Abstract
This study investigates the influence of the phosphorus-to-nickel (P:Ni) ratio on methanol steam reforming (MSR) over nickel phosphide catalysts using density functional theory (DFT) calculations. The catalytic behavior of Ni(111) and Ni12P5(001) surfaces was explored and contrasted to our previous results from research on Ni2P(001). The DFT-predicted barriers reveal that Ni(111) predominantly favors the methanol decomposition route, where methanol is converted into carbon monoxide through a stepwise pathway involving CH3OH* → CH3O* → CH2O* → CHO* → CO*. On the other hand, Ni12P5 with a P:Ni atomic ratio of 0.42 (5:12) exhibits a substantial increase in selectivity towards methanol steam reforming (MSR) relative to methanol decomposition. In this pathway, formaldehyde is transformed into CO2 through a sequence of reactions involving CH2O*→ H2COOH* → HCOOH* → HCOO* → CO2. The introduction of phosphorus into the catalyst alters the surface morphology and electronic structure, favoring the MSR pathway. However, with a further increase in the P:Ni atomic ratio to 0.5 (1:2) on Ni2P catalysts, the selectivity towards MSR decreases, resulting in a more balanced competition between methanol decomposition and MSR. These results highlight the significance of tuning the P:Ni atomic ratio in designing efficient catalysts for the selective production of CO2 through the MSR route, offering valuable insights into optimizing nickel phosphide catalysts for desired chemical transformations.
1. Introduction
Proton-exchange membrane (PEM) fuel cells, water splitting, new-generation batteries, and CO2 conversion hold great promise as cutting-edge energy technologies for the future [1,2,3,4,5,6]. However, PEM fuel cells face challenges with hydrogen storage [7,8,9]. Methanol steam reforming (MSR) shows great potential as a viable solution for addressing this challenge given its ability to generate hydrogen from liquid methanol, which offers advantages in terms of storage and handling compared to hydrogen gas [10,11,12]. MSR is a relatively simple process that can be integrated with PEM fuel cells, making it an attractive option for on-board hydrogen production [13,14]. Methanol can be used to produce hydrogen through two main reactions: methanol decomposition and steam reforming. In methanol decomposition, methanol is broken down into hydrogen and carbon monoxide via:
CH3OH → 2H2 + CO
To prevent CO poisoning in Pt-based fuel cells and further enhance hydrogen production, the water–gas shift (WGS) reaction is often used to further convert carbon monoxide to carbon dioxide via:
CO + H2O → H2 + CO2
Steam reforming provides a more efficient route to produce hydrogen directly from methanol:
thus eliminating CO production while producing a high yield of hydrogen [13,15,16,17,18,19,20,21,22].
CH3OH + H2O → 3H2 + CO2
Cu-based catalysts have been considered the traditional catalysts for MSR [9], but they face challenges related to rapid deactivation and inadequate thermal stability [22,23,24]. Recent studies have investigated other metal-based catalysts, such as Pd/ZnO and PdZn alloys [18,19], that have shown higher selectivity towards CO2 but lower catalytic activity [25,26,27,28]. The primary goal of catalyst performance enhancement for MSR is to increase the rate of hydrogen production and improve CO2 selectivity while using a thermally stable material. Transition metal phosphides (TMPs) have emerged recently as a highly versatile class of catalysts which can be used for a wide range of applications due to their unique selectivity and resistance to coke formation [29,30,31,32,33]. Among those TMPs, nickel phosphides (NixPy) have been previously examined for C–O bond cleavage in biomass-derived molecules [34,35]. These studies show that the addition of P atoms to Ni promotes cleaving the tertiary bond (3C–O) in 2-methyltetrahydrofuran relative to pure Ni in which cleaving the secondary bond (2C–O) is much more facile. Increasing the P:Ni atomic ratio (i.e., Ni → Ni12P5 → Ni2P) increases the selectivity towards 3C–O bond cleavage. The geometric or electronic effects caused by the incorporation of P atoms with Ni appear to play a role in this unique selectivity to activate hindered C–O bonds [36].
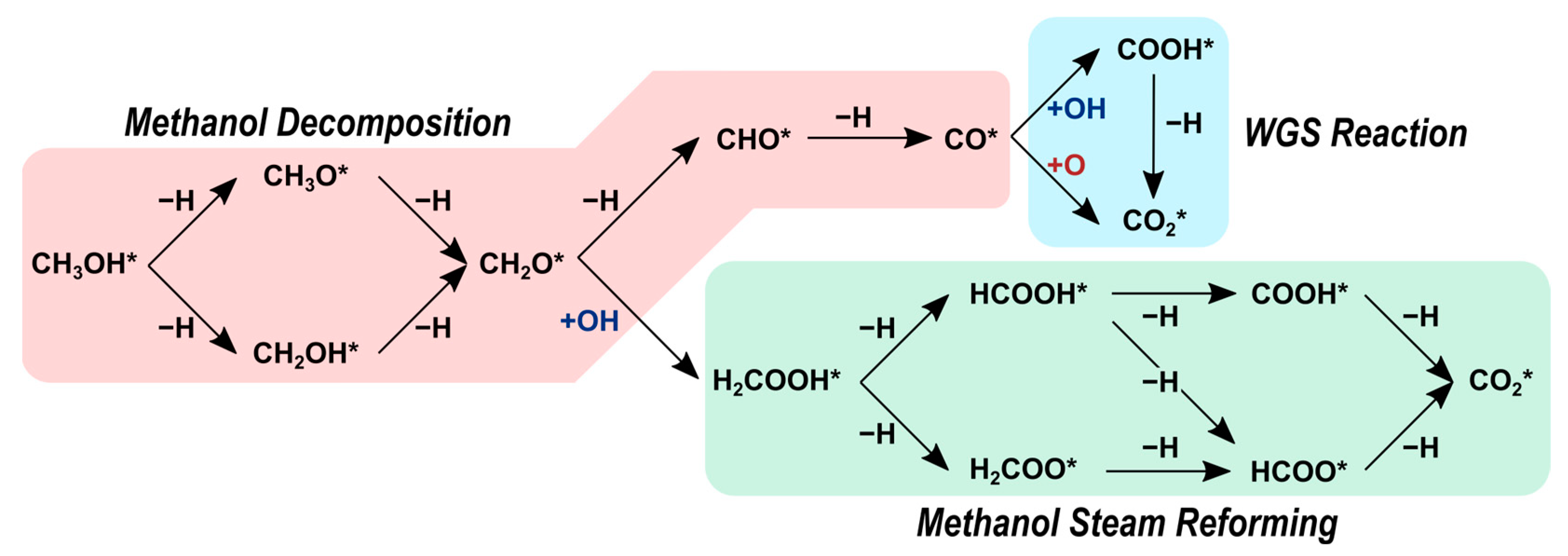
In our previous study, we explored the reaction network shown in Scheme 1 over the Ni2P(001) surface using density functional theory (DFT) calculations [32]. Our DFT-derived activation barriers indicate that methanol decomposition on the Ni2P(001) surface occurs through CH3OH* → CH3O* → CH2O* → CHO* → CO*. In the MSR pathway, formaldehyde (CH2O*) reacts with a hydroxyl (OH*) originating from a water splitting reaction to yield H2COOH*, which ultimately produces CO2 via H2COOH*→ HCOOH* → HCOO* → CO2. The activation barriers for both routes are comparable (a difference of only 5 kJ mol−1) whereas, on transition metal catalysts, methanol decomposition is more likely to dominate. The role of phosphorus and the effects of the P:Ni atomic ratio on selectivity, however, remain unclear.
