
Preparation of Novel Solid Phase Extraction Sorbents for Polycyclic Aromatic Hydrocarbons (PAHs) in Aqueous Media


 and
and
Abstract
:1. Introduction
2. Results and Discussion
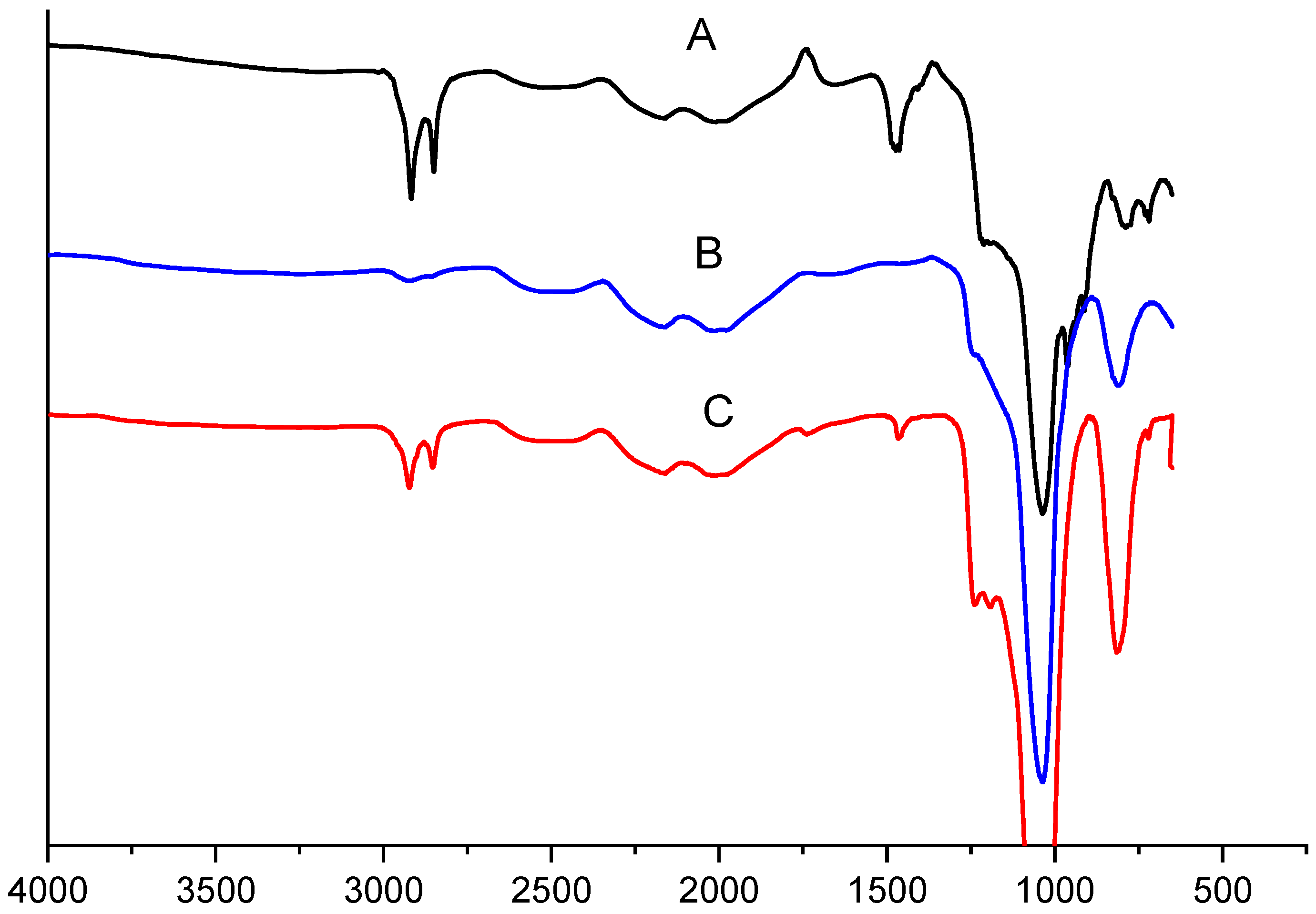
2.1. FTIR Measurements
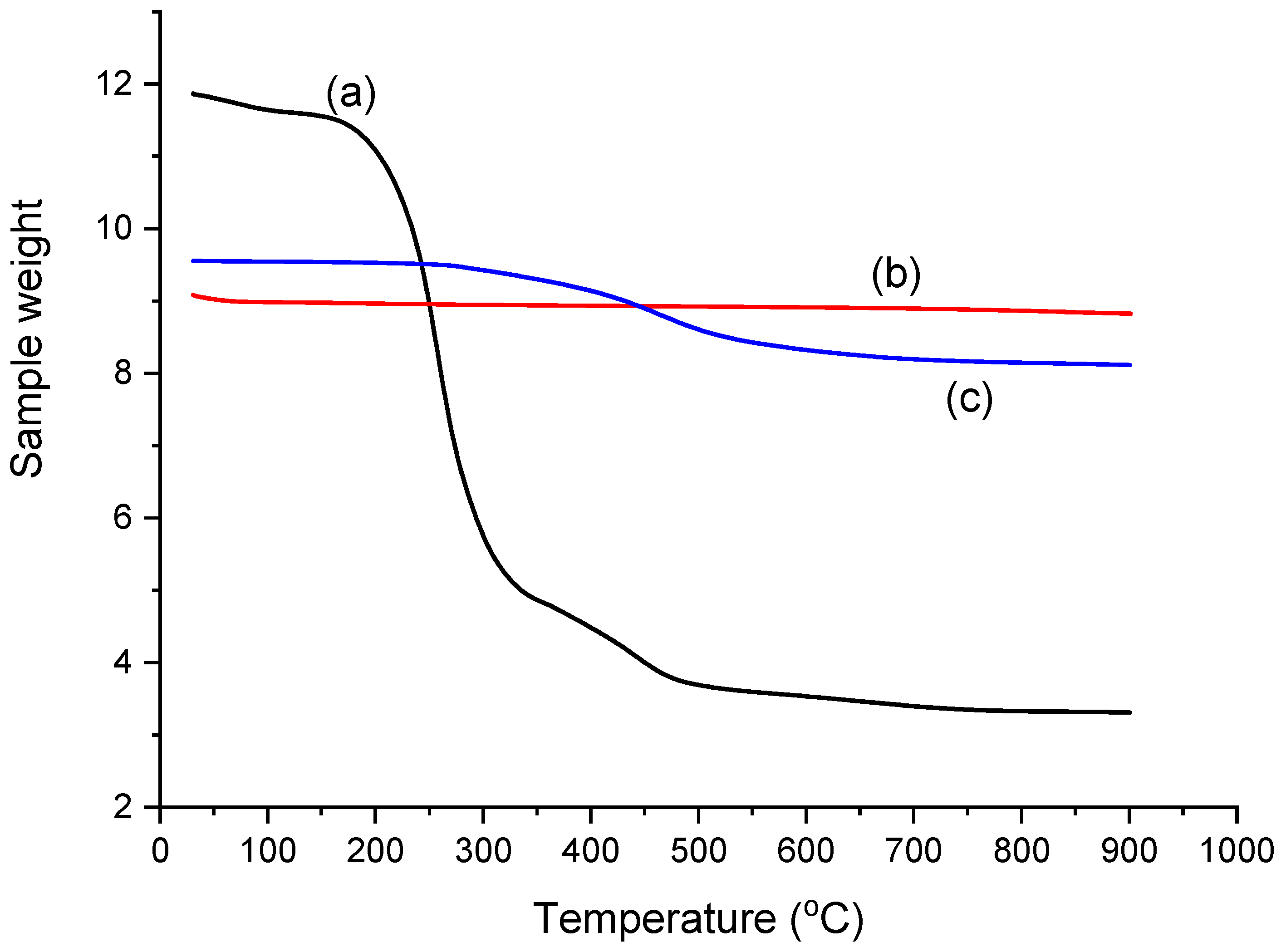
2.2. Thermogravimetric Analysis
2.3. Nitrogen Sorption Studies
2.3.1. BET Surface Area
2.3.2. Pore Size Distribution
2.4. Method Performance and Validation
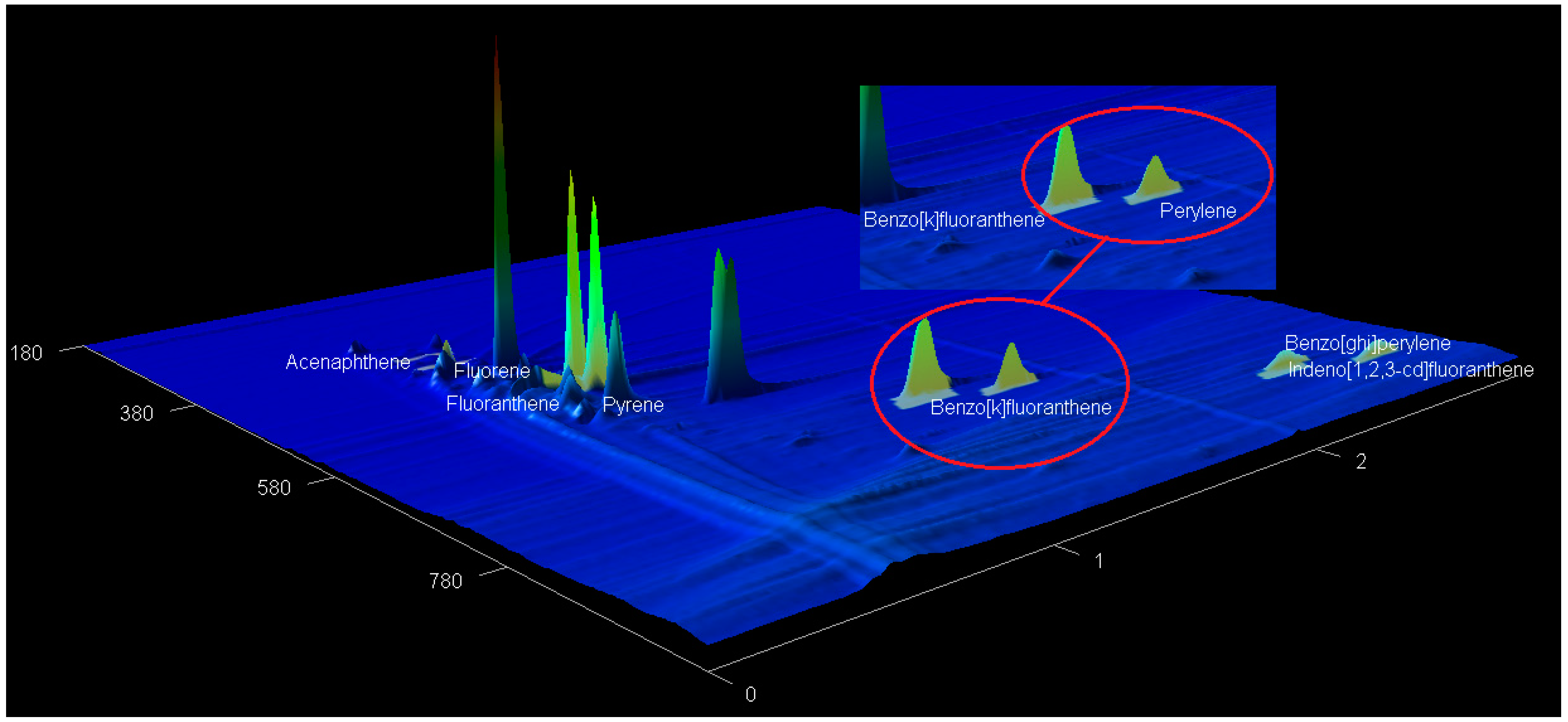
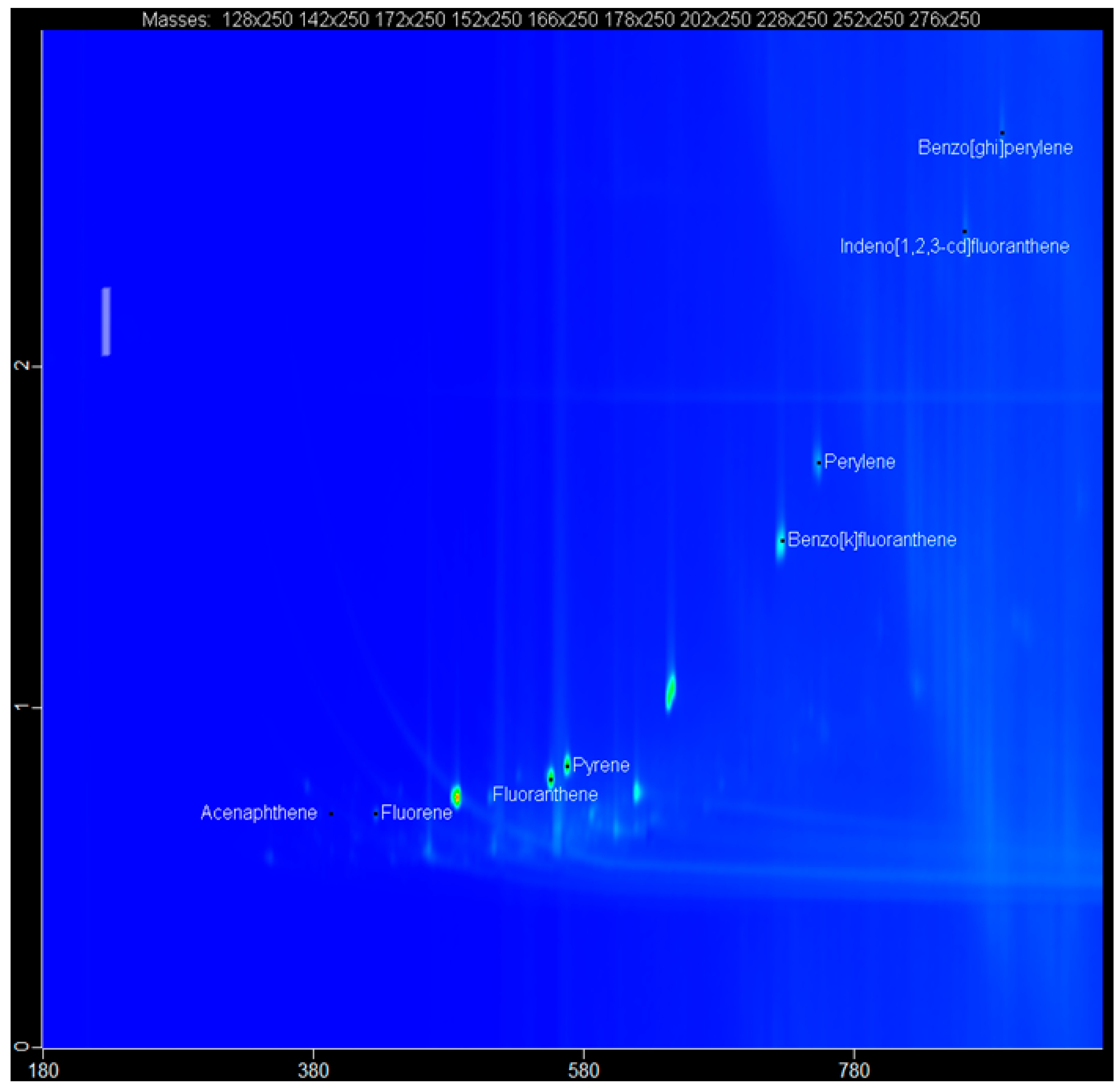
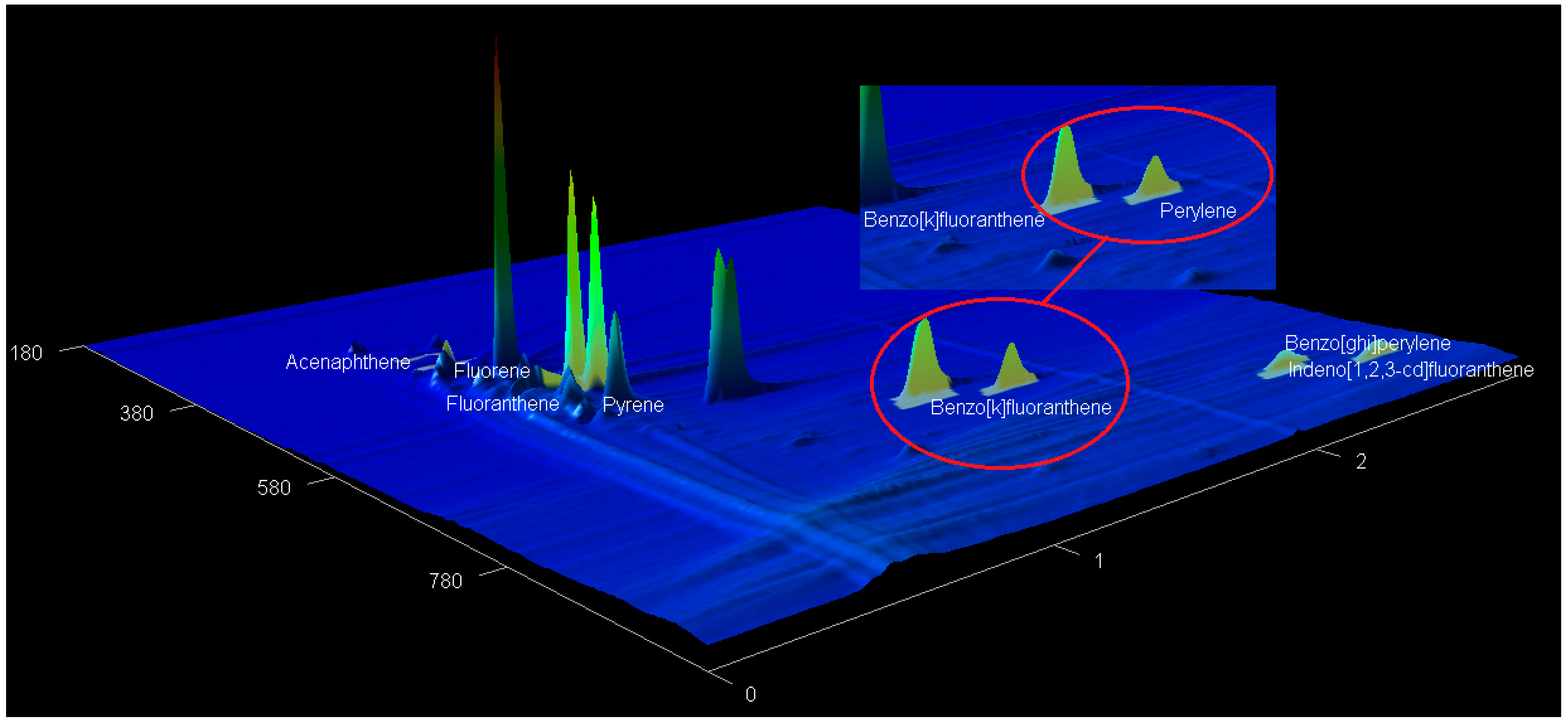
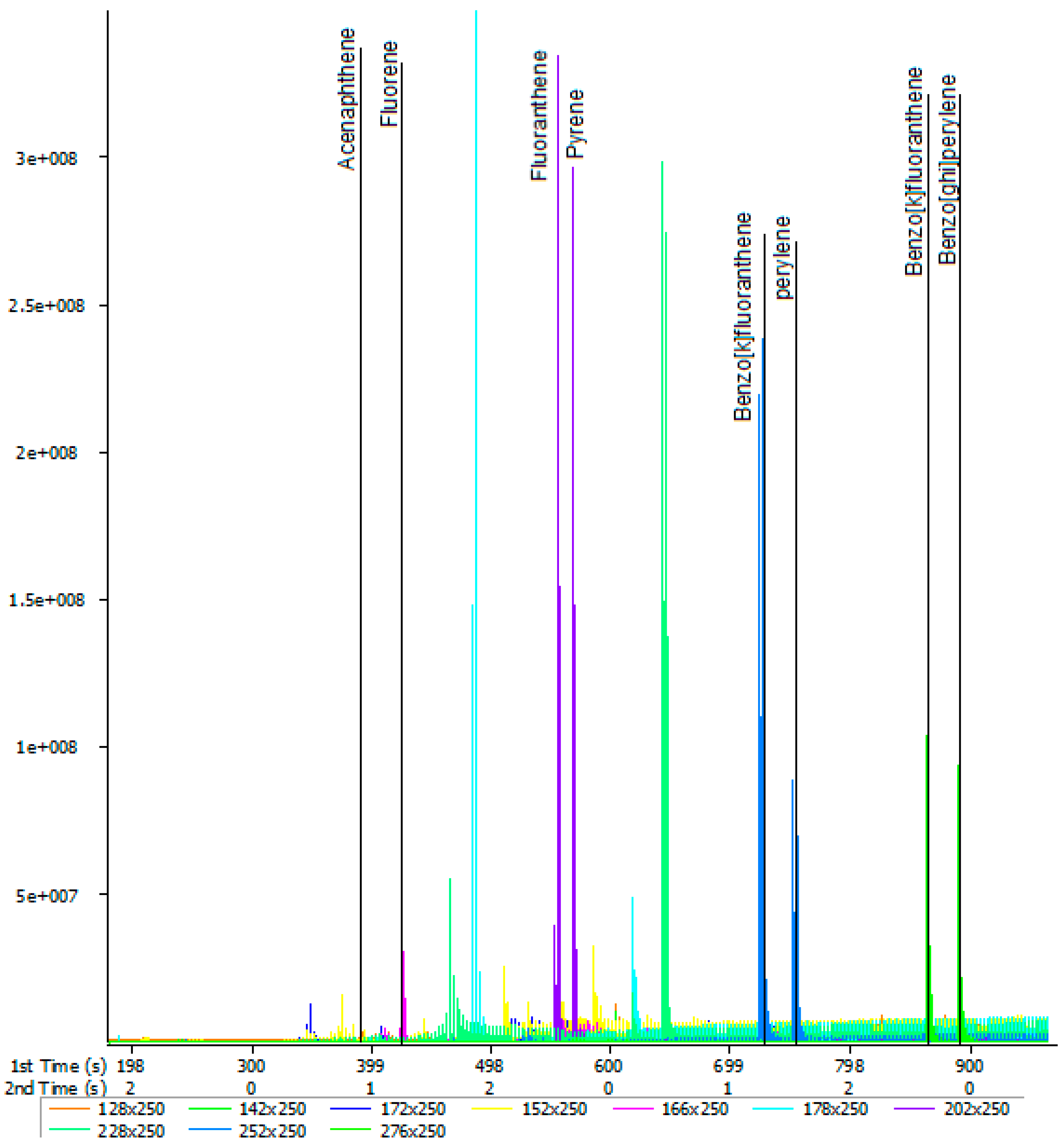
2.5. The GCxGC-TOF-MS Chromatograms of Acp, Flu, FL, Pyr, BkF, Per, InF, and BghiP
2.6. Applications on Real Water Samples
3. Experimental Section
3.1. Standard and Reagents
3.2. Sampling
3.3. Synthesis and Functionalization
3.3.1. Synthesis of Silica Materials
3.3.2. Functionalization of Silica Materials
3.4. Characterization of Silica Materials
3.4.1. FTIR Measurements
3.4.2. Thermogravimetric Analyser (TGA)
3.4.3. Nitrogen Sorption Studies
3.5. Preparation of SPE Cartridges
3.6. Preparation of Calibration Solutions
3.7. SPE Extraction Procedure
3.7.1. Extraction of Spiked Water Samples
3.7.2. Applications on Real Water Samples
3.8. Instrument and Analytical Conditions
3.9. Determination of Limits of Quantification (LOQ), Limits of Detection (LOD) and Linearity of PAHs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hoffmann, F.; Morell, J.; Fröba, M. Silica-based mesoporous organic-inorganic hybrid materials. Angew. Chemie Int. Ed. 2006, 45, 3216–3251. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Qi, L.; Ma, J.; Wu, Y.; Liu, O.; Cheng, H. Large-pore mesoporous silica spheres: Synthesis and application in HPLC. Colloids Surf. A Physicochem. Eng. Asp. 2003, 229, 1–8. [Google Scholar] [CrossRef]
- Liu, X.; Hua, Y.; Villemure, G. Preparation and characterization of thin films of amine functionalised mesoporous silica having cubic pore structures and their use for electrode surface modifications. Microporous Mesoporous Mater. 2009, 117, 317–325. [Google Scholar] [CrossRef]
