
Vinylation of N-Heteroarenes through Addition/Elimination Reactions of Vinyl Selenones

 ,
,  and
and 
Abstract
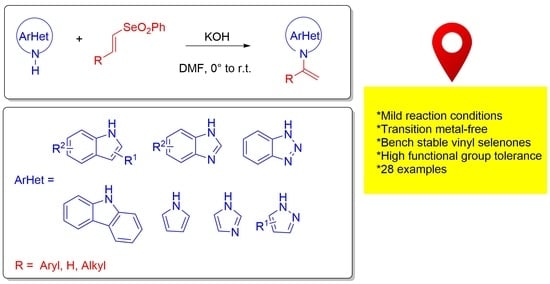
1. Introduction
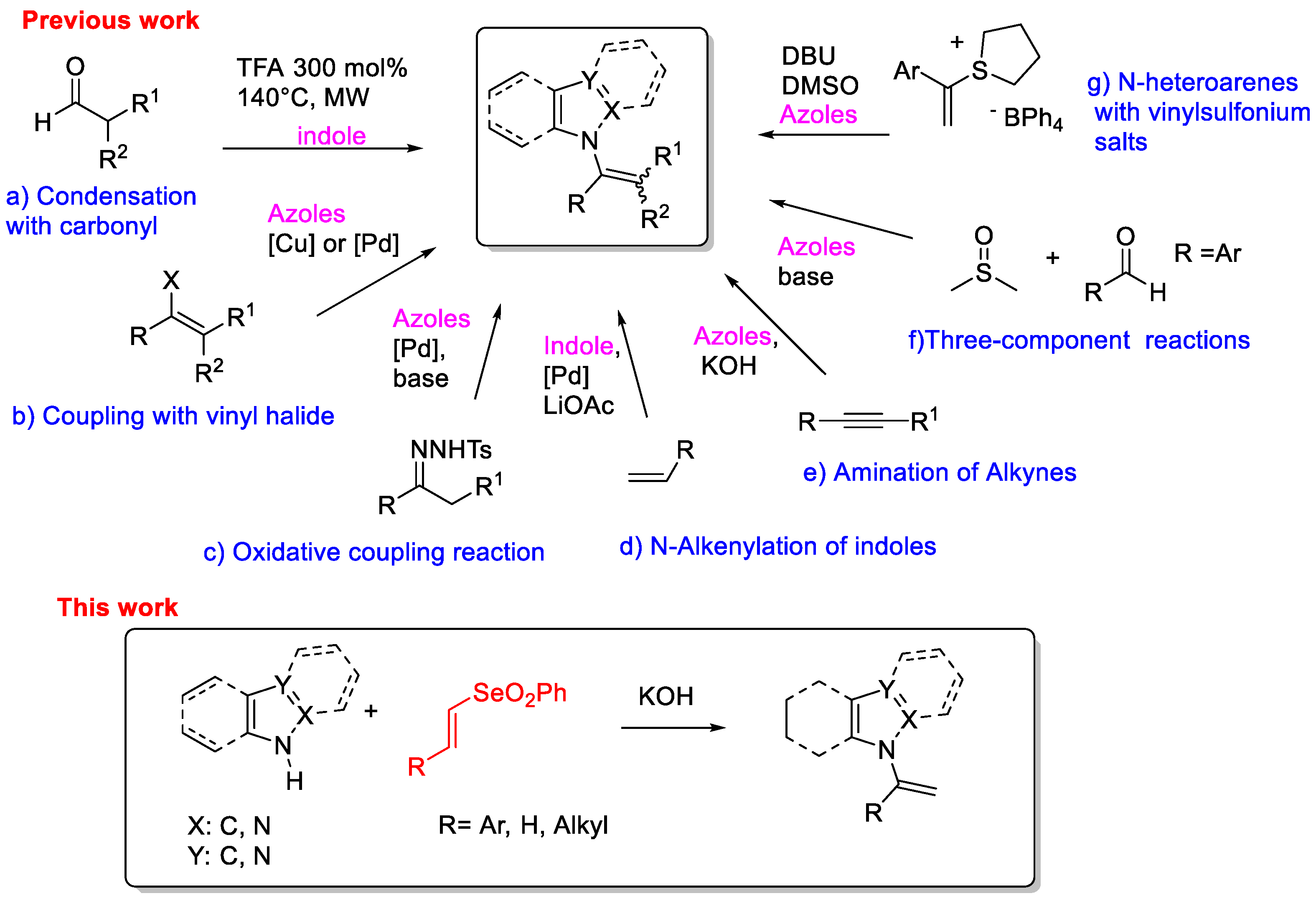
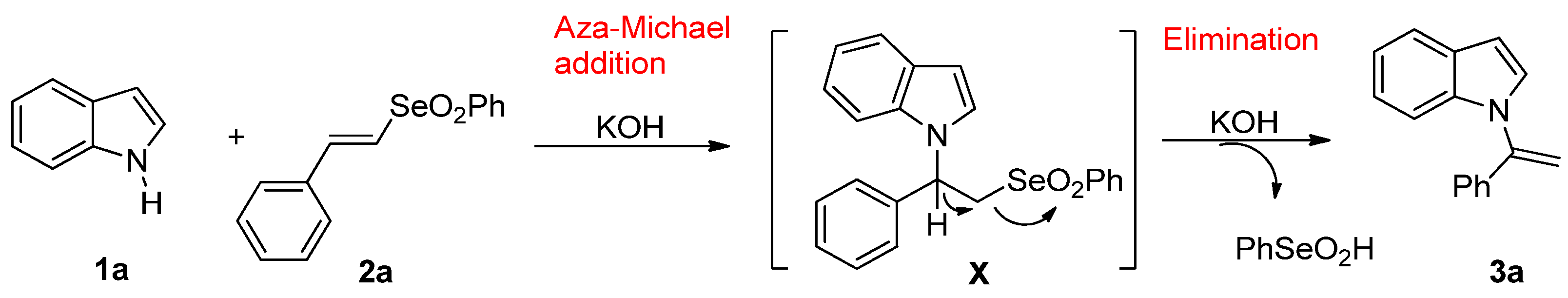
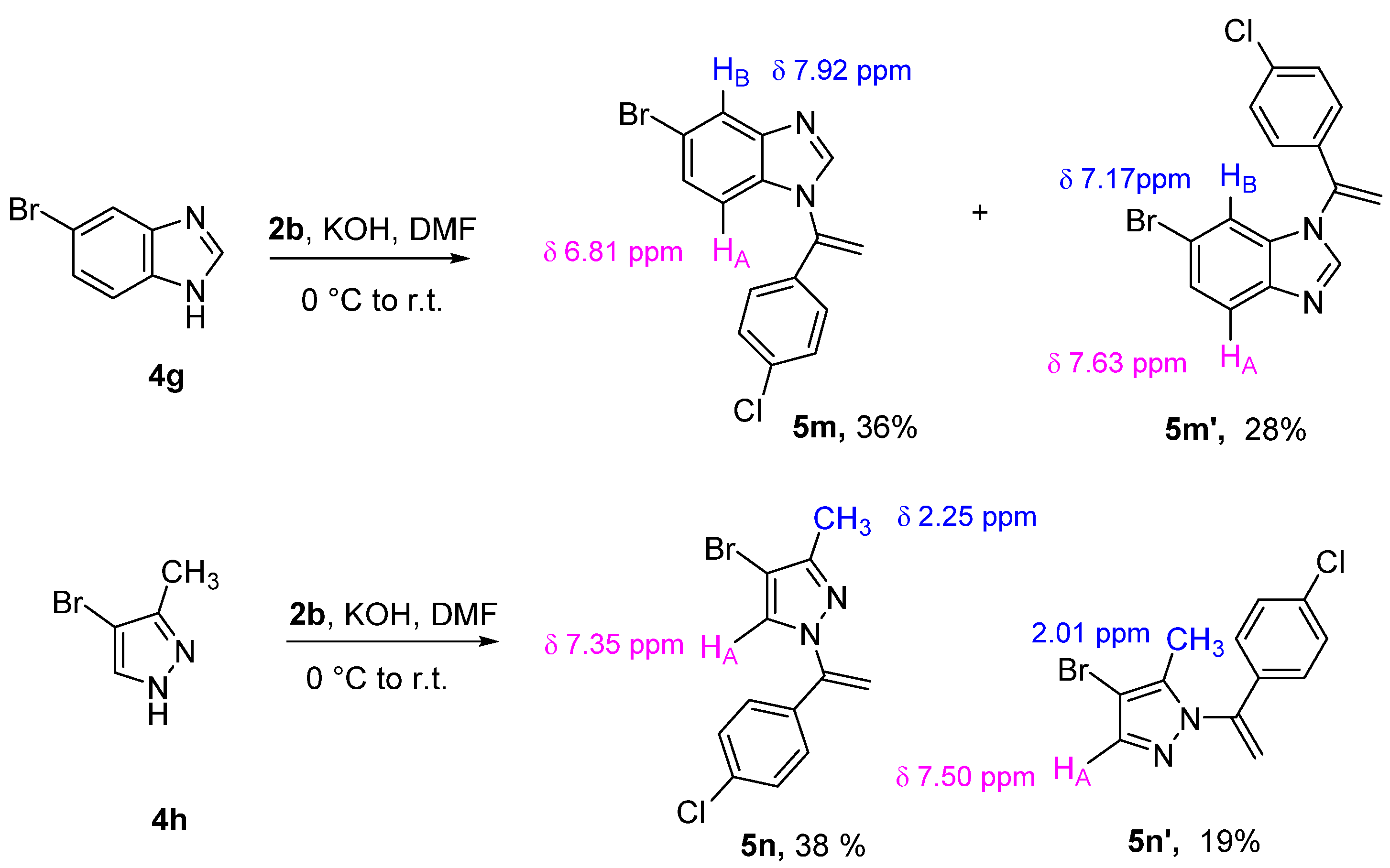
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Starting Materials
3.3. General Procedure for the Synthesis of N-Vinyl Azoles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Silva, V.L.M.; Silva, A.M.S. Revisiting the Chemistry of Vinylpyrazoles: Properties, Synthesis, and Reactivity. Molecules 2022, 27, 3493. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Matsumoto, H.; Shimizu, S.; Kida, S.; Shiro, M.; Tawara, K. Synthesis and Antifungal Activity of New 1-Vinylimidazoles. J. Med. Chem. 1987, 30, 1348–1354. [Google Scholar] [CrossRef] [PubMed]
- Radi, M.; Dreassi, E.; Brullo, C.; Crespan, E.; Tintori, C.; Bernardo, V.; Valoti, M.; Zamperini, C.; Daigl, H.; Musumeci, F.; et al. Design, Synthesis, Biological Activity, and ADME Properties of Pyrazolo[3,4-d]pyrimidines Active in Hypoxic Human Leukemia Cells: A Lead Optimization Study. J. Med. Chem. 2011, 54, 2610–2626. [Google Scholar] [CrossRef]
- Huang, W.-S.; Zhu, X.; Wang, Y.; Azam, M.; Wen, D.; Sundaramoorthi, R.; Thomas, R.M.; Liu, S.; Banda, G.; Lentini, S.P.; et al. 9-(Arenethenyl)purines as Dual Src/Abl Kinase Inhibitors Targeting the Inactive Conformation: Design, Synthesis, and Biological Evaluation. J. Med. Chem. 2009, 52, 4743–4756. [Google Scholar] [CrossRef]
- Ghost, A.; Stanley, L.M. Enantioselective hydroacylation of N-vinylindole-2-carboxaldehydes. Chem. Commun. 2014, 50, 2765–2768. [Google Scholar] [CrossRef]
