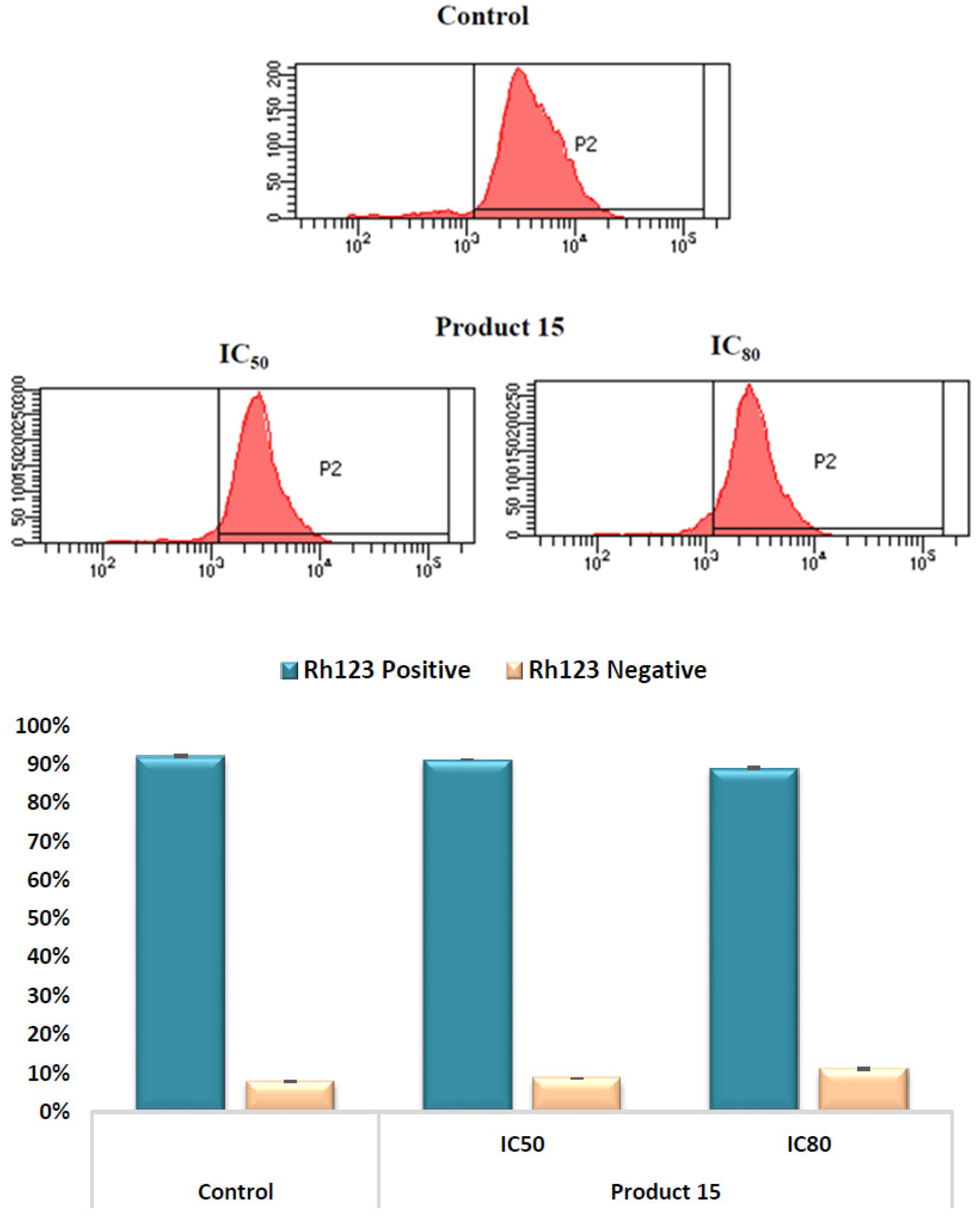
2.3.3. Mitochondrial Membrane Potential (MMP) Assay by Flow Cytometry
2.3.4. Morphological Apoptotic Changes by Fluorescence Microscopy (Hoechst Staining)
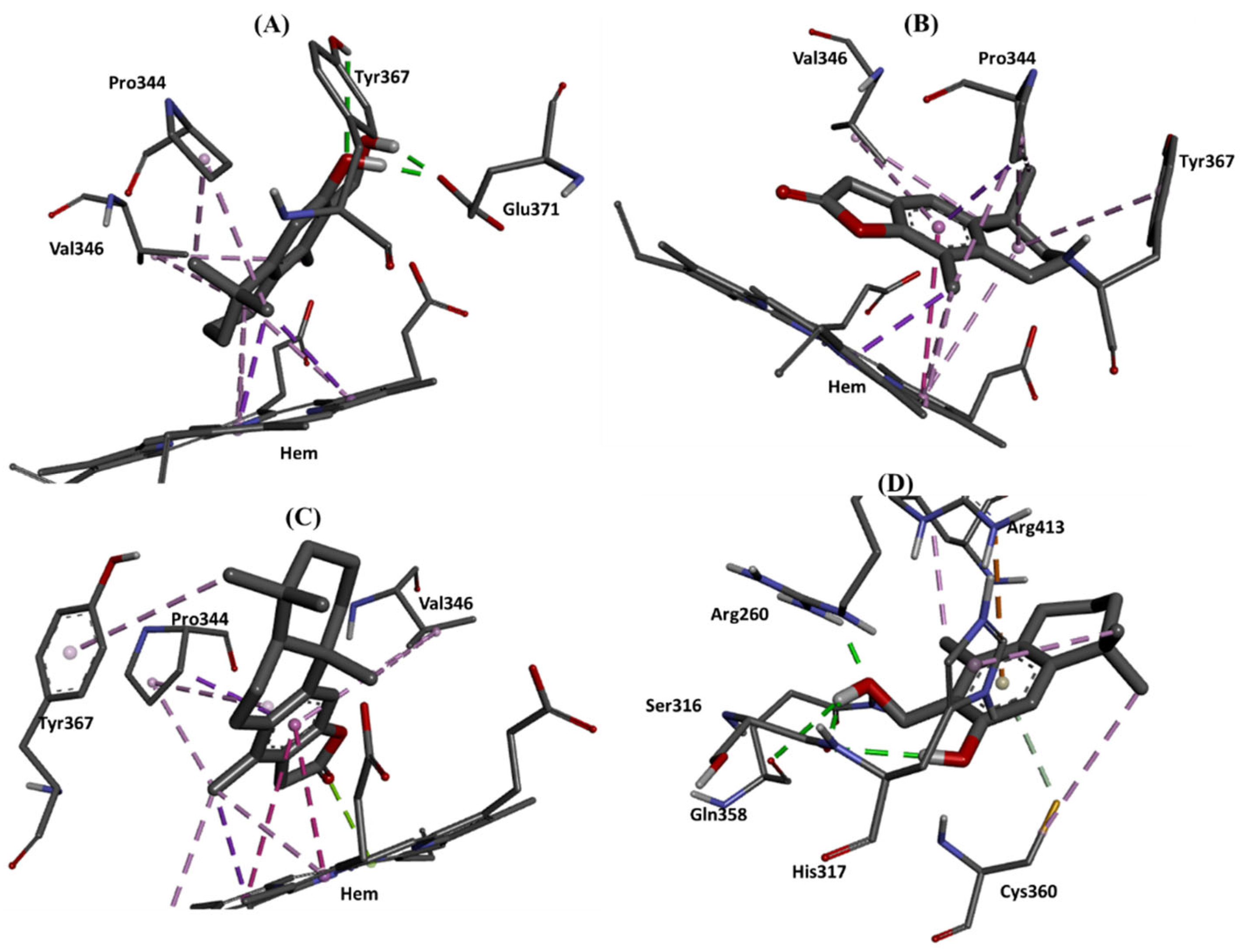
2.4. Molecular Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis
3.2. Biological Experiment
3.2.1. Test Compounds
3.2.2. Cell Cytotoxicity Test
3.2.3. Evaluation of Nitric Oxide Concentration
3.2.4. Cell–Cycle Test (RAW264.7)
3.2.5. Cell–Cycle Test (HT29)
3.2.6. Apoptosis Test (HT29)
3.2.7. Mitochondrial Membrane Potential Test (HT29)
3.2.8. Hoechst–Stained Fluorescence Microscopy Test (HT29)
3.2.9. Statistical Studies
3.2.10. Experimental Section of Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Chandrashekhar, M.; Nayak, V.L.; Ramakrishna, S.; Mallavadhani, U.V. Novel triazole hybrids of myrrhanone C, a natural polypodane triterpene: Synthesis, cytotoxic activity and cell based studies. Eur. J. Med. Chem. 2016, 114, 293–307. [Google Scholar] [CrossRef]
- Mallavadhani, U.V.; Chandrashekhar, M.; Nayak, V.L.; Ramakrishna, S. Synthesis and anticancer activity of novel fused pyrimidine hybrids of myrrhanone C, a bicyclic triterpene of Commiphora mukul gum resin. Mol. Divers. 2015, 19, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Zentar, H.; Arias, F.; Haidour, A.; Alvarez-Manzaneda, R.; Chahboun, R.; Alvarez-Manzaneda, E. Protecting-Group-Free Synthesis of Cassane-Type Furan Diterpenes via a Decarboxylative Dienone-Phenol Rearrangement. Org. Lett. 2018, 20, 7007–7010. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Zhang, X.X.; Zhou, H.; Wang, Y.; Yang, M.; Long, L.; Gao, H. Naturally Occurring Cassane Diterpenoids (CAs) of Caesalpinia: A Systematic Review of Its Biosynthesis, Chemistry, and Pharmacology. Fitoterapia 2019, 134, 226–249. [Google Scholar] [CrossRef]
- Yajima, A.; Mori, K. Diterpenoid Total Synthesis, XXXII. Synthesis and Absolute Configuration of (−)-Phytocassane D, a Diterpene Phytoalexin Isolated from the Rice Plant, Oryza sativa. Eur. J. Org. Chem. 2000, 24, 4079–4091. [Google Scholar] [CrossRef]
- Spencer, T.A.; Villarica, R.M.; Storm, D.L.; Weaver, T.D.; Friary, R.J.; Posler, J.; Shafer, P.R. Total Synthesis of Racemic Methyl Vinhaticoate. J. Am. Chem. Soc. 1967, 89, 5497–5499. [Google Scholar] [CrossRef]
