1. Introduction
ADP-ribosylation is a ubiquitous post-translational modification (PTM) found in a vast array of species [
1]. Despite being one of the first characterized PTMs [
2], the biochemical mechanisms governing ADP-ribosylation are still not fully understood. In humans, the transfer of ADP–ribose(ADPr) from nicotinamide adenine dinucleotide (NAD
+) to the protein target (
Figure 1a) is mediated by a family of seventeen diphtheria toxin-like ADP-ribosyltransferases (ARTDs) that were previously referred to as poly-ADP–ribose polymerases (or PARPs) [
3]. While the enzyme family is collectively referred to as ARTDs, the individual proteins are still identified based on their PARP numbering, a system we will adopt herein to refer to specific enzymes [
4]. The ARTD family is further subdivided based on whether a single ADPr unit is transferred (PARP3–4, 6–8, 10–12, and 14–16) or whether the initial ADPr can be elongated with multiple ADPr units (PARP1–2, TNKS1–2) [
5]. Initially discovered as DNA base repair enzymes based on the activity of PARP1 in the nucleus [
6], the ARTD family has since been linked to a growing set of biological pathways and disease states [
7,
8]. The removal of ADPr is catalyzed by a separate class of glycohydrolases [
9], which can be subdivided based on their ability to remove poly-ADP–ribose (PAR, PARG) [
10], mono-ADP–ribose (MAR;
macroD1,
macroD2, and ARH1) [
11,
12,
13], or both (TARG1, ARH3) [
12,
14,
15]. As with the ARTD family, ADPr erasers have been implicated in a number of essential biological processes [
9].
A fundamental challenge in understanding the function of MAR/PAR dependent signaling is elucidating the mechanisms of ARTD target selection. Target selection can be broadly attributed to two types of intermolecular interactions: (1) The distal interactions between the various domains attached to the catalytic fragment that help recruit and hold substrates in place for modification (such distal interactions between substrate and/or allosteric regulators can also be required to fully activate the enzymes
in vivo as seen with PARP1 and 2 during DNA damage repair [
16,
17]); and (2) the proximal interactions between the catalytic domain and the substrate itself. While the investigation of both types of interaction (proximal and distal) is required to fully understand how ARTDs identify and modify their cellular targets, the determination of specific factors that drive proximal modification would be a useful complement to ongoing efforts to identify target sequences using tandem mass-spectrometry (MS/MS) [
18,
19].
As MS/MS based efforts have vastly expanded our understanding of the ADP-ribosylome by revealing thousands of potential MAR/PAR sites in the human proteome, the task of identifying bona fide sites has become even more daunting. Bioinformatic analyses of the resultant data have identified putative site motifs [