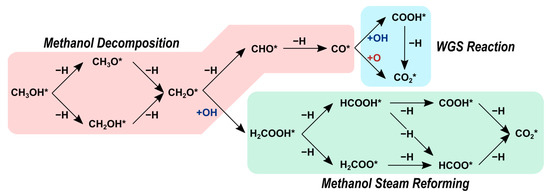
Scheme 1.
Reactions network to form CO and CO2 examined in our prior and current studies over Ni, Ni12P5, and Ni2P catalysts. Asterisk (*) denotes a surface site. Reprinted from reference [32].
In this paper, we build upon our previous study by incorporating Ni and Ni12P5 catalysts to illustrate the notable impact of the P:Ni atomic ratio (y:x in NixPy) on the selectivity and reactivity of MSR. The results demonstrate that the choice of P:Ni atomic ratio plays a crucial role in shaping catalytic performance. The moderate P:Ni of 0.42 for Ni12P5 (5:12) enhances the selectivity of MSR relative to methanol decomposition. The insights gained from these findings can guide the design of efficient catalysts tailored for use in MSR applications.
2. Results and Discussion
2.1. Optimized Adsorbates and Their Binding Energies
In this section, we study the geometries and energies of all the intermediates that form during methanol decomposition and MSR. We aim to identify the most stable configuration by exploring various adsorption modes on examined surfaces, as depicted in Figures S1 and S2 (Supplementary Materials). The binding energies presented in Table 1 were calculated using Equation (4).

Table 1.
Binding energies ΔEads (Equation (4); kJ mol−1) of examined intermediates and their preferred adsorption configuration on Ni(111), Ni12P5(001), and Ni2P(001).
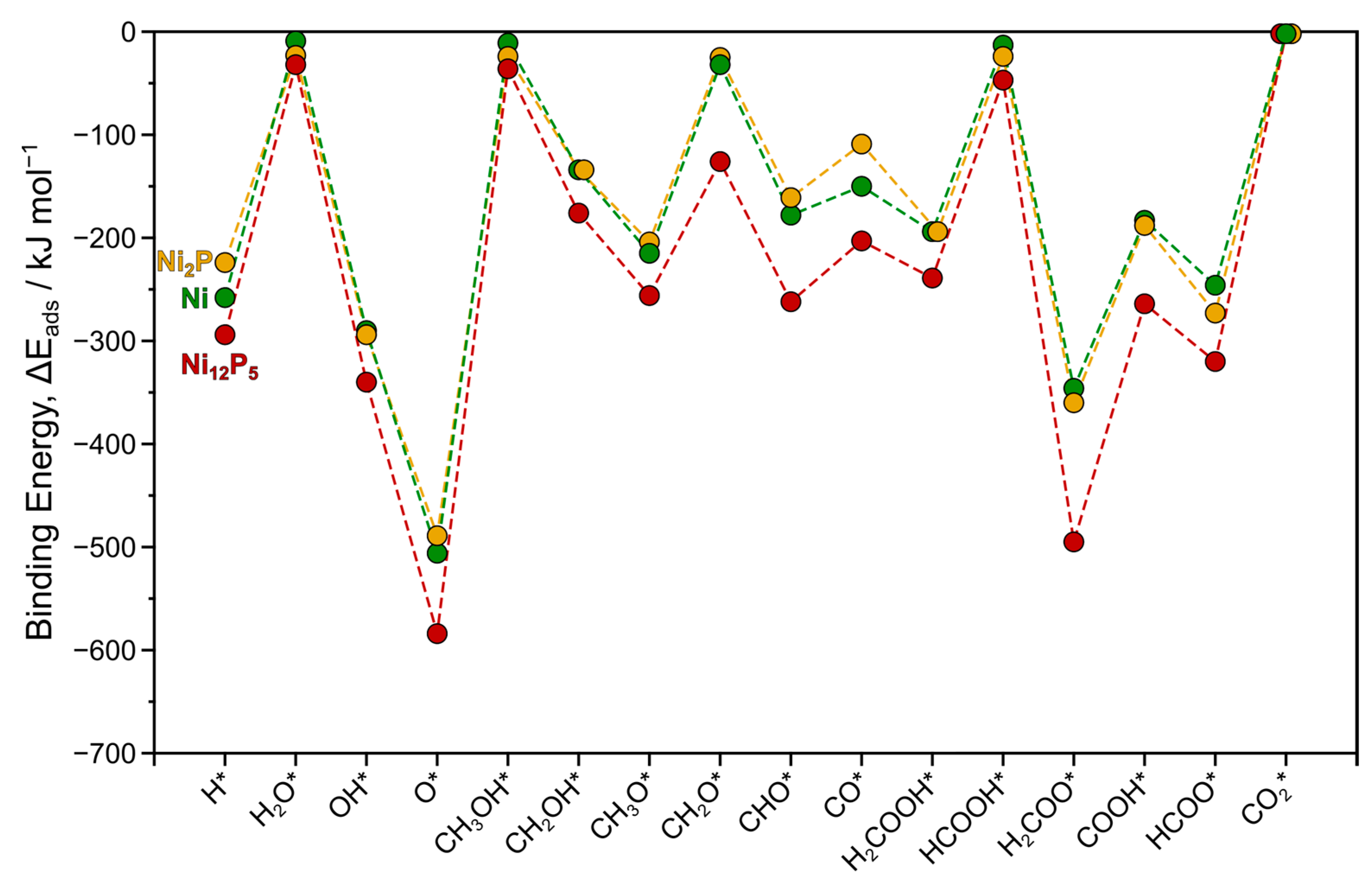
Methanol (CH3OH) binds weakly to the atop site (M1) of the Ni(111) surface through its oxygen atom (Figure S1e), with a binding energy of −11 kJ mol−1 (Table 1). This observation aligns with those found in previous theoretical and experimental studies, which also demonstrate the weak adsorption of methanol onto Ni and other transition metals [37,38,39,40]. Similarly, other physisorbed species such as water and formic acid also exhibit low binding energy to the atop site (−9 and −13 kJ mol−1, respectively). The interaction between these three intermediates and the surface metal atom likely involves the donation of the oxygen atom’s lone pair of electrons, contributing to their adsorption. Formaldehyde (CH2O; Figure S1h), on the other hand, displays a slightly stronger binding energy (−32 kJ mol−1) and preferentially binds to the 3-fold site (M3) in a top-bridge configuration. While it can also bind to the bridging site (M2) in a di-σ mode, this configuration is less favored by 18 kJ mol−1. The remaining unsaturated intermediates demonstrate stronger binding energies ranging from −134 kJ mol−1 for CH2OH* to −506 kJ mol−1 for O* (Table 1). The majority of these intermediates (H*, OH*, O*, CH3O*, CHO*, CO*, H2COOH*, and H2COO*) prefer to adsorb on the 3-fold site (M3) except CH2OH*, COOH*, and HCOO* which favorably bind to the bridging site (M2). The trends in binding energies observed here on Ni(111) closely resemble those seen in our previous study on the Ni2P(001) surface (Figure 1).