- Spencer, T.A.; Smith, R.A.J.; Storm, D.L.; Villarica, R.M. Total Syntheses of (±)-Methyl Vinhaticoate and (+)-Methyl Vouacapenate. J. Am. Chem. Soc. 1971, 93, 4856–4864. [Google Scholar] [CrossRef]
- Bernasconi, S.; Gariboldi, P.; Jommi, G.; Sisti, M.; Tavecchia, P. Synthesis of (+)-Methyl Vouacapenate from Podocarpic Acid. An Improved Route. J. Org. Chem. 1981, 46, 3719–3721. [Google Scholar] [CrossRef]
- Wang, F.; Chiba, K.; Tada, M. Stereoselective Synthesis of (±)-7β-Acetoxyvouacapane. Chem. Lett. 1993, 22, 2117–2120. [Google Scholar] [CrossRef]
- Mahdjour, S.; Harche-Kaid, M.; Haidour, A.; Chahboun, R.; Alvarez-Manzaneda, E. Short Route to Cassane-Type Diterpenoids: Synthesis of the Supposed Structure of Benthaminin 1. Org. Lett. 2016, 18, 5964–5967. [Google Scholar] [CrossRef]
- Dickson, R.A.; Houghton, P.J.; Hylands, P.J. Antibacterial and Antioxidant Cassane Diterpenoids from Caesalpinia benthamiana. Phytochemistry 2007, 68, 1436–1441. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, P.; Altarejos, J.; Linares-Palomino, P.J.; Chahboun, R.; Alvarez-Manzaneda, E. Synthesis of Cassane-Type Diterpenes from Abietane Compounds: The First Synthesis of Taepeenin F. Org. Chem. Front. 2018, 5, 2537–2546. [Google Scholar] [CrossRef]
- Zentar, H.; Jannus, F.; Gutierrez, P.; Medina-O’Donnell, M.; Lupi, A.; Reyes-Zurita, F.J.; Alvarez-Manzaneda, E.; Chahboun, R. Semisynthesis and Evaluation of Anti-Inflammatory Activity of the Cassane-Type Diterpenoid Taepeenin F and of Some Synthetic Intermediates. J. Nat. Prod. 2022, 85, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Raksat, A.; Choodej, S.; Aree, T.; Ebrahimi, S.N.; Pudhom, K. Cassane-Type Diterpenes from Roots of Pterolobium macropterum and Their Anti-Inflammatory Activity. Phytochemistry 2022, 196, 113074. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Y.R.; Yin, Y.; Song, K.R.; Long, L.P.; Li, X.Z.; Zhou, B.; Gao, H.Y. New Cassane Diterpenoids from the Seed Kernels of Caesalpinia cucullata, Exhibit Anti-Inflammatory Effect in Vitro by Inhibiting INOS Enzymatic Activity. Chin. J. Chem. 2021, 39, 1625–1634. [Google Scholar] [CrossRef]
- Zentar, H.; Jannus, F.; Medina-O’Donnell, M.; Lupi, A.; Justicia, J.; Alvarez-Manzaneda, R.; Reyes-Zurita, F.J.; Alvarez-Manzaneda, E.; Chahboun, R. Synthesis and Biological Evaluation of Cassane Diterpene (5α)-Vuacapane-8(14), 9(11)-Diene and of Some Related Compounds. Molecules 2022, 27, 5705. [Google Scholar] [CrossRef]