- Van Der Voort, P.; Esquivel, D.; De Canck, E.; Goethals, F.; Van Driessche, I.; Romero-Salguero, F.J. Periodic mesoporous organosilicas: From simple to complex bridges; A comprehensive overview of functions, morphologies and applications. Chem. Soc. Rev. 2013, 42, 3913–3955. [Google Scholar] [CrossRef]
- García-Peñas, A.; Gómez-Ruiz, S.; Pérez-Quintanilla, D.; Paschke, R.; Sierra, I.; Prashar, S.; del Hierro, I.; Kaluđerović, G.N. Study of the cytotoxicity and particle action in human cancer cells of titanocene-functionalized materials with potential application against tumors. J. Inorg. Biochem. 2012, 106, 100–110. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, S.; Pérez-Quintanilla, D.; Gómez-Ruiz, S. Synthesis and photocatalytic applications of nano-sized zinc-doped mesoporous titanium oxide. Mater. Res. Bull. 2013, 48, 250–255. [Google Scholar] [CrossRef]
- Pérez, Y.; Quintanilla, D.P.; Fajardo, M.; Sierra, I.; del Hierro, I. Immobilization of titanium chiral alkoxides on SBA-15 and modelling the active sites of heterogeneous catalyst using titanium silsesquioxane complexes. J. Mol. Catal. A Chem. 2007, 271, 227–237. [Google Scholar] [CrossRef]
- Ciesla, U.; Schüth, F. Ordered mesoporous materials. Microporous Mesoporous Mater. 1999, 27, 131–149. [Google Scholar] [CrossRef]
- Trewyn, B.G.; Slowing, I.I.; Giri, S.; Chen, H.T.; Lin, V.S.Y. Synthesis and functionalization of a mesoporous silica nanoparticle based on the sol-gel process and applications in controlled release. Acc. Chem. Res. 2007, 40, 846–853. [Google Scholar] [CrossRef]
- Taib, N.I.; Endud, S.; Katun, M.N. Functionalization of Mesoporous Si-MCM-41 by Grafting with Trimethylchlorosilane. Int. J. Chem. 2011, 3, 2. [Google Scholar] [CrossRef]