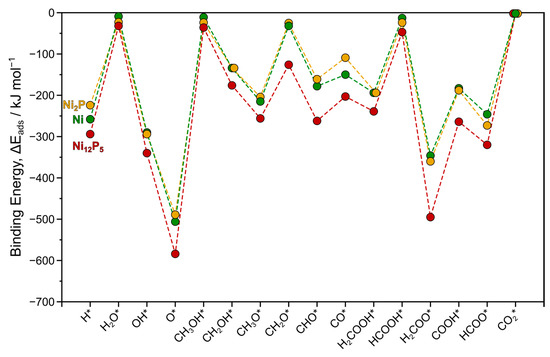
Figure 1.
Trends in binding energies ΔEads (kJ mol−1) for all surface intermediates examined in this study over Ni (green) and Ni12P5 (red) surfaces compared to Ni2P (yellow) examined in our previous study [32]. Dashed lines are provided as visual guides. Asterisk (*) denotes a surface site.
Compared to the Ni(111) and Ni2P(001) surfaces, the Ni12P5(001) surface exhibits an overall increase in binding strength for all surface intermediates (Figure 1). While the differences in binding energies among different species remain generally similar, the average binding energy on Ni12P5(001) is notably higher, with an average binding energy of approximately −230 kJ mol−1 on Ni12P5, compared to −170 kJ mol−1 on both Ni(111) and Ni2P(001). For example, methanol, water, and formic acid also bind to the metal atop site (M1), but with stronger affinities relative to Ni and Ni2P surfaces. Formaldehyde, a crucial intermediate in the MSR pathway, exhibits a binding energy of −126 kJ mol−1 when interacting with the metal 4-fold site on Ni12P5(001), which is over 100 kJ mol−1 stronger than its binding energy on Ni2P(001). This is consistent with the differences in calculated binding energies on Ni, Ni12P5, and Ni2P reported in previous studies for the intermediates involved in the hydrodeoxygenation reaction of oxygenated compounds [34]. Ni12P5(001) has two unique 4-fold sites denoted by M4a and M4b (discussed in Section 3). Notably, the M4b site displays higher reactivity compared to M4a, as evidenced by the preference of most intermediates examined here, with the exception of H2COOH*. For instance, formaldehyde (CH2O) binds more strongly to the M4b site by around 75 kJ mol−1 compared to the M4a site. These findings collectively suggest that Ni12P5 exhibits higher reactivity compared to Ni and Ni2P surfaces.
2.2. Reaction Pathways
Next, we present a thorough analysis of the reaction pathways shown in Scheme 1 on Ni and Ni12P5. We compare these results with our prior work on Ni2P to elucidate the effects of the P:Ni atomic ratio on selectivity and reactivity [32]. The reaction network, as illustrated in Scheme 1, serves as the basis for our investigation. To ensure that the most stable transition states are identified, we extensively explored various configurations on distinct surface sites for each elementary step. Table 2 displays the respective forward activation barriers and reaction energies. These values do not encompass the diffusion steps, as these steps are typically assumed to be relatively fast and have negligible influence on the overall reaction pathways.
Table 2.
Forward activation enthalpy (ΔHact = HTS − Hreactants) and the reaction energy (ΔHrxn = Hproducts − Hreactants) in kJ mol−1 for the elementary reactions analyzed in this study at 573 K.
2.2.1. H2O Dissociation
On the Ni(111) surface, the direct water dissociation (reaction 17; Figure S3) has an enthalpic activation barrier (ΔHact) of 74 kJ mol−1 and is an exothermic reaction with a reaction energy (ΔHrxn) of −25 kJ mol−1 (reaction 17; Table 2). Further dehydrogenation of hydroxyl (OH*) adsorbed onto the M3 site takes place with a higher activation barrier of 97 kJ mol−1 and a reaction energy of −18 kJ mol−1. These values agree with previously reported data on the Ni(111) surface [41], and they are consistent with our earlier findings on the Ni2P(001) surface, where OH* activation demonstrated a larger barrier. The disproportion of two adjacent OH* species can produce O* and H2O* with an activation barrier of only 28 kJ mol−1. Our previous study revealed that H2O* dissociation can be promoted by hydrogen bonding with another vicinal H2O* molecule. Here, on Ni(111), we observe a slight decrease in the activation barrier of H2O-assisted water dissociation (reaction 20; Table 2) by 6 kJ mol−1. The structures of the reactants, transition states, and products for all reactions examined in this study are shown in Figures S3 and S4 (Supplementary Materials).
On Ni12P5(001), we observe a similar trend, albeit with lower activation barriers compared to both Ni(111) and Ni2P(001) surfaces. For example, H2O* dissociates with an activation barrier of 49 kJ mol−1 (reaction 17; Table 2), which is 25 and 42 kJ mol−1 lower than the calculated barriers on Ni and Ni2P, respectively. Concerted water dissociation (reaction 20) is also more facile, with an activation barrier of only 35 kJ mol−1. Thus, we believe that hydrogen bonding plays an important role in H2O dissociation over nickel phosphide catalysts.