- Wender, P.A.; Verma, V.A.; Paxton, T.J.; Pillow, T.H. Function-Oriented Synthesis, Step Economy, and Drug Design. Acc. Chem. Res. 2008, 41, 40–49. [Google Scholar] [CrossRef]
- Chatzopoulou, M.; Antoniou, A.; Pitsinos, E.N.; Bantzi, M.; Koulocheri, D.; Haroutounian, S.A.; Giannis, A. A Fast Entry to Furanoditerpenoid-Based Hedgehog Signaling Inhibitors: Identifying Essential Structural Features. Org. Lett. 2014, 16, 3344–3347. [Google Scholar] [CrossRef]
- Antoniou, A.; Chatzopoulou, M.; Bantzi, M.; Athanassopoulos, C.M.; Giannis, A.; Pitsinos, E.N. Identification of Gli-Mediated Transcription Inhibitors through Synthesis and Evaluation of Taepeenin D Analogues. MedChemComm 2016, 7, 2328–2331. [Google Scholar] [CrossRef]
- Huynh, K.Q.; Seizert, C.A.; Ozumerzifon, T.J.; Allegretti, P.A. Platinum-Catalyzed α,β-Unsaturated Carbene Formation in the Formal Syntheses of Frondosin B and Liphagal. Org. Lett. 2017, 19, 294–297. [Google Scholar] [CrossRef]
- Hollinshead, D.M.; Howell, S.C.; Ley, S.V.; Mahon, M.; Ratcliffe, N.M.; Worthington, P.A. The Diels-Alder Route to Drimane Related Sesquiterpenes; Synthesis of Cinnamolide, Polygodial, Isodrimeninol, Drimenin and Warburganal. J. Chem. Soc. Perkin Trans. 1983, 1, 1579–1588. [Google Scholar] [CrossRef]
- Loperfido, J.C. Pyrolytic Aromatization of Dimethyl 3,5,6,7,8,8a-Hexahydro-5,5,8a-trimethyl-l,2-naphthalenedicarboxylate. J. Org. Chem. 1973, 38, 399. [Google Scholar] [CrossRef]
- Tanis, S.P.; Abdallah, Y.M. A Study of the Diels-Alder Regioselectivity of Substituted Vinyl Cyclohexenes. Synth. Commun. 1986, 16, 251–259. [Google Scholar] [CrossRef]
- Abad, A.; Agulló, C.; Castelblanque, L.; Cuñat, A.C.; Navarro, I.; Ramírez de Arellano, M.C. Synthesis of Terpenoid Unsaturated 1,4-Dialdehydes. π-Facial Selectivity in the Diels-Alder Reaction of the 1-Vinyl-2-Methylcyclohexene Moiety of Polycyclic Systems with DMAD. J. Org. Chem. 2000, 65, 4189–4192. [Google Scholar] [CrossRef] [PubMed]
- Tanis, S.P.; Nakanishi, K. Stereospecific total synthesis of (+/−)-warburganal and related compounds. J. Am. Chem. Soc. 1979, 101, 4398–4400. [Google Scholar] [CrossRef]