- Neimark, A.V.; Ravikovitch, P.I.; Grün, M.; Schüth, F.; Unger, K.K. Pore size analysis of MCM-41 type adsorbents by means of nitrogen and argon adsorption. J. Colloid Interface Sci. 1998, 207, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, T.; Müller, K. Synthesis and characterization of surface modified SBA-15 silica materials and their application in chromatography. J. Chromatogr. A 2011, 1218, 6464–6475. [Google Scholar] [CrossRef] [PubMed]
- Mureseanu, M.; Reiss, A.; Cioatera, N.; Trandafir, I.; Hulea, V. Mesoporous silica functionalized with 1-furoyl thiourea urea for Hg(II) adsorption from aqueous media. J. Hazard. Mater. 2010, 182, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jeon, J.B.; Chang, J.Y. Selectively functionalized mesoporous silica particles with the PEGylated outer surface and the doxorubicin-grafted inner surface: Improvement of loading content and solubility. Microporous Mesoporous Mater. 2013, 172, 118–124. [Google Scholar] [CrossRef]
- Sánchez, A.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I.; Del Hierro, I. Determination of Hg(II) in natural waters using a carbon paste electrode modified with hybrid mesostructured silica nanoparticles. Sens. Actuators B Chem. 2012, 163, 38–43. [Google Scholar] [CrossRef]
- Sánchez, A.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Del Hierro, I.; Sierra, I. A comparative study on carbon paste electrodes modified with hybrid mesoporous materials for voltammetric analysis of lead (II). J. Electroanal. Chem. 2013, 689, 76–82. [Google Scholar] [CrossRef]
- Mureseanu, M.; Reiss, A.; Stefanescu, I.; David, E.; Parvulescu, V.; Renard, G.; Hulea, V. Modified SBA-15 mesoporous silica for heavy metal ions remediation. Chemosphere 2008, 73, 1499–1504. [Google Scholar] [CrossRef]
- Pal, N.; Bhaumik, A. Soft templating strategies for the synthesis of mesoporous materials: Inorganic, organic-inorganic hybrid and purely organic solids. Adv. Colloid Interface Sci. 2013, 189, 21–41. [Google Scholar] [CrossRef]
- Yasmin, T.; Müller, K. Synthesis and surface modification of mesoporous mcm-41 silica materials. J. Chromatogr. A 2010, 1217, 3362–3374. [Google Scholar] [CrossRef]