2.2.2. Methanol Decomposition
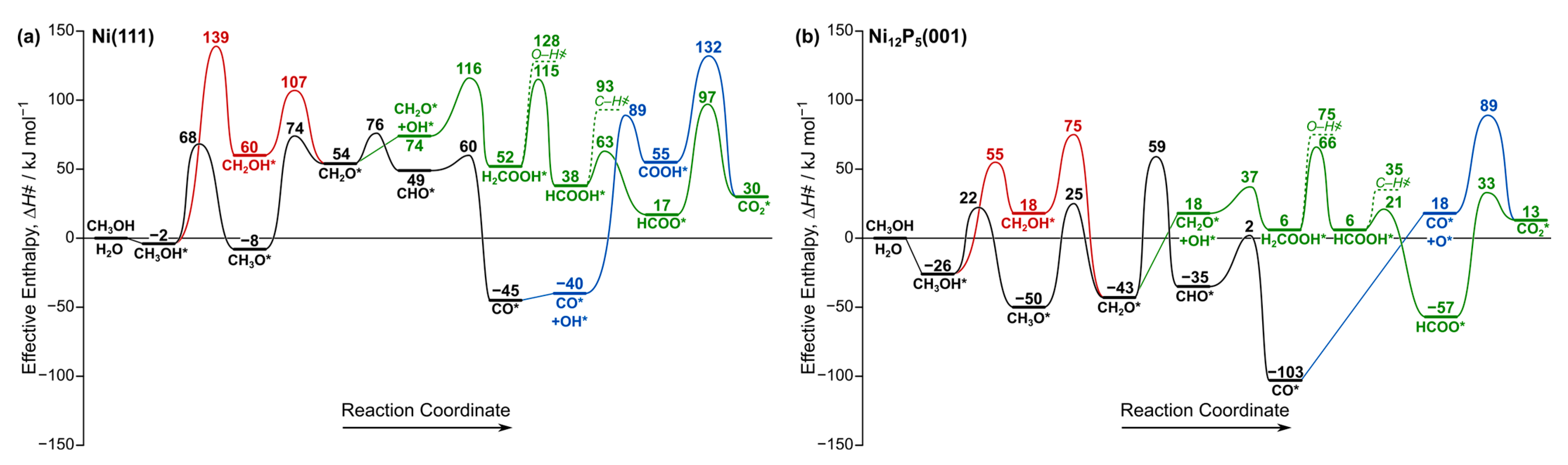
Methanol (CH3OH) can either undergo C–H or O–H bond activation (reactions 1 and 2; Table 2) on the Ni(111) surface. However, the activation barrier of the C–H bond to form CH2OH* (141 kJ mol−1) is twice that of O–H bond activation required to form CH3O* (70 kJ mol−1), suggesting that the methoxy formation route is more favorable. Similarly, on the Ni12P5(001) surface, O–H activation in methanol is more favorable by 33 kJ mol−1. In order to assess the relative preference of the various reaction routes shown in Scheme 1, we next analyze the effective enthalpy barriers (ΔH҂; Equation (5)) as shown in Figure 2. These ΔH҂ values are referenced to the energies of gas-phase methanol and water, which is particularly relevant when the reaction involves water.
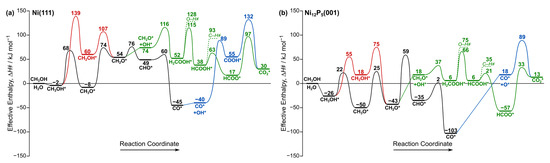
Figure 2.
Effective enthalpy (Equation (5); 573 K) diagram for the reactions network shown in Scheme 1 on (a) Ni(111) and (b) Ni12P5(001). Abstracted H atoms are desorbed as H2(g). Unfavorable routes are displayed using dashed lines. A similar diagram for Ni2P(001) is shown in Figure S5 (Supplementary Materials). Asterisk (*) denotes a surface site. Black and red represent methane decomposition pathway, green represents MSR pathway, and blue represents WGS reaction pathway.
On the Ni(111) surface, the O–H bond of physisorbed CH3OH* is initially cleaved to from CH3O* with an effective barrier (ΔH҂) of 68 kJ mol−1 (Figure 2a). A reaction cleaving the C–H bond in CH3O* then requires a barrier of 74 kJ mol−1 to form formaldehyde (CH2O*), and the reverse path to form CH3O* requires only 20 kJ mol−1. Methoxy is a stable intermediate relative to other intermediates in the methanol decomposition pathway, as shown in Figure 2a. This suggests that CH3O* is more likely the dominant species on Ni(111) as reported in previous experimental studies [42,43]. Subsequent dehydrogenation of CH2O* leads to the formation of CHO* and finally CO* with activation barriers of 76 and 60 kJ mol−1, respectively. For Ni12P5(001), the methanol decomposition steps followed to form CH3O* and then CH2O* are both facile, with relatively similar activation barriers (22–25 kJ mol−1; Figure 2b). Once CH2O* is formed, it can undergo C–H bond activation to form CHO* with a barrier of 59 kJ mol−1, which eventually forms CO* with a barrier of only 2 kJ mol−1. Both CH3O* and CH2O* are stable intermediates on Ni12P5, in contrast to Ni and Ni2P, where CH3O* is more dominant.
2.2.3. Methanol Steam Reforming
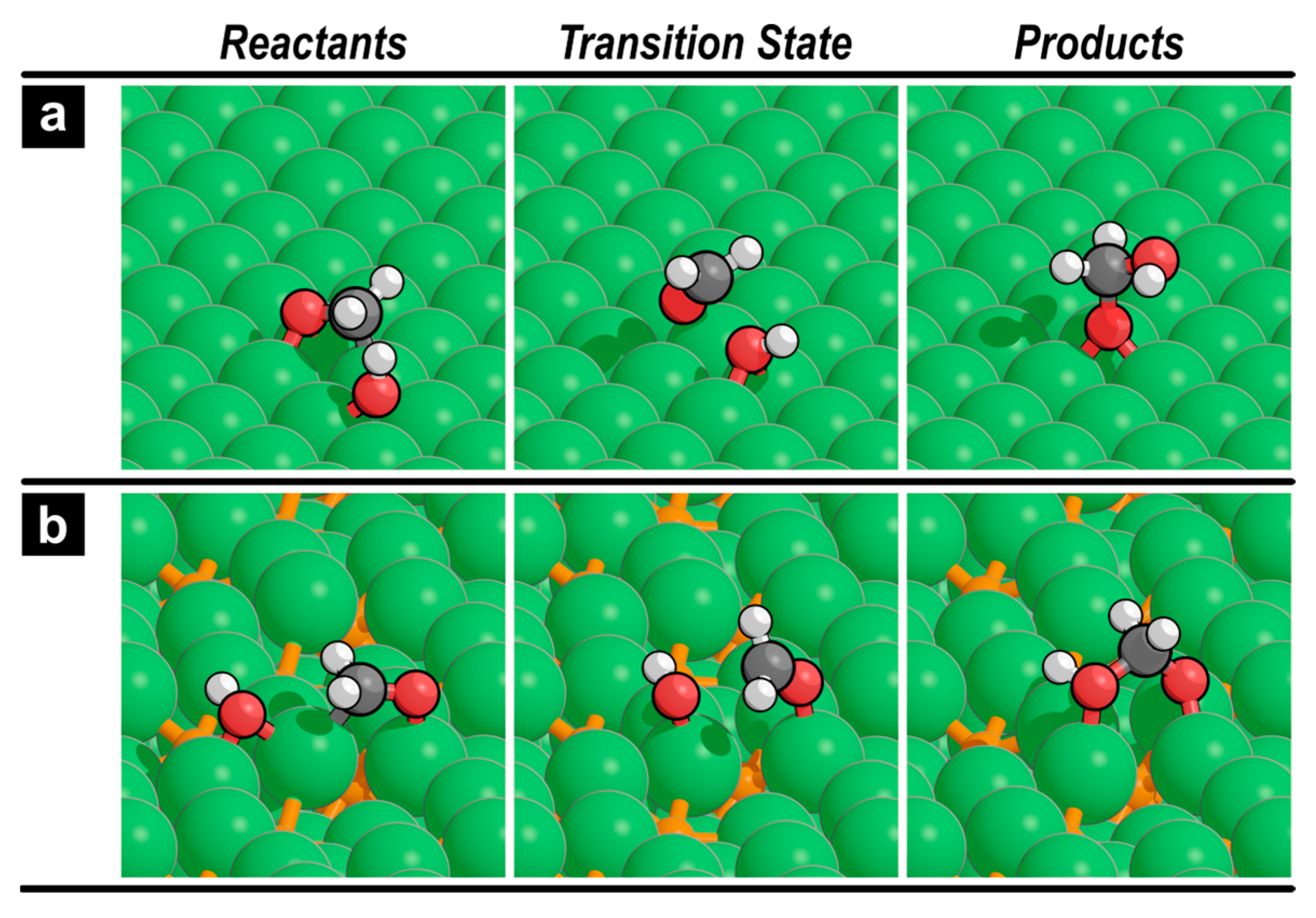
Here, we examine the possibility of reacting formaldehyde (CH2O*) with co-adsorbed OH* to form H2COOH*. On the Ni(111) surface, this reaction requires an effective activation barrier of 116 kJ mol−1 (Figure 2a), which is much larger than the activation barrier of further decomposition to CHO* (76 kJ mol−1). This is consistent with the results of the previous studies that demonstrated the preference of transition metal catalysts to decompose methanol into CO, except for Cu, which shows a distinct selectivity towards H2COOH* formation via MSR. In contrast, Ni12P5(001) can form H2COOH* from CH2O* and OH* with a barrier of 37 kJ mol−1 (Figure 2b), which is 22 kJ mol−1 lower than that of CH2O* decomposition. In our previous study, we found that the barriers of CH2O* decomposition to CHO* and CH2O* reaction with OH* are within ~5 kJ mol−1 on the Ni2P(001) surface (Figure S5), indicating that both pathways are kinetically relevant. Figure 3 shows the reactant, transition state, and product structures for reaction 7 on Ni(111) and Ni12P5(001) surfaces. On both surfaces, the carbon atom in CH2O* needs to undergo partial desorption from the surface before reacting with the adjacent OH* to form H2COOH*.