- Ghosh, A.; Bainbridge, D.T.; Stanley, L.M. Enantioselective model synthesis and progress toward the Putative structure of Yuremamine. J. Org. Chem. 2016, 81, 7945–7951. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Boonnak, N.; Padwa, A. N-Alkenyl Indoles as Useful Intermediates for Alkaloid Synthesis. J. Org. Chem. 2011, 76, 9488–9496. [Google Scholar] [CrossRef]
- Maki, Y.; Mori, H.; Endo, T. Xanthate-Mediated Controlled Radical Polymerization of N-Vinylindole Derivatives. Macromolecules 2007, 40, 6119–6130. [Google Scholar] [CrossRef]
- Brustolin, F.; Castelvetro, V.; Ciardelli, F.; Ruggeri, G.; Colligiani, A. Synthesis and Characterization of Different Poly(1-vinylindole)s for Photorefractive Materials. J. Polym. Sci. Part A Polym. Chem. 2001, 39, 253–262. [Google Scholar] [CrossRef]
- Bekkar, F.; Bettahar, F.; Moreno, I.; Meghabar, R.; Hamadouche, M.; Hernáez, E.; Vilas-Vilela, J.L.; Ruiz-Rubio, L. Polycarbazole and Its Derivatives: Synthesis and Applications. A Review of the Last 10 Years. Polymers 2020, 12, 2227. [Google Scholar] [CrossRef] [PubMed]
- Fridkin, G.; Boutard, N.; Lubell, W.D. β,β-Disubstituted C- and N-vinylindoles from One-Step Condensations of Aldehydes and Indole Derivatives. J. Org. Chem. 2009, 74, 5603–6603. [Google Scholar] [CrossRef] [PubMed]
- Taillefer, M.; Ouali, A.; Renard, B.; Spindler, J.-F. Mild Copper-Catalyzed Vinylation Reactions of Azoles and Phenols with Vinyl bromides. Chem. Eur. J. 2006, 12, 5301–5313. [Google Scholar] [CrossRef] [PubMed]
- Liao, Q.; Wang, Y.; Zhang, L.; Xi, C. A General Copper—Catalyzed Coupling of azoles with Vinyl Bromide. J. Org. Chem. 2009, 74, 6371–6373. [Google Scholar] [CrossRef] [PubMed]
- Lebedev, A.Y.; Izmer, V.V.; Kazyul’kin, D.N.; Beletskaya, I.P.; Voskoboynikov, A.Z. Palladium-Catalyzed Stereocontrolled Vinylation of Azoles and Phenothiazine. Org. Lett. 2002, 4, 623–626. [Google Scholar] [CrossRef]