- Titato, G.M.; Lanças, F.M. Optimization and validation of HPLC-UV-DAD and HPLC-APCI-MS methodologies for the determination of selected PAHs in water samples. J. Chromatogr. Sci. 2006, 44, 35–40. [Google Scholar] [CrossRef]
- Gañán, J.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Comparison of different mesoporous silicas for off-line solid phase extraction of 17β-estradiol from waters and its determination by HPLC-DAD. J. Hazard. Mater. 2013, 260, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Bispo, J.R.L.; Navickiene, S.; Dórea, H.S. Method Validation for SPE Applied to Determination of PAH in Petroliferous Industry Effluent Water. Am. J. Anal. Chem. 2011, 2, 971. [Google Scholar] [CrossRef]
- Sibiya, P.; Potgieter, M.; Cukrowska, E.; Jönsson, J.Å.; Chimuka, L. Development and Application of Solid Phase Extraction Method for Polycyclic Aromatic Hydrocarbons in Water Samples in Johannesburg Area, South Africa. S. Afr. J. Chem. 2012, 65, 206–213. [Google Scholar]
- Kanchanamayoon, W.; Tatrahun, N. Determination of Polycyclic Aromatic Hydrocarbons in Water Samples by Solid Phase Extraction and Gas Chromatography. World J. Chem. 2008, 3, 51–54. [Google Scholar]
- Kiss, G.; Varga-Puchony, Z.; Hlavay, J. Determination of polycyclic aromatic hydrocarbons in precipitation using solid-phase extraction and column liquid chromatography. J. Chromatogr. A 1996, 725, 261–272. [Google Scholar] [CrossRef]
- Xie, M.X.; Xie, F.; Deng, Z.W.; Zhuang, G.S. Determination of polynuclear aromatic hydrocarbons in aerosol by solid-phase extraction and gas chromatography-mass spectrum. Talanta 2003, 60, 1245–1257. [Google Scholar] [CrossRef]
- Wang, W.; Meng, B.; Lu, X.; Liu, Y.; Tao, S. Extraction of polycyclic aromatic hydrocarbons and organochlorine pesticides from soils: A comparison between Soxhlet extraction, microwave-assisted extraction and accelerated solvent extraction techniques. Anal. Chim. Acta 2007, 602, 211–222. [Google Scholar] [CrossRef]