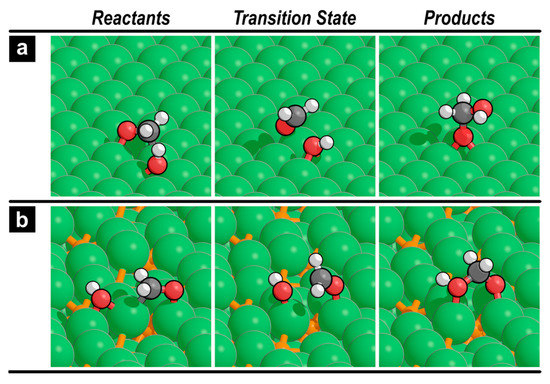
Figure 3.
Reactants, transition state, and products for CH2O* reaction with OH* (reaction 7) on (a) Ni(111) and (b) Ni12P5(001) surfaces.
Once H2COOH* is formed on the Ni12P5(001) surface, it can either cleave its C–H bond or its O–H bond, but C–H bond cleavage with an activation barrier of 66 kJ mol−1 is slightly more favorable (Figure 2b). Formic acid (HCOOH*) then forms HCOO*, which eventually forms CO2*, the product of the MSR reaction. Taken together, these findings indicate that the P:Ni atomic ratio can dictate the selectivity towards CO2 formation via MSR relative to methanol decomposition into CO. The moderate P:Ni atomic ratio in Ni12P5 (P:Ni = 0.42) favors the MSR pathway compared to Ni2P (P:Ni = 0.5), in which both methanol decomposition and MSR are competitive. Ni (P:Ni = 0), on the other hand, predominantly decomposes methanol to carbon monoxide.
The impact of the P:Ni atomic ratio on the differences in selectivity stems from a complex interplay between geometric and electronic factors, each contributing to the observed effects. For example, the Ni atoms exposed on the Ni2P(001) surface form 3-fold sites similar to those found in Ni(111), only with larger Ni–Ni bond distances (3.16 Å versus 2.49 Å). This may explain the similarities in the reaction coordinate diagram between Ni (Figure 2a) and Ni2P (Figure S5), except that both MSR and methanol decomposition pathways start to compete on Ni2P, whereas methanol decomposition is more dominant on Ni. The Ni12P5(001) surface, however, exposes unique metal 4-fold sites (M4) that predominantly favor MSR. Another factor to consider here is the electronic effects. We have previously performed charge analysis for these three surfaces and we have shown that the Ni atoms become more positively charged with an increasing P:Ni atomic ratio [35]. This may indicate that the moderate Lewis acidity on Ni12P5 enhances the selectivity of MSR relative to methanol decomposition.
2.2.4. Water–Gas Shift Reaction
CO* generated through the methanol decomposition route can be transformed into CO2 via the water–gas shift reaction. It can either react with a co-adsorbed OH* to form COOH* in the carboxyl mechanism, which subsequently dehydrogenates to produce CO2 (CO* + OH* → COOH* → CO2* + H*), or it can react with O* to directly produce CO2 via the redox mechanism (CO* + O* → CO2*), as shown in Scheme 1. The carboxyl mechanism was found to be more favorable on Ni2P than the redox mechanism, which is similar to what we found here on the Ni(111) surface (Table 2). However, for Ni12P5(001), we could not find a stable transition state for the carboxyl mechanism. Instead, Ni12P5 exclusively forms CO2 via the redox mechanism with a large effective barrier of 89 kJ mol−1 (Figure 2b). WGR reaction pathways involve substantially higher barriers than those observed for the MSR pathway across all catalysts studied here. Thus, CO2 formation via the WGS reaction can be ruled out.