- Roche, M.; Frison, G.; Brion, J.-D.; Provot, O.; Hamze, A.; Alami, M. Csp2-N bond Formation via Ligand-Free Pd-Catalyzed Oxidative Coupling Reaction of N-Tosylhydrazones and Indole derivatives. J. Org. Chem. 2013, 78, 8485–8495. [Google Scholar] [CrossRef]
- Zeng, X.; Cheng, G.; Shen, J.; Cui, X. Palladium–catalyzed Oxidative Cross-Coupling of N-Tosylhydrazones with Indoles: Synthesis of N-Vinylindoles. Org. Lett. 2013, 15, 3022–3025. [Google Scholar] [CrossRef]
- Wu, G.; Su, W. Regio- and Stereoselective Direct N-Alkenylation of Indoles via Pd-Catalyzed Aerobic Oxidation. Org. Lett. 2013, 15, 5278–5281. [Google Scholar] [CrossRef]
- Rodygin, K.R.; Bogachenkov, A.S.; Ananikov, V.P. Vinylation of a Secondary Amine Core with Calcium Carbide for Efficient Post-Modification and Access to Polymeric Materials. Molecules 2018, 23, 648. [Google Scholar] [CrossRef]
- Verma, A.K.; Joshi, M.; Singh, V.P. Base–Mediated Regio– and Stereoselective Intermolecular Addition of Alkynes to N-Heterocycles. Org. Lett. 2011, 13, 1630–1633. [Google Scholar] [CrossRef]
- Garg, V.; Kumar, P.; Verma, A.K. Substrate-Controlled Regio- and Stereoselective Synthesis of (Z)- and (E)-N- Styrylated Carbazoles, Aza-carbazoles, and γ-Carbolines via Hydroamination of Alkynes. J. Org. Chem. 2018, 83, 11686–11702. [Google Scholar] [CrossRef]
- Zhou, M.; Tan, X.; Hu, Y.; Shen, H.C.; Qian, X. Highly Chemo- and Regioselective Vinylation of N-Heteroarenes with Vinylsulfonium salts. J. Org. Chem. 2018, 83, 8627–8635. [Google Scholar] [CrossRef]
- Zou, L.-H.; Liu, B.; Wang, C.; Shao, Z.; Zhou, J.; Shao, A.; Wen, J. Selective synthesis of alkyl amines and N- vinylazoles from vinyl sulfonium salts with N-nucleophiles. Org. Chem. Front. 2022, 9, 3231–3236. [Google Scholar] [CrossRef]
- Nie, Z.; Lv, H.; Li, H.; Su, M.; Yang, T.; Luo, W.; Liu, Q.; Guo, C. Synthesis of terminal N-Vinylazoles from Aromatic Aldehydes, DMSO, and Azoles based DMSO as Terminal Carbon Synthon. Adv. Synth Catal. 2021, 363, 4621–4626. [Google Scholar] [CrossRef]