20], but those sites are often weakly enriched and not fully validated. The lack of a sequence motif could be due to: (1) A real lack of a consensus for a target sequence within the ARTD family; (2) the presence of weak motif signatures within an otherwise promiscuous enzyme class; and/or (3) the artificial enrichment of non-physiological sites due to the various interventions required to enrich MAR/PAR sites in MS/MS procedures. Understanding the proximal factors that influence ARTD target selection will help distinguish between these three possibilities. Additionally, the same factors that dictate ADPr transfer could impact ADPr hydrolysis, thereby expanding the importance of sequence motifs more broadly to the glycohydrolases.
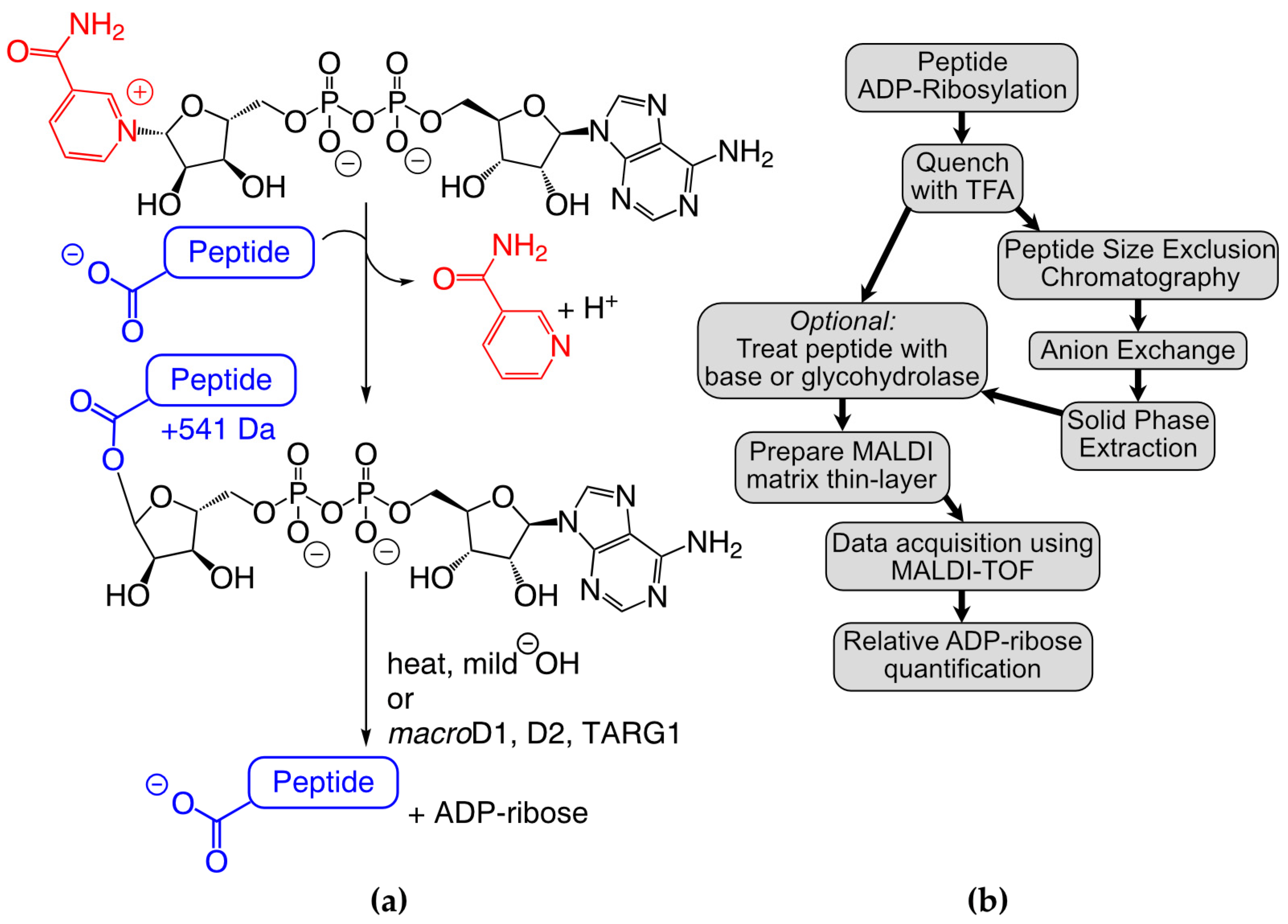
Ultra-thin layer chromatography matrix-assisted laser-desorption/ionization time-of-flight (TLC-MADLI) analysis provides a unique opportunity to interrogate proximal ARTD–substrate interactions. Unlike MS/MS, where it can be difficult and/or expensive to determine the relative levels of MAR/PAR for each identified site, TLC-MALDI allows for the direct comparison of specific sequences and the quantification of their ADP-ribosylation
in vitro. The pre-application of a thin-layer of matrix on the steel objective enhances crystal formation in the presence of common contaminants and improves the resultant signal intensity and resolution [
21,
22,
23]. Further, the ability to rapidly and inexpensively probe peptide substrates in isolation facilitates the characterization of each amino acid’s contribution to ARTD activity without the confounding input from multiple MAR/PAR sites found on whole proteins. By applying TLC-MALDI analysis to peptides identified through MS/MS screening methods, experimentalists can make detailed assessments of the relative contribution of each sequence to ADP–ribose activity, providing a complementary method to identify bona fide MAR/PAR sites. Each of these features of TLC-MALDI is vital for uncovering proximal preferences that guide ADP-ribosylation.
We previously used TLC-MALDI to identify an 18-mer peptide (P14p1) found on PARP13 that was preferentially labeled by the catalytic fragment of PARP14 (hereafter referred to as P14c)
in vitro [
24]. Herein, we describe the expansion of our efforts to quantitatively assess the proximal preferences of P14c for this peptide (
Figure 1b). We demonstrate that a truncated five amino acid sequence is sufficient to drive modification via P14c and we identify the positions within that sequence that are required for maximal ADPr transfer. We develop a three-step purification strategy to chemoenzymatically synthesize homogenous MARylated peptides using our findings. We use the resultant MAR-peptides to assess ADPr removal and describe the effects of specific sequences on both enzymatic and non-enzymatic hydrolysis. We observe that the non-enzymatic hydrolysis of MAR occurs within hours in mild conditions, a result which highlights the potential reversibility of ADP-ribosylation
in vivo and impacts future site identification methods. Taken together, our findings elucidate proximal preferences for P14 for these peptidyl substrates and demonstrate the wider importance of proximal sequences in ADPr-dependent signaling.