- Bartolomé, L.; Cortazar, E.; Raposo, J.C.; Usobiaga, A.; Zuloaga, O.; Etxebarria, N.; Fernández, L.A. Simultaneous microwave-assisted extraction of polycyclic aromatic hydrocarbons, polychlorinated biphenyls, phthalate esters and nonylphenols in sediments. J. Chromatogr. A 2005, 1068, 229–236. [Google Scholar] [CrossRef]
- Letellier, M.; Budzinski, H. Microwave assisted extraction of organic compounds. Analusis 1999, 27, 259–270. [Google Scholar] [CrossRef]
- Pena, T.; Pensado, L.; Casais, C.; Mejuto, C.; Phan-Tan-Luu, R.; Cela, R. Optimization of a microwave-assisted extraction method for the analysis of polycyclic aromatic hydrocarbons from fish samples. J. Chromatogr. A 2006, 1121, 163–169. [Google Scholar] [CrossRef]
- Lund, M.; Duedahl-Olesen, L.; Christensen, J.H. Extraction of polycyclic aromatic hydrocarbons from smoked fish using pressurized liquid extraction with integrated fat removal. Talanta 2009, 79, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.K.; Wang, J. The accumulation of polycyclic aromatic hydrocarbons in lubricating oil over time—A comparison of supercritical fluid and liquid–liquid extraction methods. Environ. Pollut. 2001, 112, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Garcıa-Falcón, M.S.; Cancho-Grande, B.; Simal-Gándara, J. Stirring bar sorptive extraction in the determination of PAHs in drinking waters. Water Res. 2004, 38, 1679–1684. [Google Scholar] [CrossRef]
- King, A.J.; Readman, J.W.; Zhou, J.L. The application of solid-phase micro-extraction (SPME) to the analysis of polycyclic aromatic hydrocarbons (PAHs). Environ. Geochem. Health 2003, 25, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Comas, A. Analysis of Polycyclic Aromatic Hydrocarbons in Tobacco Related Samples via High Performance Liquid Chromatography and Laser Excited Time Resolved Shpol’Skii Spectroscopy. Ph.D. Thesis, University of Central Florida Orlando, Orlando, FL, USA, 2023. [Google Scholar]