- Bagnoli, L.; Santi, C. Organoselenium compound as chiral building blocks. In Chiral Building Blocks in Asymmetric Synthesis: Synthesis and Application, 1st ed.; WILEY–VCH: Hoboken, NJ, USA, 2022; Chapter 14; pp. 463–488. ISBN 978-3-527-34946-3. [Google Scholar]
- Torres-Ochoa, R.O.; Buyck, T.; Wang, Q.; Zhu, J. Heteroannulation of arynes with α-amino imides: Synthesis of 2,2-disubstituted indolin-3-ones and application to the enantioselective total synthesis of (+)-Hinckdentine A. Angew. Chem. Int. Ed. 2018, 57, 5679–5683. [Google Scholar] [CrossRef]
- Palomba, M.; De Monte, E.; Mambrini, A.; Bagnoli, L.; Santi, C.; Marini, F. A three-component [3 + 2]-cycloaddition/ elimination cascade for the synthesis of spirooxindole-pyrrolizines. Org. Biomol. Chem. 2021, 19, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, A.; Samanta, S.; Pathak, T. Enantiopure 1,4,5-trisubstituted 1,2,3-triazoles from carbohydrates: Applications of organoselenium chemistry. J. Org. Chem. 2014, 79, 6895–6904. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Casini, S.; Marini, F.; Santi, C.; Testaferri, L. Vinyl selenones: Annulation agents for the synthesis of six-membered benzo-1,4-heterocyclic compounds. Tetrahedron 2013, 69, 481–486. [Google Scholar] [CrossRef]
- Palomba, M.; Vinti, E.; Marini, F.; Santi, C.; Bagnoli, L. Synthesis of oxazino[4,3-a]indoles by domino addition-cyclization reactions of 1H-indol-2-yl)methanols and vinyl selenones in the presence of 18-crown-6. Tetrahedron 2016, 72, 7059–7064. [Google Scholar] [CrossRef]
- Palomba, M.; Sancineto, L.; Marini, F.; Santi, C.; Bagnoli, L. A domino approach to pyrazino-indoles and pyrroles using vinyl selenones. Tetrahedron 2018, 74, 7156–7163. [Google Scholar] [CrossRef]