2. Results and Discussion
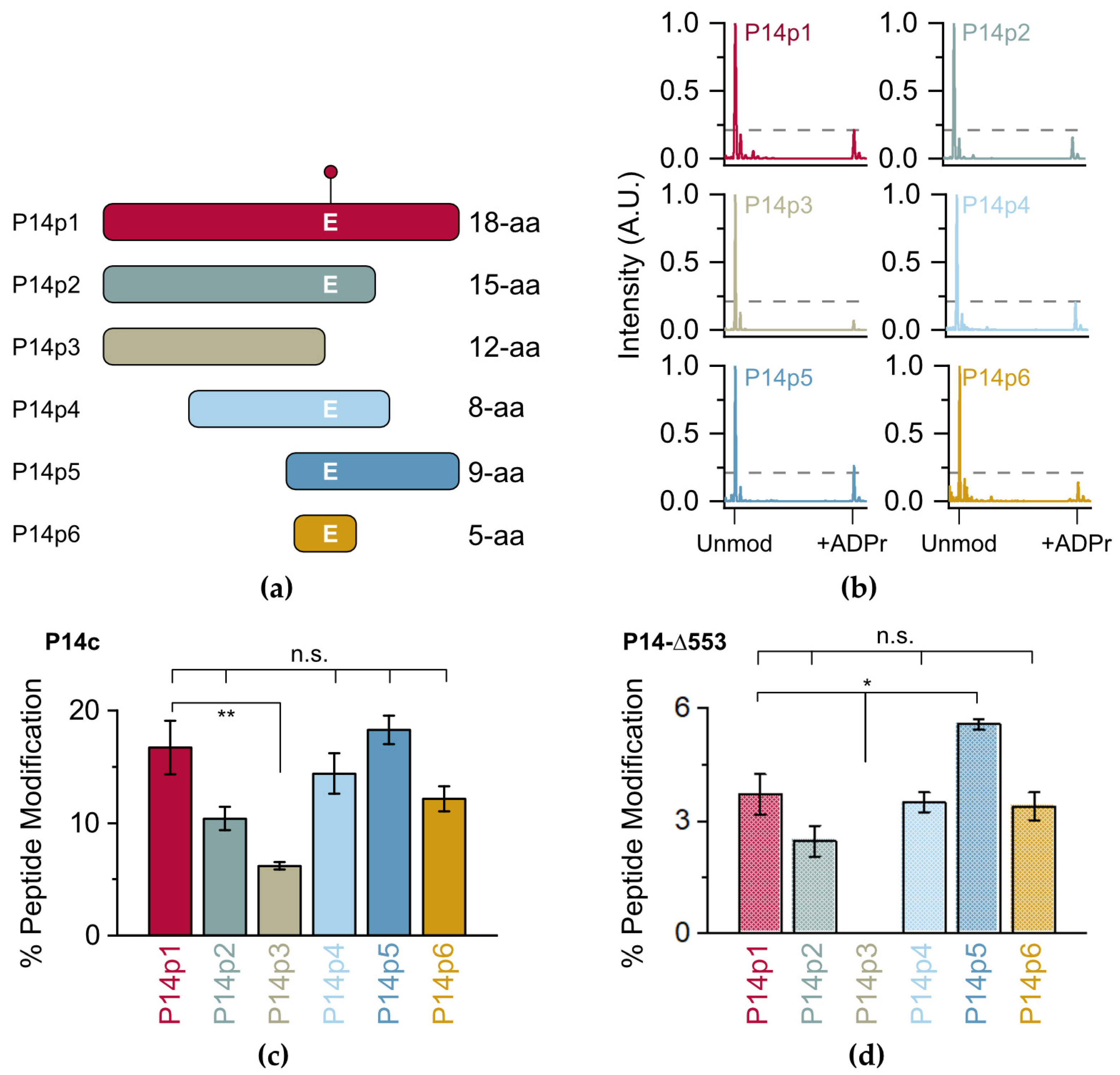
Initially, a series of P14p1 truncations were designed (P14p2-6) to identify the necessary and sufficient sequence elements required for P14c preferential modification (
Figure 2a, full sequences in
Table S1). Each of the peptides were incubated in the presence of P14c and NAD
+ and subjected to TLC-MALDI. All of the resulting spectra displayed varying levels of modification by MAR as indicated by the expected mass shift of +541 Da (
Figure 1a and
Figure 2b). Integration of the unmodified and modified peaks in the MS spectra was used to quantify the relative levels of ADP-ribosylation for each of the truncated peptides (
Figure 2c). Dividing the integrated area of the modified peptide by the total area of both the modified and unmodified peptide allowed for the determination of the total percentage of modified peptide produced in the experiment. The loss of C-terminal residues to the modified glutamate (E) resulted in a decrease in ADPr transfer, though this effect is not significant until the acceptor residue is lost (P14p3), leaving behind a tyrosine (Y) or a number of serines (S) as potential acceptors. These data are consistent with recent studies identifying Y as a modified site on PARP14, but the ARTD responsible for modification of Y on PARP14 is still unknown [
25]. However, it appears that neither Y nor the other residues are the preferred acceptor sites for P14c within this sequence, as their presence as alternative P14c targets resulted in a nearly three-fold decrease in modification (from 16.7% to 6.2% of the total peptide). By contrast, the N-terminal portion of the 18-mer can be truncated to an overlap of two amino acids with no apparent loss in activity (compare P14p1 to P14p5). While a minor contribution of the final three C-terminal residues in P14c targeting cannot be ruled out, a truncated five amino acid sequence surrounding the acceptor E residue showed no significant loss in P14c activity.
As the catalytic fragment of P14c is likely more promiscuous than the full-length protein, it was important to confirm that the trends observed with P14c were representative of the native protein. Previous work by Barbarulo and colleagues showed that a longer construct (with a 553 amino acid N-terminal truncation of P14) complemented the activity of full-length P14 in a P14 knock-out model [
26]. This nearly full-length construct (P14-∆553) is compatible with bacterial expression and was selected as a proxy for full-length activity. While the level of modification was lower overall with P14-∆553 than that observed with P14c, a nearly identical pattern of ADP-ribosylation occurred with the tested peptides (
Figure 2d, representative spectra in
Figure S1). These data demonstrate that, while P14c is possibly more promiscuous than longer forms of P14, the proximal factors that guide P14 targeting are recapitulated in the shorter construct. Given the higher activity of P14c, its ability to be expressed and purified more readily in bacteria, and its utility as a proxy for P14 full-length activity, it was selected as the primary construct going forward. Importantly, the incubation of any of the tested peptides with PARP15, a closely related P14 ortholog, resulted in almost no transfer of ADPr (
Figure S2). Given the low levels of MARylation in the presence of P15, the activity of P15 was confirmed using an auto-modification assay (
Figure S2). As P15 and P14 are closer orthologs than the other ARTD family-members, it is interesting to note that they do not share a preference for the same minimal peptide sequence. While determining if the remaining ARTD family members are active in the presence of this minimal sequence is a motivating topic for future investigation, these findings nonetheless demonstrate that the 5-mer sequence is preferentially targeted by P14c and will be an optimal minimal fragment to assess the effects of proximal sequence on P14 activity.