- Li, H.P.; Ai, H.J.; Qi, X.; Peng, J.B.; Wu, X.F. Palladium-Catalyzed Carbonylative Synthesis of Benzofuran-2(3H)-ones from 2-Hydroxybenzyl Alcohols Using Formic Acid as the CO Source. Org. Biomol. Chem. 2017, 15, 1343–1345. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Kone, B.C.; Kuncewicz, T.; Zhang, W.; Yu, Z. Protein Interactions with Nitric Oxide Synthases: Controlling the Right Time, the Right Place, and the Right Amount of Nitric Oxide. Am. J. Physiol. Renal Physiol. 2023, 285, 178–190. [Google Scholar] [CrossRef]
- Elisia, I.; Pae, H.B.; Lam, V.; Cederberg, R.; Hofs, E.; Krystal, G. Comparison of RAW264. 7, Human Whole Blood and PBMC Assays to Screen for Immunomodulators. J. Immunol. Methods 2018, 452, 26–31. [Google Scholar] [CrossRef]
- Taciak, B.; Białasek, M.; Braniewska, A.; Sas, Z.; Sawicka, P.; Kiraga, Ł.; Rygiel, T.; Król, M. Evaluation of Phenotypic and Functional Stability of RAW 264.7 Cell Line through Serial Passages. PLoS ONE 2018, 13, e0198943. [Google Scholar] [CrossRef]
- Bryan, N.S.; Grisham, M.B. Methods to Detect Nitric Oxide and Its Metabolites in Biological Samples. Free. Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef]
- Galisteo, A.; Jannus, F.; García-García, A.; Aheget, H.; Rojas, S.; Lupiañez, J.A.; Rodríguez-Diéguez, A.; Reyes-Zurita, F.J.; del Moral, F.J.Q. Diclofenac N-Derivatives as Therapeutic Agents with Anti-Inflammatory and Anti-Cancer Effect. Int. J. Mol. Sci. 2021, 22, 5067. [Google Scholar] [CrossRef] [PubMed]
- Navas, A.; Jannus, F.; Cepeda, J.; O’Donnell, M.M.; Díaz-Ruiz, L.; Cristina, S.; Llopis, J.; Seco, J.M.; Rufino-Palomares, E.; Lupiáñez, J.A. Designing Single-Molecule Magnets as Drugs with Dual Anti-Inflammatory and Anti-Diabetic Effects. Int. J. Mol. Sci. 2020, 21, 3146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Chen, Z.H.; Xu, J.; Li, J.; Tan, Y.F.; Zhou, J.H.; Ye, W.C.; Tian, H.Y.; Jiang, R.W. New Structures, Chemotaxonomic Significance and COX-2 Inhibitory Activities of Cassane-Type Diterpenoids from The Seeds of Caesalpinia minax. RSC Adv. 2015, 5, 76567–76574. [Google Scholar] [CrossRef]
- Overbeeke, R.; Steffens-Nakken, H.; Vermes, I.; Reutelingsperger, C.; Haanen, C. Early Features of Apoptosis Detected by Four Different Flow Cytometry Assays. Apoptosis 1998, 3, 115–121. [Google Scholar] [CrossRef]