- Buyck, T.; Wang, Q.; Zhu, J. Catalytic enantioselective Michael addition of α-aryl-α-isocyanoacetates to vinyl selenone: Synthesis of α,α-disubstituted α-amino acids and (+)- and (–)-trigonoliimine A. Angew. Chem. Int. Ed. 2013, 52, 12714–12718. [Google Scholar] [CrossRef]
- Buyck, T.; Wang, Q.; Zhu, J. Triple role of phenylselenonyl group enabled a one-pot synthesis of 1,3-oxazinan-2-ones from α-isocyanoacetates, phenyl vinyl selenones, and water. J. Am. Chem. Soc. 2014, 136, 11524–11528. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Scarponi, C.; Testaferri, L.; Tiecco, M. Preparation of both enantiomers of cyclopropane derivatives from the reaction of vinyl selenones with di-(-)-bornyl malonate. Tetrahedron Asymmetry 2009, 20, 1506–1514. [Google Scholar] [CrossRef]
- Zhang, T.; Cheng, L.; Hameed, S.; Liu, L.; Wang, D.; Chen, Y.-J. Highly enantioselective Michael addition of 2-oxindoles to vinyl selenone in RTILs catalyzed by a Cinchona alkaloid-based thiourea. Chem. Commun. 2011, 47, 6644–6646. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, L.; Scarponi, C.; Rossi, M.G.; Testaferri, L.; Tiecco, M. Synthesis of enantiopure 1,4-dioxanes, morpholines, and piperazines from the reaction of chiral 1,2-diols, amino alcohols, and diamines with vinyl selenones. Chem. Eur. J. 2011, 17, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Sternativo, S.; Walczak, O.; Battistelli, B.; Testaferri, L.; Marini, F. Organocatalytic Michael addition of indanone carboxylates to vinyl selenone for the asymmetric synthesis of polycyclic pyrrolidines. Tetrahedron 2012, 68, 10536–10541. [Google Scholar] [CrossRef]
- Bhaumik, A.; Das, A.; Pathak, T. Vinyl selenones derived from d-Fructose: A new platform for fructochemistry. Asian J. Org. Chem. 2016, 5, 1048–1062. [Google Scholar] [CrossRef]
- Bhaumik, A.; Pathak, T. Methyl-α D 2-selenonyl Pent-2-enofuranoside: A reactive selenosugar for the diversity-oriented synthesis of enantiomerically pure heterocycles, carbocycles, and isonucleosides. J. Org. Chem. 2015, 80, 11057–11064. [Google Scholar] [CrossRef]