- Yang, X.; Yin, Y.; Zong, Y.; Wan, T.; Liao, X. Magnetic nanocomposite as sorbent for magnetic solid phase extraction coupled with high performance liquid chromatography for determination of polycyclic aromatic hydrocarbons. Microchem. J. 2019, 145, 26–34. [Google Scholar] [CrossRef]
- Kailasam, K.; Müller, K. Physico-chemical characterization of MCM-41 silica spheres made by the pseudomorphic route and grafted with octadecyl chains. J. Chromatogr. A 2008, 1191, 125. [Google Scholar] [CrossRef] [PubMed]
- Puanngam, M.; Unob, F. Preparation and use of chemically modified MCM-41 and silica gel as selective adsorbents for Hg(II) ions. J. Hazard. Mater. 2008, 154, 578–587. [Google Scholar] [CrossRef]
- Wirth, M.J.; Fatunmbi, H.O. Horizontal Polymerization of Mixed Trifunctional Silanes on Silica. 2. Application to Chromatographic Silica Gel. Anal. Chem. 1993, 65, 822–826. [Google Scholar] [CrossRef]
- Unger, K.K.; Skudas, R.; Schulte, M.M. Particle packed columns and monolithic columns in high-performance liquid chromatography-comparison and critical appraisal. J. Chromatogr. A 2008, 1184, 393–415. [Google Scholar] [CrossRef]
- Delhomme, O.; Rieb, E.; Millet, M. Solid-phase extraction and LC with fluorescence detection for analysis of PAHs in rainwater. Chromatographia 2007, 65, 163–171. [Google Scholar] [CrossRef]
- Pensado, L.; Blanco, E.; Casais, M.C.; Mejuto, M.C.; Martinez, E.; Carro, A.M.; Cela, R. Strategic sample composition in the screening of polycyclic aromatic hydrocarbons in drinking water samples using liquid chromatography with fluorimetric detection. J. Chromatogr. A 2004, 1056, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, M.; Assadi, Y.; Hosseini, M.-R.M.; Aghaee, E.; Ahmadi, F.; Berijani, S. Determination of organic compounds in water using dispersive liquid–liquid microextraction. J. Chromatogr. A 2006, 1116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Galarneau, A.; Di Renzo, F.; Brunel, D.; Fajula, F.; Heinisch, S.; Crétier, G.; Rocca, J.L. Great Improvement of Chromatographic Performance Using MCM-41 Spheres as Stationary Phase in HPLC. Chem. Mater. 2004, 16, 1725–1731. [Google Scholar] [CrossRef]