- Vermes, I.; Haanen, C.; Steffens-Nakken, H.; Reutellingsperger, C. Novel Assay for Apoptosis Flow Cytometric Detection of Phosphatidylserine Expression on Early Apoptotic Cells Using Fluorescein Labelled Annexin V. J. Immunol. Methods 1995, 184, 39–51. [Google Scholar] [CrossRef]
- Heiskanen, K.M.; Bhat, M.B.; Wang, H.W.; Ma, J.; Nieminen, A.L. Mitochondrial Depolarization Accompanies Cytochrome cRelease during Apoptosis in PC6 Cells. J. Biol. Chem. 1999, 274, 5654–5658. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Diguiseppi, J.; Nieminen, A.L.; Herman, B. Blebbing, Free Ca2+ and Mitochondrial Membrane Potential Preceding Cell Death in Hepatocytes. Nature 1987, 325, 5–8. [Google Scholar] [CrossRef]
- Galluzzi, L.; Zamzami, N.; Rouge, T.D.L.M.; Lemaire, C.; Brenner, C.; Kroemer, G. Methods for The Assessment of Mitochondrial Membrane Permeabilization in Apoptosis. Apoptosis 2007, 12, 803–813. [Google Scholar] [CrossRef]
- Emaus, R.K.; Grunwald, R.; Lemasters, J.J. Rhodamine 123 as a Probe of Transmembrane Potential in Isolated Rat-Liver Mitochondria: Spectral and Metabolic Properties. Biochim. Biophys. Acta. 1986, 850, 436–448. [Google Scholar] [CrossRef]
- Reyes, F.J.; Centelles, J.J.; Lupiáñez, J.A.; Cascante, M. (2α, 3β)-2, 3-Dihydroxyolean-12-en-28-oic Acid, a New Natural Triterpene from Olea europea, Induces Caspase Dependent Apoptosis Selectively in Colon Adenocarcinoma Cells. FEBS Lett. 2006, 580, 6302–6310. [Google Scholar] [CrossRef] [PubMed]
- Senwar, K.R.; Sharma, P.; Reddy, T.S.; Jeengar, M.K.; Nayak, V.L.; Naidu, V.G.M.; Kamal, A.; Shankaraiah, N. Spirooxindole-derived Morpholine-Fused-1, 2, 3-Triazoles: Design, Synthesis, Cytotoxicity, and Apoptosis Inducing Studies. Eur. J. Med. Chem. 2015, 102, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The Concept of Intrinsic versus Extrinsic Apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Sayers, T.J. Targeting the Extrinsic Apoptosis Signaling Pathway for Cancer Therapy. Cancer Immunol. Immunother. 2011, 60, 1173–1180. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K. Extrinsic versus Intrinsic Apoptosis Pathways in Anticancer Chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Suhaili, S.H.; Karimian, H.; Stellato, M.; Lee, T.H. Mitochondrial Outer Membrane Permeabilization: A Focus on The Role of Mitochondrial Membrane Structural Organization. Biophys. Rev. 2017, 9, 443–457. [Google Scholar] [CrossRef]