- Palomba, M.; Trappetti, F.; Bagnoli, L.; Santi, C.; Marini, F. Oxone mediated oxidation of vinyl selenides in water. Eur. J. Org. Chem. 2018, 2018, 3914–3919. [Google Scholar] [CrossRef]
- Bandini, M.; Melloni, A.; Umani-Ronchi, A. New versatile Pd-Catalyzed Alkylation of Indoles via Nucleophilic Allylic Substitution: Controlling the Regioselectivity. Org. Lett. 2004, 6, 3199–3202. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Young, M.W.; Lauer, R.F. Reactions of Selenoxides: Thermal Syn-Elimination and H218O Exchange. Tetrahedron Lett. 1973, 14, 1979–1982. [Google Scholar] [CrossRef]
- Palomba, M.; Dias, I.F.C.; Rosati, O.; Marini, F. Modern Synthetic Strategies with Organoselenium Reagents. Molecules 2021, 26, 3148. [Google Scholar] [CrossRef]
- Azaz, T.; Mourya, H.; Singh, V.; Ram, B.; Tiwari, B. Reductive Alkenylation of Ketimines via Hydride Transfer from Aldehydes. J. Org. Chem. 2023, 88, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, A.; Azaz, T.; Singh, V.; Khatana, A.K.; Tiwari, B. Carbene/Base–Mediated Redox Alkenylation of Isatins using β-Substituted Organoselenenones and Aldehydes. J. Org. Chem. 2019, 84, 14898–14903. [Google Scholar] [CrossRef] [PubMed]
- Tsuchimoto, T.; Aoki, K.; Wagatsuma, T.; Suzuki, Y. Lewis Acid Catalyzed Addition of Pyrazoles to Alkynes: Selective Synthesis of Double and Single Addition Products. Eur. J. Org. Chem. 2008, 4035–4040. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Base | Equivalent | Solvent | Temp. | Time | 3a, Yields % |
| 1 | Cs2CO3 | 2.5 | Toluene | reflux | 12 h | - |
| 2 | Li2CO3 | 2.5 | CH2Cl2 | r.t. | 48 h | - |
| 3 | KOH | 2.5 | CH2Cl2 | 0 °C to r.t. | 9 h | - |
| 4 | KOH | 2.5 | EtOH | 0 °C to r.t. | 60 h | - |
| 5 | KOH | 2.5 | DMF | 0 °C to r.t. | 2 h | 86 |