3. Computational Methods
All periodic DFT calculations were carried out using the Vienna ab initio simulation package (VASP) [44,45,46,47] and employing the computational catalysis interface (CCI) [48]. Projector augmented-wave (PAW) potentials with an energy cutoff of 396 eV were used to construct the plane waves [49,50]. The exchange and correlation energies were described using the revised Perdew–Burke–Ernzerhof (RPBE) form of the generalized gradient approximation [51,52,53]. For modeling gaseous species, 15 × 15 × 15 Å unit cells were used. Available crystallographic data were used to obtain the bulk unit cells of Ni and Ni12P5, which were then optimized using DFT to determine their lattice parameters, as reported in more detail in our previous work [34]. Spin polarization was used for all Ni calculations, whereas Ni12P5 does not exhibit ferromagnetic properties.
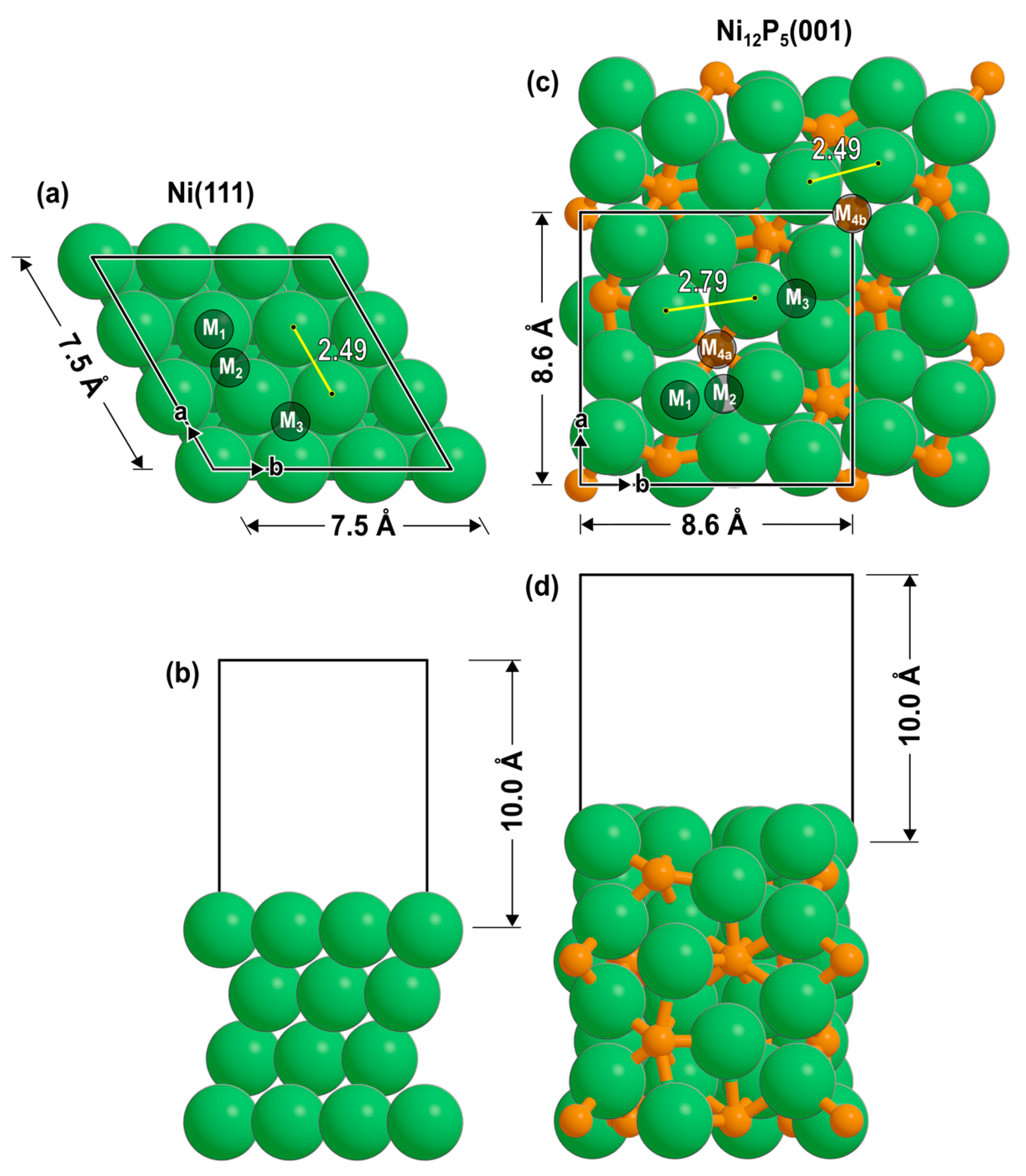
A 3 × 3 periodic lattice was used to model the Ni(111) surface (Figure 4a), with four atomic layers and 10 Å of vacuum orthogonal to the surface (Figure 4b). For geometric convergence, a k-point mesh of 3 × 3 × 1 was employed [54,55]. This was followed by a single-point calculation with a k-point mesh of 6 × 6 × 1 to compute the electronic energy. For the Ni12P5 catalyst, the Ni-terminated (001) surface was used (Figure 4c) due to its favorable surface formation energy, as shown previously [34], with two repeating units (8 atomic layers) and 10 Å of vacuum orthogonal to the surface (Figure 4d). This termination does not expose any phosphorus atoms on the surface. It has atop sites (M1), bridging sites (M2), 3-fold sites (M3), and two unique 4-fold sites (M4a and M4b). The M4a site has a P atom directly beneath it (in the second layer), whereas the P atom beneath M4b is in the fourth layer. A k-point mesh of 3 × 3 × 1 was used for Ni12P5 calculations. The bottom half of each catalyst model was constrained during optimizations.
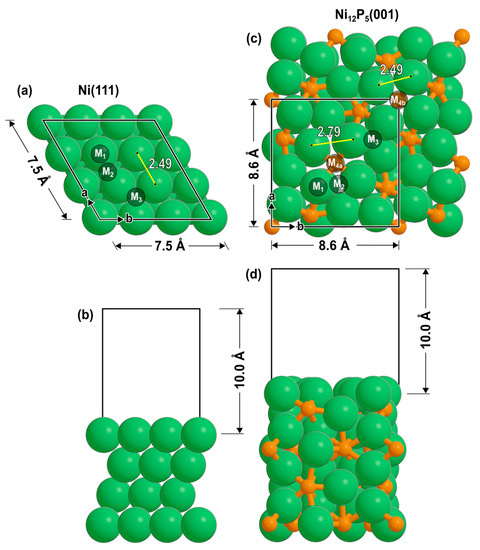
Figure 4.
(a,b) Ni(111) and (c,d) Ni12P5(001) slabs as seen from top and side views. Labels denote the binding sites (M1: atop; M2: bridge; M3: 3-fold hollow; M4: 4-fold hollow).