- Rajput, M.S.; Nirmal, N.P.; Nirmal, S.J.; Santivarangkna, C. Bio-actives from Caesalpinia sappan L.: Recent Advancements in Phytochemistry and Pharmacology. S. Afr. J. Bot. 2022, 151, 60–74. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, M.; Yan, Y.; Zhang, X.X.; Li, X.Z.; Gao, H.Y. Bridged Cassane Derivatives from The Seeds of Caesalpinia sappan L. and Their Cytotoxic Activities. Phytochemistry 2022, 197, 113111. [Google Scholar] [CrossRef]
- Huang, G.D.; Chen, F.F.; Ma, G.X.; Li, W.P.; Zheng, Y.Y.; Meng, X.B.; Li, Z.Y.; Chen, L. Cassane Diterpenoid Derivative Induces Apoptosis in IDH1 Mutant Glioma Cells through The Inhibition of Glutaminase in Vitro and in Vivo. Phytomedicine 2021, 82, 153434. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, L.L.; Liu, Q.Y.; Feng, L.; Ye, Y.; Lu, J.J.; Lin, L.G. Cytotoxic and Pro-Apoptotic Effects of Cassane Diterpenoids from The Seeds of Caesalpinia sappan in Cancer Cells. Molecules 2016, 21, 791–804. [Google Scholar] [CrossRef]
- Li, Z.Y.; Li, Q.Z.; Ma, G.X.; Chen, L.; Zhang, C.; Chen, B.D.; Yang, J.S.; Li, W.P. Cassane-Type Diterpenes from Caesalpinia minax Induce Apoptosis in Pituitary Adenoma: Structure–Activity Relationship, ER Stress and Wnt/β-Catenin Pathways. J. Asian Nat. Prod. Res. 2016, 19, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Tran, M.H.; Nguyen, M.T.T.; Nguyen, H.D.; Nguyen, T.D.; Phuong, T.T. Cytotoxic Constituents from The Seeds ff Vietnamese Caesalpinia sappan. Pharm. Biol. 2015, 53, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, J.; Horiguchi, K.; Wong, C.P.; Hosoya, T.; Iihoshi, A.; Kaneda, T.; Morita, H. Sucutinirane-Diterpene Derivatives Induce Apoptosis via Oxidative Stress in HL-60 Cells. J. Nat. Med. 2014, 68, 723–729. [Google Scholar] [CrossRef]
- Kim, H.J.; Seo, B.G.; Kim, K.D.; Yoo, J.; Lee, J.H.; Min, B.S.; Lee, J.H.; Hwangbo, C. C5, a Cassaine Diterpenoid Amine, Induces Apoptosis via The Extrinsic Pathways in Human Lung Cancer Cells and Human Lymphoma Cells. Int. J. Mol. Sci. 2020, 21, 1298. [Google Scholar] [CrossRef]
- Zhang, H.; Zan, J.; Yu, G.; Jiang, M.; Liu, P. A Combination of 3D-QSAR, Molecular Docking and Molecular Dynamics Simulation Studies of Benzimidazole-Quinolinone Derivatives as Inos Inhibitors. Int. J. Mol. Sci. 2012, 13, 11210–11227. [Google Scholar] [CrossRef]