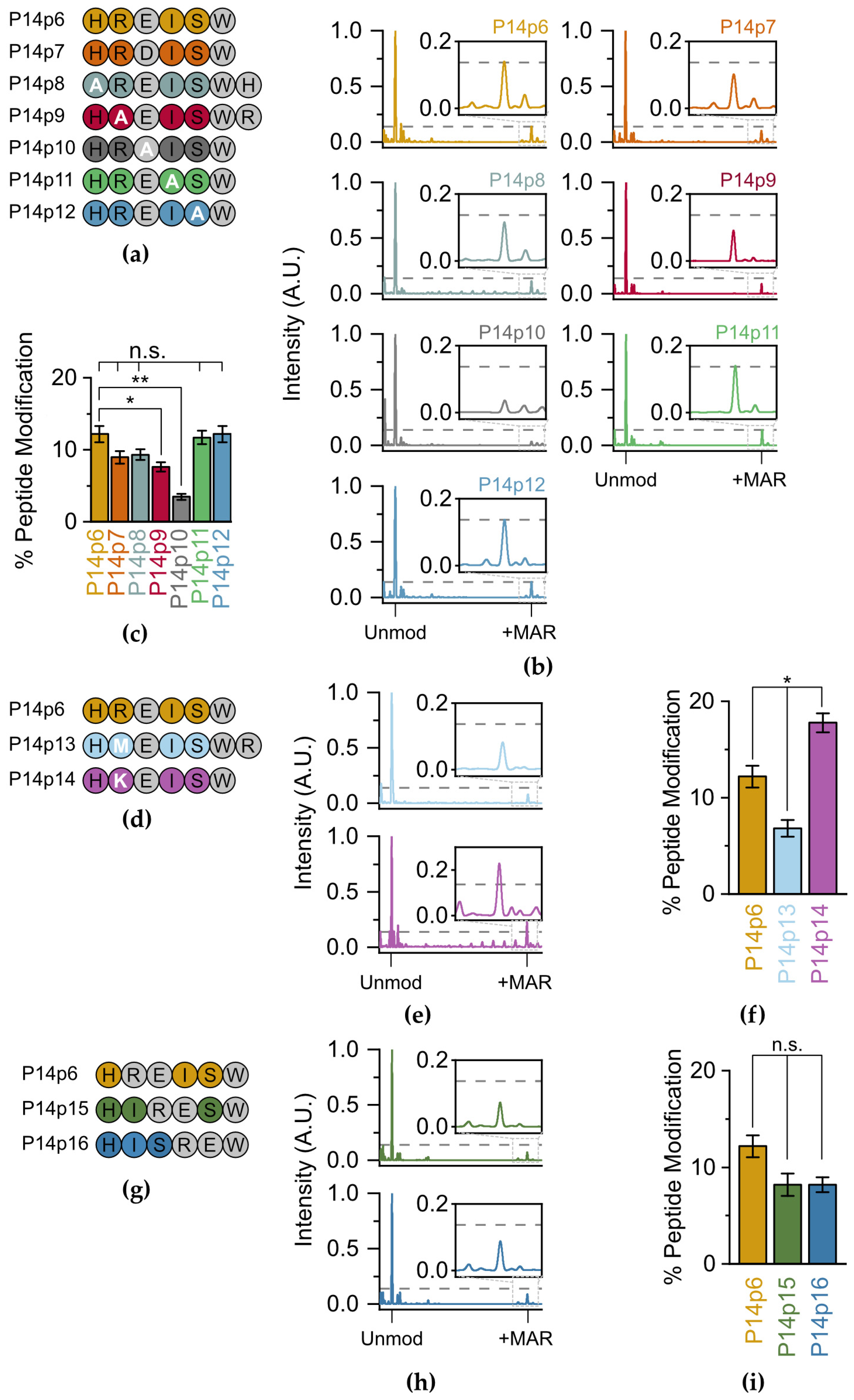
Next, we assessed the relative contribution of each of the non-acceptor amino acids on P14 selection. We systematically replaced each of the residues surrounding the E acceptor (and the acceptor itself) with alanine (A) and performed TLC-MALDI with P14c as described above (
Figure 3a). Our initial experiments with peptide variants that involved the substitution of a charged residue with alanine resulted in spectra that had severely diminished signal intensities compared to the parent peptide. As ionization in the positive mode used in our MALDI-TOF is dependent on the overall charge of the molecule, we theorized that this lack of signal was caused by the loss of a +1 charge on these peptides. To avoid artifacts due to charge imbalances in the peptides tested, we added back the lost residue to the C-terminus of the peptide while maintaining the alanine substitution (e.g., P14p8 and P14p9). This allowed us to maintain the same charge on each peptide and allowed for direct comparisons in spectral intensity across the screened mutants (
Figure 3b). Replacing the glutamate acceptor with alanine resulted in an expected significant decrease in MARylation (3.5% versus 12.2%), confirming this as the primary acceptor site (
Figure 3c). The 3.5% modification rate is therefore the background level of non-preferential transfer in this
in vitro setting. A decrease in MARylation similarly occurred when the acceptor minus two (P14p8) and minus one (P14p9) positions were altered to alanine as compared to the native sequences in P14p6 (9.3% or 7.6% peptide modification versus 12.2%, respectively). However, the change with P14p8 was not significant. As both histidine (H) and arginine (R) are positively charged, these data suggest that P14 prefers either a basic and/or larger residue in the two N-terminal positions to the acceptor residue. However, as there was no change in ADPr transfer with an alanine substitution at the two C-terminal positions to the acceptor residue, they are non-essential for P14 targeting.