- Gañán, J.; Morante-Zarcero, S.; Pérez-Quintanilla, D.; Sierra, I. Evaluation of functionalized mesoporous silicas for reverse phase high performance liquid chromatography: An application for the separation of steroids. Microchem. J. 2014, 114, 53–58. [Google Scholar] [CrossRef]
- Wanda, E.; Nyoni, H.; Mamba, B.; Msagati, T. Occurrence of emerging micropollutants in water systems in Gauteng, Mpumalanga, and North West Provinces, South Africa. Int. J. Environ. Res. Public Health 2017, 14, 79. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | SBET (m2/g) | BJH Dpore (nm) |
|---|---|---|
| Mesoporous silica | 1321.0 | 3.8 |
| Mesoporous silica modified with n-octadecyltrimethoxysilane | 1269.4 | 2.8 |
| Mesoporous silica modified and end-capped with HMDS | 991.8 | 2.3 |
| Compound | Linearity (R2) | %RSD (n = 3) | Recovery (%) (n = 3) | LOD (µg/L) | LOQ (µg/L) |
|---|---|---|---|---|---|
| Acp | 0.99 | 8.72 | 81.53 | 0.19 | 0.58 |
| Flu | 0.98 | 6.53 | 84.53 | 1.04 | 3.14 |
| FL | 0.98 | 2.92 | 80.80 | 0.31 | 0.94 |
| Pyr | 0.99 | 8.83 | 91.07 | 0.57 | 1.72 |
| BkF | 0.98 | 5.27 | 82.47 | 0.51 | 1.53 |
| Per | 0.98 | 8.39 | 95.67 | 0.04 | 0.14 |
| InF | 0.99 | 6.52 | 89.73 | 0.02 | 0.05 |
| BghiP | 0.99 | 1.85 | 79.87 | 0.75 | 2.26 |
| Preconcentration Technique | Analytical Technique | Sample Volume (mL) | LOD (µg/L) | LOQ (µg/L) | RSD (%) | Recovery (%) | Reference |
|---|---|---|---|---|---|---|---|
| SPE | HPLC-Flu a | 50 | <0.02 | 0.7–56.2 | 2–5 | 67–99 | [41] |
| SPE | GC-MS | 10 | 50–500 | NR b | 1.56–4.47 | 87.3–97.4 | [20] |
| LLE c | HPLC-Flu | 250 | NR | 0.1–4.4 | NR | 51–104 | [42] |
| SBSE d | HPLC-Flu | 39.5 | 0.0005–0.007 | 0.001–0.022 | <9 | 43–57 | [33] |
| SPE | GC-FID e | 105 | 0.01–0.04 | 0.05–0.16 | 3.8–22.2 | 30.9–119 | [23] |
| DLLME f | GC-FID | 5 | 0.007–0.030 | 23.3–100 | 1.4–10.2 | 60–111 | [43] |