- Garcin, E.D.; Arvai, A.S.; Rosenfeld, R.J.; Kroeger, M.D.; Crane, B.R.; Andersson, G.; Andrews, G.; Hamley, P.J.; Mallinder, P.R.; Nicholls, D.J.; et al. Anchored Plasticity Opens Doors for Selective Inhibitor Design in Nitric Oxide Synthase. Nat. Chem. Biol. 2008, 4, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Minhas, R.; Bansal, Y.; Bansal, G. Inducible Nitric Oxide Synthase Inhibitors: A Comprehensive Update. Med. Res. Rev. 2019, 40, 823–855. [Google Scholar] [CrossRef] [PubMed]
- Badroon, N.; Majid, N.A.; Al-suede, F.S.R.; Nazari, V.M.; Giribabu, N.; Majid, A.M.S.A.; Eid, E.E.M.; Alshawsh, M.A. Cardamonin Exerts Antitumor Effect on Human Hepatocellular Carcinoma Xenografts in Athymic Nude Mice through Inhibiting NF-κβ Pathway. Biomedicines 2020, 8, 586. [Google Scholar] [CrossRef]
- Janowska, S.; Khylyuk, D.; Bielawska, A.; Szymanowska, A.; Gornowicz, A.; Bielawski, K.; Noworól, J.; Mandziuk, S.; Wujec, M. New 1, 3, 4-Thiadiazole Derivatives with Anticancer Activity. Molecules 2022, 27, 1814. [Google Scholar] [CrossRef]
- He, B.; Zhu, Z.; Zhang, T.; Chen, F.; Zhang, R.; Chen, W.; Zhang, T.; Wang, T.; Lei, J. Synthesis and Antitumor Potential of New Arylidene Ursolic Acid Derivatives via Caspase-8 Activation. Archiv. Der. Pharm. 2021, 354, 2000448. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | IC50 (μg/mL) | ¼ IC50 (μg/mL) | ½ IC50 (μg/mL) | ¾ IC50 (μg/mL) |
|---|---|---|---|---|
| 10 | 69.86 ± 0.21 | 17.46 ± 0.57 | 34.93 ± 0.33 | 52.39 ± 0.33 |
| 11 | 81.10 ± 0.93 | 20.27 ± 1.67 | 40.55 ± 0.55 | 60.82 ± 0.55 |
| 12 | 71.67 ± 0.87 | 17.91 ± 3.03 | 35.83 ± 0.43 | 53.75 ± 0.43 |
| 13 | 90.14 ± 1.26 | 22.53 ± 2.77 | 45.07 ± 0.34 | 67.60 ± 0.34 |
| 14 | 47.56 ± 0.25 | 11.89 ± 0.62 | 23.87 ± 1.63 | 35.67 ± 1.63 |
| 15 | 3.98 ± 0.94 | 0.99 ± 0.25 | 1.99 ± 0.27 | 2.98 ± 0.27 |
| 16 | 35.24 ± 0.94 | 8.81 ± 1.97 | 17.62 ± 0.26 | 26.43 ± 0.26 |
| Pterolobirin H (3) | 45.34 ± 1.33 | 11.33 ± 0.36 | 22.67 ± 0.77 | 34.00 ± 1.02 |
| Product | IC50 NO (μg/mL) | IC50 NO DCF/ IC50 NO Compound # |
|---|---|---|
| DCF | 47.12 ± 4.85 | 1.0 |
| 10 | 20.37 ± 0.01 | 2.32 |
| 11 | 20.34 ± 0.30 | 2.32 |
| 12 | 19.44 ± 0.12 | 2.42 |
| 13 | 27.60 ± 0.13 | 1.71 |
| 14 | 11.18 ± 0.09 | 4.21 |
| 15 | 0.62 ± 0.21 | 76.0 |
| 16 | 7.49 ± 0.15 | 6.29 |
| Pterolobirin H (3) | 17.57 ± 0.31 | 2.68 |