P14 is thought to modify aspartate (D) as well as E, so a final swap from E to D in the 5-mer was similarly tested for P14c activity. There was a non-significant decrease (8.9%) in labeling when D was the primary acceptor, confirming the ability of P14c to modify both D and E within these minimal peptide substrates. The activity of P14-∆553 was also assessed to confirm that the significant decreases in labeling observed with P14p9 and P14p10 were not attributable to a promiscuity difference in P14c. For both peptides, there was no detectable labeling in the presence of P14-∆553 (
Figure S3). These results match the observations made with P14c and confirm that both the catalytic fragment and the longer PARP14 proteins share a similar set of proximal preferences when identifying a target sequence
in vitro.
To determine whether the 37.7% decrease in P14c activity we observed for the P14p9 peptide was due to the positive charge in the arginine (R) or its larger size compared to A, two new peptide substrates were designed with either methionine (M, P14p13) or lysine (K, P14p14) at the acceptor minus one site (
Figure 3d). As with the A substitution, the presence of the larger, though uncharged, M residue results in a 44% decrease in P14 activity. Therefore, the decrease observed with an A substitution was likely not due to the difference in size at this position. The K substitution—which maintains its charge at this position—resulted in a 46% increase in P14 activity. It appears that, while R is found at this position in the original sequence, a K residue further enhances activity. Interestingly, the preference for a lysine in the acceptor minus one site has been observed for PARP1 [
27] and was one of the putative motifs suggested in prior proteomic analyses of the ADP-ribosylome [
20].
To analyze the potential requirement for a minimum of two C-terminal residues for ADP–ribose transfer, a set of peptides were designed with the R-E sequence slid towards the C-terminus (P14p15 and P14p16) (
Figure 3g). In both cases, the loss of amino acids C-terminal to the E acceptor resulted in a non-significant decrease in overall ADP-ribosylation (8.2% for both P14p15 and P14p16 versus 12.2% for P14p6,
Figure 3h,i). Therefore, while a minimal overhang is required for maximal transfer, it is not as essential as the preference for a K/R in the position N-Terminal for the acceptor residue. Taken together, these results demonstrate a general promiscuity in P14c targeting and validate a preference for sequences with a basic–K/R–D/E–X–X signature.
Following the identification of a putative motif for P14-dependent ADPr transfer, we determined whether this motif influenced enzymatic and non-enzymatic MAR hydrolysis. Like other ester bonds, the peptide–MAR linkage is base labile [