Transition state structures were identified using the combination of the nudged elastic band (NEB) method and the dimer method [56,57,58]. Electronic energies converged to within 10−6 eV, and forces converged to less than 0.05 eVÅ−1. Frequency calculations were performed, with all catalysts’ atoms constrained, to estimate enthalpies at 573 K. The binding energy (ΔEads) is defined as:
and effective enthalpy barriers (ΔH҂) are calculated using:
where λ is the number of H2 molecules desorbed from the surface as a result of the hydrogen removal steps. Section S1 in the Supplementary Materials provides more details regarding the computational methods.
4. Conclusions
The effects of the P:Ni ratio on selectivity towards the methanol steam reforming reaction were investigated using periodic density functional theory calculations. These calculations showed that Ni(111) surface (P:Ni = 0) predominantly decomposes methanol into carbon monoxide through: CH3OH* → CH3O* → CH2O* → CHO* → CO*. These results are in agreement with those of prior studies on various transition metals. On the other hand, Ni12P5(001) with a P:Ni atomic ratio of 0.42 favors the methanol steam reforming pathway of CO2 production from formaldehyde via CH2O* + OH* → H2COOH*→ HCOOH* → HCOO* → CO2. Increasing the P:Ni atomic ratio to 0.5 in Ni2P(001) decreases the selectivity of MSR relative to methanol decomposition, but both pathways remain competitive. The catalytic surfaces analyzed in this study displayed negligible activity towards WGS reaction pathways, as indicated by the substantial activation barriers observed. These findings shed light on the catalytic behavior of nickel phosphide catalysts and provide valuable insights into the distinctive pathways of CO2 generation, guiding the development of efficient and selective catalytic materials for MSR.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28166079/s1, Section S1: Computation Methods Details; Figure S1: Images for adsorbed intermediates on the Ni(111) surface; Figure S2: Images for adsorbed intermediates on the Ni12P5(001) surface; Figure S3: Reaction images on the Ni(111) surface; Figure S4: Reaction images on the Ni12P5(001) surface; Figure S5: Effective enthalpy diagram for Ni2P(001).
Funding
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia (Grant No. 3910).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
Sample Availability
Not applicable.
References
- Manoharan, Y.; Hosseini, S.E.; Butler, B.; Alzhahrani, H.; Senior, B.T.F.; Ashuri, T.; Krohn, J. Hydrogen Fuel Cell Vehicles; Current Status and Future Prospect. Appl. Sci. 2019, 9, 2296. [Google Scholar] [CrossRef]
- Kurnia, J.C.; Chaedir, B.A.; Sasmito, A.P.; Shamim, T. Progress on Open Cathode Proton Exchange Membrane Fuel Cell: Performance, Designs, Challenges and Future Directions. Appl. Energy 2021, 283, 116359. [Google Scholar] [CrossRef]
- Guan, D.; Xu, H.; Zhang, Q.; Huang, Y.; Shi, C.; Chang, Y.; Xu, X.; Tang, J.; Gu, Y.; Pao, C.; et al. Identifying A Universal Activity Descriptor and a Unifying Mechanism Concept on Perovskite Oxides for Green Hydrogen Production. Adv. Mater. 2023, 2305074. [Google Scholar] [CrossRef]
- Lv, Z.; Xu, H.; Xu, W.; Peng, B.; Zhao, C.; Xie, M.; Lv, X.; Gao, Y.; Hu, K.; Fang, Y.; et al. Quasi-Topological Intercalation Mechanism of Bi0.67 NbS2 Enabling 100 C Fast-Charging for Sodium-Ion Batteries. Adv. Energy Mater. 2023, 13, 2300790. [Google Scholar] [CrossRef]
- Xiao, W.; Kiran, G.K.; Yoo, K.; Kim, J.; Xu, H. The Dual-Site Adsorption and High Redox Activity Enabled by Hybrid Organic-Inorganic Vanadyl Ethylene Glycolate for High-Rate and Long-Durability Lithium–Sulfur Batteries. Small 2023, 19, 2206750. [Google Scholar] [CrossRef]
- Xu, H.; Guan, D.; Ma, L. The Bio-Inspired Heterogeneous Single-Cluster Catalyst Ni100–Fe4S4 for Enhanced Electrochemical CO2 Reduction to CH4. Nanoscale 2023, 15, 2756–2766. [Google Scholar] [CrossRef] [PubMed]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Xu, X.; Shuai, K.; Xu, B. Review on Copper and Palladium Based Catalysts for Methanol Steam Reforming to Produce Hydrogen. Catalysts 2017, 7, 183. [Google Scholar] [CrossRef]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for Methanol Steam Reforming—A Review. Appl. Catal. B 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Rivard, E.; Trudeau, M.; Zaghib, K. Hydrogen Storage for Mobility: A Review. Materials 2019, 12, 1973. [Google Scholar] [CrossRef]
- Eberle, U.; Felderhoff, M.; Schüth, F. Chemical and Physical Solutions for Hydrogen Storage. Angew. Chem. Int. Ed. 2009, 48, 6608–6630. [Google Scholar] [CrossRef] [PubMed]
- Schlapbach, L.; Züttel, A. Hydrogen-Storage Materials for Mobile Applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef]
- Agrell, J.; Birgersson, H.; Boutonnet, M. Steam Reforming of Methanol over a Cu/ZnO/Al2O3 Catalyst: A Kinetic Analysis and Strategies for Suppression of CO Formation. J. Power Sources 2002, 106, 249–257. [Google Scholar] [CrossRef]
- Agrell, J. Production of Hydrogen from Methanol over Cu/ZnO Catalysts Promoted by ZrO2 and Al2O3. J. Catal. 2003, 219, 389–403. [Google Scholar] [CrossRef]
- Luo, W.; Asthagiri, A. Density Functional Theory Study of Methanol Steam Reforming on Co(0001) and Co(111) Surfaces. J. Phys. Chem. C 2014, 118, 15274–15285. [Google Scholar] [CrossRef]
- Lin, S.; Johnson, R.S.; Smith, G.K.; Xie, D.; Guo, H. Pathways for Methanol Steam Reforming Involving Adsorbed Formaldehyde and Hydroxyl Intermediates on Cu(111): Density Functional Theory Studies. Phys. Chem. Chem. Phys. 2011, 13, 9622–9631. [Google Scholar] [CrossRef]
- Sutton, J.E.; Panagiotopoulou, P.; Verykios, X.E.; Vlachos, D.G. Combined DFT, Microkinetic, and Experimental Study of Ethanol Steam Reforming on Pt. J. Phys. Chem. C 2013, 117, 4691–4706. [Google Scholar] [CrossRef]