| 6 | KOH | 2.5 | DMSO | r.t. | 12 h | 82 |
| 7 | KOH | 2.5 | CH3CN | 0 °C to r.t. | 12 h | 50 |
| 8 | Cs2CO3 | 2.5 | DMF | 0 °C to r.t. | 72 h | 51 |
| 9 | NaH 60% | 2.5 | DMF | 0 °C to r.t. | 48 h | 49 |
| 10 | DBU | 2.5 | DMF | 0 °C to r.t. | 12 h | - |
| 11 | t-BuOK | 2.5 | DMF | 0 °C to r.t. | 12 h | 79 |
| 12 | KOH | 1.5 | DMF | 0 °C to r.t. | 72 h | 40 |
| 13 | - | - | DMF | 0 °C to r.t. | 12 h | - |
 | |||
 |  |  |  |
| 3a, 82% | 3b, 86% | 3c, 87% | 3d, 60% |
 |  |  |  |
| 3e, 65% | 3f, 45% | 3g, 71% | 3h, 50% |
 |  |  |  |
| 3i, 60% | 3j, 35% | 3k, 50% | 3l, 36% Oxazine *, 22% |
 | |||
|---|---|---|---|
 |  |  |  |
| 5a, 40% | 5b, 50% | 5c, 77% | 5d, 66% |
 |  |  |  |
| 5e, 30% | 5f, 53% | 5g, 58% | 5h, 63% |
 |  |  |  |
| 5i, 75% | 5j, 61% | 5k, 74% | 5l, 73% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palomba, M.; Dias, I.F.C.; Cocchioni, M.; Marini, F.; Santi, C.; Bagnoli, L. Vinylation of N-Heteroarenes through Addition/Elimination Reactions of Vinyl Selenones. Molecules 2023, 28, 6026. https://doi.org/10.3390/molecules28166026
Palomba M, Dias IFC, Cocchioni M, Marini F, Santi C, Bagnoli L. Vinylation of N-Heteroarenes through Addition/Elimination Reactions of Vinyl Selenones. Molecules. 2023; 28(16):6026. https://doi.org/10.3390/molecules28166026
Chicago/Turabian StylePalomba, Martina, Italo Franco Coelho Dias, Michelangelo Cocchioni, Francesca Marini, Claudio Santi, and Luana Bagnoli. 2023. "Vinylation of N-Heteroarenes through Addition/Elimination Reactions of Vinyl Selenones" Molecules 28, no. 16: 6026. https://doi.org/10.3390/molecules28166026
APA StylePalomba, M., Dias, I. F. C., Cocchioni, M., Marini, F., Santi, C., & Bagnoli, L. (2023). Vinylation of N-Heteroarenes through Addition/Elimination Reactions of Vinyl Selenones. Molecules, 28(16), 6026. https://doi.org/10.3390/molecules28166026