28]. However, previous studies on the reversal of the MAR ester bond routinely used basic conditions well above the pH range observed in the cell (pH > 9) [
5,
29], making it difficult to interpret how stable this modification is
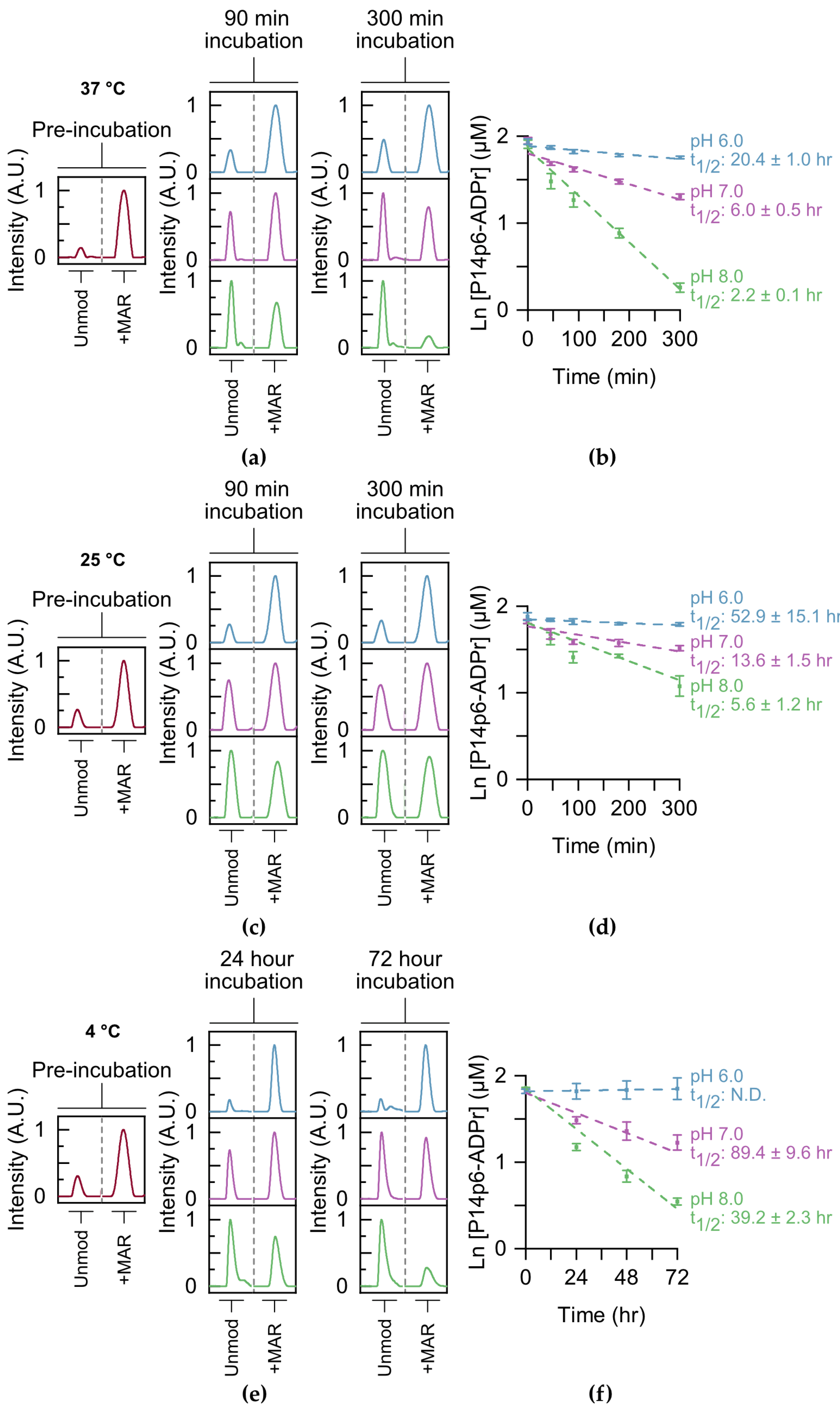
in vivo. The presence of multiple MAR/PAR sites on proteins further complicates the analysis of single-site hydrolysis. We reasoned that our TLC-MALDI approach would be well suited to studying the kinetics of single-site MAR removal without these complications. P14c was used to label both P14p6 and P14p9 homogenously, and MARylated peptide was purified using a three-step method. The resultant P14p6–MAR peptide was equilibrated in a range of pH conditions (pH 6–8) and the removal kinetics at 37 °C were monitored using TLC-MALDI (
Figure 4a). The hydrolysis of ADP–ribose was best described by a pseudo first-order reaction, which was used to determine the half-life of MAR hydrolysis. The half-life of MAR–peptide at pH 8 is 2.2 h and is only slightly longer at pH 7 (t
1/2 = 6.0 h) (
Figure 4b). Only by equilibrating the MAR–peptide in slightly acidic conditions were half-lives in the day range (t
1/2 = 20.4 h) observed. The hydrolysis assays were repeated at room temperature (25 °C) to investigate the effect of temperature on the stability of the MAR–peptide bond. Predictably, lowering the temperature slowed the loss of MAR at all pHs tested, though there is still appreciable removal within the mild base (t
1/2 = 5.6 h at a pH of 8) (
Figure 4c). However, equilibration in slightly acidic conditions significantly slows the hydrolysis of MAR and results in a half-life of greater than two days (
Figure 4d). Lowering the temperature even further to 4 °C stabilized the modification, though it did not prevent hydrolysis at either pH 8 or 7 (
Figure 4e). Of all the conditions tested, only the equilibration of MAR–P14p6 in acidic conditions on ice seemed to halt non-enzymatic removal (
Figure 4f). These results highlight the dynamics of ADP-ribosylation
in vitro and have important implications for the methodologies utilized to study ADP-ribosylation (as discussed further below).
Next, we wanted to determine whether the observed specificity was attributable to either (1) a specific interaction between the acceptor minus one site and P14, or (2) a stabilizing intramolecular interaction between the positive charge on the adjacent R residue and MAR that slows hydrolysis. To discern this, hydrolysis assays were performed with P14p9, and the rates of hydrolysis for P14p9 were compared to those for P14p6 (
Figure S4). No significant difference was observed in any of the tested conditions between the non-enzymatic rates of hydrolysis for P14p6 and P14p9. As such, the observed increase in MARylation activity seen for P14p6 is likely due to a specific interaction with P14 rather than its susceptibility to hydrolysis.