- Smith, G.K.; Lin, S.; Lai, W.; Datye, A.; Xie, D.; Guo, H. Initial Steps in Methanol Steam Reforming on PdZn and ZnO Surfaces: Density Functional Theory Studies. Surf. Sci. 2011, 605, 750–759. [Google Scholar] [CrossRef]
- Lin, S.; Xie, D.; Guo, H. Pathways of Methanol Steam Reforming on Pdzn and Comparison with Cu. J. Phys. Chem. C 2011, 115, 20583–20589. [Google Scholar] [CrossRef]
- Papavasiliou, J.; Paxinou, A.; Słowik, G.; Neophytides, S.; Avgouropoulos, G. Steam Reforming of Methanol over Nanostructured Pt/TiO2 and Pt/CeO2 Catalysts for Fuel Cell Applications. Catalysts 2018, 8, 544. [Google Scholar] [CrossRef]
- Wu, H.; La Parola, V.; Pantaleo, G.; Puleo, F.; Venezia, A.; Liotta, L. Ni-Based Catalysts for Low Temperature Methane Steam Reforming: Recent Results on Ni-Au and Comparison with Other Bi-Metallic Systems. Catalysts 2013, 3, 563–583. [Google Scholar] [CrossRef]
- Köpfle, N.; Mayr, L.; Schmidmair, D.; Bernardi, J.; Knop-Gericke, A.; Hävecker, M.; Klötzer, B.; Penner, S. A Comparative Discussion of the Catalytic Activity and CO2-Selectivity of Cu-Zr and Pd-Zr (Intermetallic) Compounds in Methanol Steam Reforming. Catalysts 2017, 7, 53. [Google Scholar] [CrossRef]
- Twigg, M.V. Deactivation of Copper Metal Catalysts for Methanol Decomposition, Methanol Steam Reforming and Methanol Synthesis. Top. Catal. 2003, 22, 191–203. [Google Scholar] [CrossRef]
- Kurtz, M.; Wilmer, H.; Genger, T.; Hinrichsen, O.; Muhler, M. Deactivation of Supported Copper Catalysts for Methanol Synthesis. Catal. Lett. 2003, 86, 77–80. [Google Scholar] [CrossRef]
- Takezawa, N.; Iwasa, N. Steam Reforming and Dehydrogenation of Methanol: Difference in the Catalytic Functions of Copper and Group VIII Metals. Catal. Today 1997, 36, 45–56. [Google Scholar] [CrossRef]
- Iwasa, N.; Masuda, S.; Ogawa, N.; Takezawa, N. Steam Reforming of Methanol over Pd/ZnO: Effect of the Formation of PdZn Alloys upon the Reaction. Appl. Catal. A Gen. 1995, 125, 145–157. [Google Scholar] [CrossRef]
- Iwasa, N.; Mayanagi, T.; Nomura, W.; Arai, M.; Takezawa, N. Effect of Zn Addition to Supported Pd Catalysts in the Steam Reforming of Methanol. Appl. Catal. A Gen. 2003, 248, 153–160. [Google Scholar] [CrossRef]
- Iwasa, N. New Supported Pd and Pt Alloy Catalysts for Steam Reforming and Dehydrogenation of Methanol. Top. Catal. 2003, 22, 215–224. [Google Scholar] [CrossRef]
- Al-Ali, L.I.; Elmutasim, O.; Al Ali, K.; Singh, N.; Polychronopoulou, K. Transition Metal Phosphides (TMP) as a Versatile Class of Catalysts for the Hydrodeoxygenation Reaction (HDO) of Oil-Derived Compounds. Nanomaterials 2022, 12, 1435. [Google Scholar] [CrossRef]
- Li, W.; Dhandapani, B.; Oyama, S.T. Molybdenum Phosphide: A Novel Catalyst for Hydrodenitrogenation. Chem. Lett. 1998, 27, 207–208. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H.; Li, D.; Rodríguez-Castellón, E.; Jiménez López, A. Studies of the Synthesis of Transition Metal Phosphides and Their Activity in the Hydrodeoxygenation of a Biofuel Model Compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Almithn, A.; Alhulaybi, Z. A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst. Catalysts 2022, 12, 1174. [Google Scholar] [CrossRef]
- Almithn, A.; Alghanim, S.N.; Mohammed, A.A.; Alghawinim, A.K.; Alomaireen, M.A.; Alhulaybi, Z.; Hossain, S.S. Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts 2023, 13, 531. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Triezenberg, M.D.; Hibbitts, D.D.; Flaherty, D.W. In Situ Methods for Identifying Reactive Surface Intermediates during Hydrogenolysis Reactions: C–O Bond Cleavage on Nanoparticles of Nickel and Nickel Phosphides. J. Am. Chem. Soc. 2019, 141, 16671–16684. [Google Scholar] [CrossRef] [PubMed]
- Waldt, C.; Montalvo-Castro, H.; Almithn, A.; Loaiza-Orduz, Á.; Plaisance, C.; Hibbitts, D. Role of Phosphorous in Transition Metal Phosphides for Selective Hydrogenolysis of Hindered C–O Bonds. J. Catal. 2023, 421, 403–418. [Google Scholar] [CrossRef]
- Öström, H.; Zhang, B.; Vallejo, T.; Merrill, B.; Huang, J.; Larue, J. Methanol Decomposition on Ni(111) and O/Ni(111). J. Chem. Phys. 2022, 156, 024704. [Google Scholar] [CrossRef]
- Wang, G.C.; Zhou, Y.H.; Morikawa, Y.; Nakamura, J.; Cai, Z.S.; Zhao, X.Z. Kinetic Mechanism of Methanol Decomposition on Ni(111) Surface: A Theoretical Study. J. Phys. Chem. B 2005, 109, 12431–12442. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Competitive Paths for Methanol Decomposition on Pt(111). J. Am. Chem. Soc. 2004, 126, 3910–3919. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Methanol Decomposition on Cu(111): A DFT Study. J. Catal. 2002, 208, 291–300. [Google Scholar] [CrossRef]
- Mohsenzadeh, A.; Bolton, K.; Richards, T. DFT Study of the Adsorption and Dissociation of Water on Ni(111), Ni(110) and Ni(100) Surfaces. Surf. Sci. 2014, 627, 1–10. [Google Scholar] [CrossRef]
- Yang, H.; Whitten, J.L.; Friend, C.M. Adsorption of CH3O on Ni(111). Surf. Sci. 1994, 313, 295–307. [Google Scholar] [CrossRef]
- Schaff, O.; Hess, G.; Fritzsche, V.; Fernandez, V.; Schindler, K.-M.; Theobald, A.; Hofmann, P.; Bradshaw, A.M.; Davis, R.; Woodruff, D.P. The Structure of the Surface Methoxy Species on Ni(111). Surf. Sci. 1995, 331–333, 201–206. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Hafner, J. Ab initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kravchenko, P.; Plaisance, C.; Hibbitts, D. A New Computational Interface for Catalysis. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—A Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Classical and Quantum Dynamics in Condensed Phase Simulations; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).