| SPE | GCxGC-TOF-MS | 10 | 0.03–0.04 | 0.05–3.14 | 1.85–8.83 | 80–96 | This work |
| Compound | Abbreviation | Surface Water (µg/L) |
|---|---|---|
| Acenaphthene | Acp | 8.57 |
| Fluorene | Flu | 4.28 |
| Fluoranthene | FL | 1.48 |
| Pyrene | Pyr | 3.13 |
| Benzo[k]fluoranthene | BkF | 5.54 |
| Perylene | Per | 0.57 |
| Indeno[1,2,3-cd]fluoranthene | InF | 8.77 |
| Benzo[ghi]perylene | BghiP | 12.31 |
| Σ PAHs | 44.67 | |
| Σ PAHs = sum of polycyclic aromatic hydrocarbons | ||
| Compound | Abbreviation | Retention Time (min) | Quantified Masses |
|---|---|---|---|
| Acenaphthene | Acp | 6.40 | 154 |
| Fluorene | Flu | 7.10 | 166 |
| Fluoranthene | FL | 9.25 | 202 |
| Pyrene | Pyr | 9.45 | 202 |
| Benzo[k]fluoranthene | BkF | 12.10 | 252 |
| Perylene | Per | 12.55 | 252 |
| Indeno[1,2,3-cd]fluoranthene | InF | 14.35 | 276 |
| Benzo[ghi]perylene | BghiP | 14.80 | 276 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiga, D.T.; Kibechu, R.W.; Mamba, B.B.; Msagati, T.A.M.; Phadi, T.T. Preparation of Novel Solid Phase Extraction Sorbents for Polycyclic Aromatic Hydrocarbons (PAHs) in Aqueous Media. Molecules 2023, 28, 6129. https://doi.org/10.3390/molecules28166129
Maiga DT, Kibechu RW, Mamba BB, Msagati TAM, Phadi TT. Preparation of Novel Solid Phase Extraction Sorbents for Polycyclic Aromatic Hydrocarbons (PAHs) in Aqueous Media. Molecules. 2023; 28(16):6129. https://doi.org/10.3390/molecules28166129
Chicago/Turabian StyleMaiga, Deogratius T., Rose W. Kibechu, Bhekie B. Mamba, Titus A. M. Msagati, and Terence T. Phadi. 2023. "Preparation of Novel Solid Phase Extraction Sorbents for Polycyclic Aromatic Hydrocarbons (PAHs) in Aqueous Media" Molecules 28, no. 16: 6129. https://doi.org/10.3390/molecules28166129
APA StyleMaiga, D. T., Kibechu, R. W., Mamba, B. B., Msagati, T. A. M., & Phadi, T. T. (2023). Preparation of Novel Solid Phase Extraction Sorbents for Polycyclic Aromatic Hydrocarbons (PAHs) in Aqueous Media. Molecules, 28(16), 6129. https://doi.org/10.3390/molecules28166129