After surveying non-enzymatic hydrolysis, we moved on to study the effects of proximal sequence on glycohydrolase activity. We limited our study to removal enzymes that had previously been validated as being both D/E and mono-ADPr selective:
macroD1,
macroD2, and TARG1 [
11,
12,
13,
14,
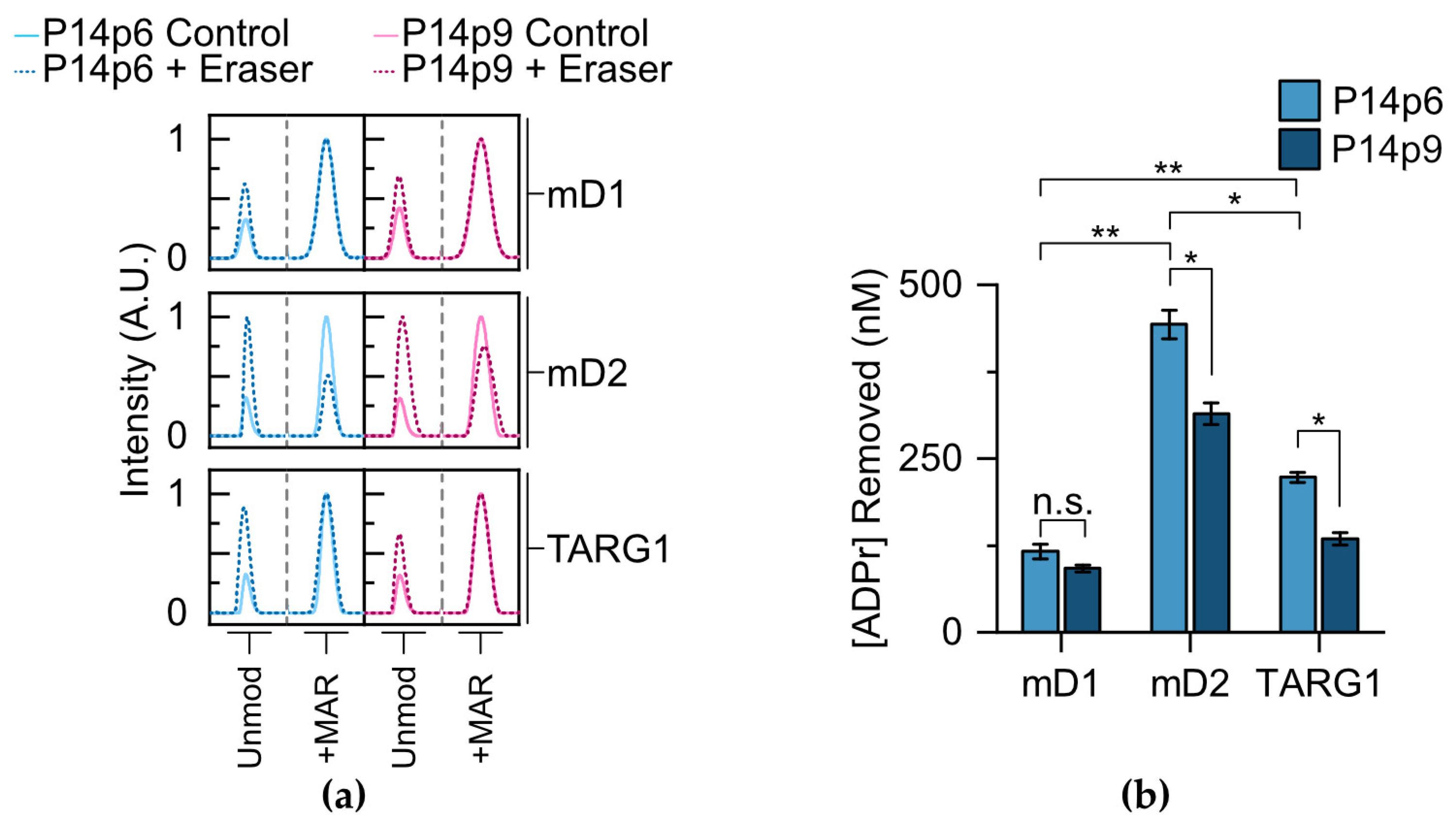
15]. Each of the glycohydrolases were incubated with either P14p6 or P14p9, and the amount of MAR hydrolysis present was compared to a non-enzymatic control reaction (
Figure 5a). All three of the erasers showed activity for the P14p6 peptide, with
macroD2 displaying a 3.8-fold increase in activity compared to
macroD1 and a 2.0-fold increase compared to TARG1 (
Figure 5b).
MacroD2 is, therefore, the most active enzyme in our experimental conditions with peptidyl substrates; however, it is important to note that the effects of removal on full-length proteins in a cell-based context requires further investigation to determine how they differ from
in vitro conditions with a simple peptide.
Next, each of the erasers was incubated with the P14p9 peptide and MAR removal was assessed to determine the effects of the acceptor minus one position on glycohydrolase activity. As with P14p6, each of the enzymes tested was active in the presence of the MARylated P14p9 peptide (
Figure 5a). However, differential sensitivities were observed for the substitution at the acceptor adjacent position, with
macroD1 displaying no significant difference in activity for P14p6 versus P14p9, while both
macroD2 and TARG1 had a 30–40% decrease in activity with the altered sequence (
Figure 5b). While potential differences between the activities of the glycohydrolases
in vitro and in a cellular context could account for some of this discrepancy, it is interesting to find a relative difference in sequence preference at the proximal site for these erasers. Taken together, this investigation of glycohydrolase activity with P14c generated MAR–peptides reveals differences in substrate preference and sensitivity to proximal sequences while demonstrating the utility of TLC-MALDI in elucidating fundamental ADP–ribose biochemistry.