| Product | IC50 (μg/mL) | ||
|---|---|---|---|
| HT29 | Hep-G2 | B16–F10 | |
| 10 | 30.38 ± 2.53 | 52.86 ± 0.07 | 45.26 ± 1.87 |
| 11 | 93.24 ± 1.86 | 69.97 ± 3.40 | N/A |
| 12 | N/A | 78.42 ± 1.07 | 69.89 ± 3.17 |
| 13 | 92.94 ± 0.79 | 66.28 ± 0.07 | 53.24 ± 1.05 |
| 14 | 32.15 ± 0.40 | 38.27 ± 0.67 | 43.40 ± 10.42 |
| 15 | 2.45 ± 0.29 | 3.25 ± 0.33 | 3.84 ± 0.45 |
| 16 | 19.20 ± 0.34 | 27.08 ± 1.39 | 27.06 ± 1.31 |
| Pterolobirin H (3) | 70.07 ± 1.57 | 45.31 ± 1.81 | 39.71 ± 0.28 |
| Protein | Product | Estimated Binding Energy (kcal/mol) | Hydrogen Bonds Interactions | Electrostatic Interactions | Hydrophobic Interactions |
|---|---|---|---|---|---|
| iNOS (4UX6) | 3 | −7.89 | HEM | PRO344 (x3) HEM (x6) VAL346 TYR367 | |
| iNOS (4UX6) | 15 | −8.15 | GLU371 (x2) TYR367 | - | PRO344 (x2) HEM (x5) VAL346 (x2) |
| iNOS (4UX6) | 16 | −7.61 | - | - | PRO344 (x4) HEM (x5) VAL346 (x3) TYR367 |
| Caspase 8 (3KJN) | 15 | −7.07 | ARG260 SER316 (x2) GLN358 | ARG413 | CYS360 ARG413 HIS317 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zentar, H.; Jannus, F.; Medina-O’Donnell, M.; El Mansouri, A.-e.; Fernández, A.; Justicia, J.; Alvarez-Manzaneda, E.; Reyes-Zurita, F.J.; Chahboun, R. Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations. Molecules 2023, 28, 6208. https://doi.org/10.3390/molecules28176208
Zentar H, Jannus F, Medina-O’Donnell M, El Mansouri A-e, Fernández A, Justicia J, Alvarez-Manzaneda E, Reyes-Zurita FJ, Chahboun R. Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations. Molecules. 2023; 28(17):6208. https://doi.org/10.3390/molecules28176208
Chicago/Turabian StyleZentar, Houda, Fatin Jannus, Marta Medina-O’Donnell, Az-eddine El Mansouri, Antonio Fernández, José Justicia, Enrique Alvarez-Manzaneda, Fernando J. Reyes-Zurita, and Rachid Chahboun. 2023. "Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations" Molecules 28, no. 17: 6208. https://doi.org/10.3390/molecules28176208
APA StyleZentar, H., Jannus, F., Medina-O’Donnell, M., El Mansouri, A.-e., Fernández, A., Justicia, J., Alvarez-Manzaneda, E., Reyes-Zurita, F. J., & Chahboun, R. (2023). Synthesis of Tricyclic Pterolobirin H Analogue: Evaluation of Anticancer and Anti-Inflammatory Activities and Molecular Docking Investigations. Molecules, 28(17), 6208. https://doi.org/10.3390/molecules28176208