4. Conclusions
Herein, we demonstrated the applicability of the TLC-MALDI approach in the identification and validation of site motifs within the ARTD family. Working with a minimal motif containing a single acceptor site facilitated the determination of the effects of the surrounding amino acids on the activity of P14c. While P14c is broadly promiscuous, there is a distinct preference
in vitro for sites with a basic–K/R–D/E signature. As we have presented a method for generating ADP-ribosylated peptides, it will be interesting to include these reagents in structure-based efforts to determine how the proximal basic–K/R signature influences ADP–ribose transfer. It will be important to discover if the proximal sequence directly interacts with P14 or if it plays a more indirect role in stabilizing the negative charge present within the attached phosphodiester. Based on our prior work with P14, we were also curious how prevalent this signature was within previously identified P14 targets. Unfortunately, the number of validated P14 specific target sites is still limited (eight sites total on PARP13). Of those sites, a single site has this specific signature, though this was the site that was modified the most rapidly in the prior study [
31]. There is a clear need for further work identifying ARTD family-member specific targets, and it will be interesting to compare how the
in vitro preferences found herein can be compared to future modification sites identified in ongoing proteomic efforts. It will also be worthwhile to apply this technique to the remaining ARTD family members with their own unique target sequences. Completion of this comparative analysis will help reveal the level of specificity within the family and build on the mechanism for ARTD targeting. Regardless, our current results can immediately be used to screen putative sites from proteomic surveys to identify bona fide
in vivo P14 sites.
It is important to note that there are inherent limitations to the work we have described herein. Our study was designed to explore the minimal motifs required to guide proximal targeting by PARP14, which means we were only considering linear sequences and not considering the effects of folded proteins on targeting. It is certainly possible that the folded nature of various sites could influence their accessibility at the PARP14 active site. However, the recently proposed structure of PARP13 on AlphaFold has localized the P14p1 site to a disordered loop, which would be similar in structure to our analyzed peptides [
32,
33]. We also focused our study primarily on a single peptide. While we would argue that we have selected one of the more compelling PARP14 sites based on the previous literature, it will be important to explore how the proximal interactions we have identified are shared between future PARP14 sites and those found for other ARTD family members. Finally, P14c and the longer PARP14 variants undergo significant MARylation both
in vitro and
in vivo [
25,
31]. The presence of MARylation on these proteins could certainly alter target selection, and it would be interesting to compare how proximal preferences are impacted by the loss of auto-modification sites.
One of the main advantages of the described work is the ability to interrogate both the attachment and removal of MAR at a single minimal acceptor site. TLC-MALDI reveals that the esterification of peptide with MAR results in a far more labile modification than previously described [
5,
29]. While certain factors in the cell (e.g., burying of sites within protein–protein interaction surfaces, intracellular compartmentalization, etc.) could certainly alter the hydrolysis dynamics we have uncovered, understanding how the innate stability of MAR at D/E residues impacts signal persistence will be of particular interest going forward. Moreover, recent studies have shown that MAR/PAR transfer is not restricted to a single type of amino acid, with PARP activity observed on arginine (R), lysine (K), cysteine (C), histidine (H), serine (S), tyrosine (Y), and the aforementioned D/E [
25,
34,
35,
36,
37,
38]. Expanding this technique to additional types of MAR/PAR–peptide bond chemistries will help uncover how the unique chemistry at the acceptor site affects PTM lability. Investigation of the interplay between site chemistry and MAR/PAR stability could have important implications for how the kinetics of non-enzymatic removal impact MAR/PAR signal transduction pathways. Recent work completed by Tashiro and colleagues demonstrated a similar effect of non-enzymatic hydrolysis on synthetically derived MARylated peptides using an orthogonal HPLC method [
39]. Combined with our findings, these data suggest that esterification of acidic residues by ADP–ribose is a highly reversible process in mild conditions. These results similarly hint at a larger potential pool of underrepresented MAR/PAR sites that have been overlooked due to the instability of the ester bond. When preparing protein libraries for MS/MS analysis, it is fairly common to equilibrate the digests in pH neutral conditions at elevated temperatures for more than a day (e.g., overnight treatment with trypsin). Based on the current findings, it is possible that treatment of peptide libraries in this manner would result in a significant loss of MAR/PAR from acidic acceptor sites. In a complex pool of acceptor site chemistries, this could result in the over-enrichment of non-ester linkages and an apparent absence of D/E modifications. Therefore, the exploration of the impact of temperature and pH on MS/MS-based site discovery has the potential to identify an underrepresented population of sites.
Finally, the TLC-MALDI method has been successfully adapted from quantifying ADPr transfer to ADPr removal using several different glycohydrolases. As this class of enzymes were thought to be fairly substrate agnostic, it was surprising to find differential substrate preferences based on proximal interactions [
39,
40]. For example,
macroD1 has been previously shown to remove ADPr from a range of substrates (e.g., DNA, RNA, and protein) and can function as an O-acetyl-ADPr deacetylase [
41]. The data support the role of
macroD1 as a promiscuous removal enzyme, but this lack of specificity might contribute to a lower activity for the peptidyl substrates analyzed. Recent work by Žaja and co-workers identified an enrichment of
macroD2 in neuronal cells [
42]. Further, similar efforts have highlighted specific neurological phenotypes in mice models associated with a loss of
macroD2 [
43]. Coupled with the observation that
macroD2 is the most active eraser of the P14c generated MAR–peptides, this could hint at a potential antagonistic role for P14 and
macroD2 in the brain. Obviously, it is not possible in this study to definitively state that any of the tested glycohydrolases are P14 specific in the cell. Further work will be required to determine if the preferences observed
in vitro with peptidyl substrates will match the
in vivo preferences with whole proteins. Combined, these results have expanded the role TLC-MALDI can have in analyzing ADP-ribosylation while providing new insights into the mechanism of target selection, and we suggest this as a complement to ongoing efforts to examine the function of ADPr-dependent signaling.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




