
Fluoroquinolone Analogs, SAR Analysis, and the Antimicrobial Evaluation of 7-Benzimidazol-1-yl-fluoroquinolone in In Vitro, In Silico, and In Vivo Models
 , , ,
, , ,  , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
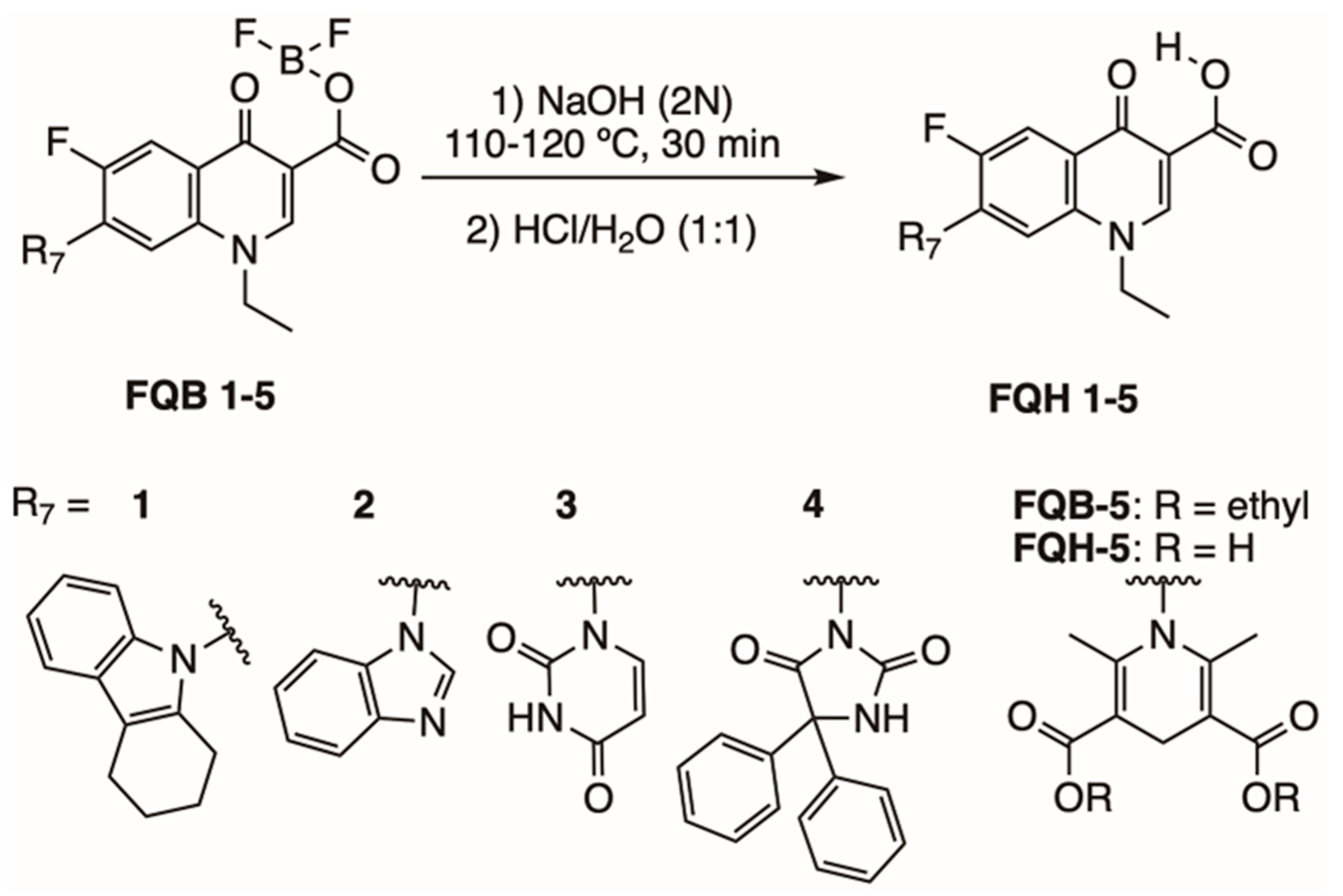
2.1. Synthesis and Chemical Evaluation
2.2. Structure–Activity Relationship (SAR) of the Fluoroquinolone Analogs FQH 1–5 against Gram-Positive and Gram-Negative Reference Bacteria
2.3. In Silico Model by Molecular Docking to Confirm the Antimicrobial Activity of FQH 1–5 and the Mechanism of Action of FQH-2
2.4. Non-Cytotoxic Effect of FQH-2 on Non-Bacterial Cells by Flow Cytometry Assay
2.5. Evaluation of the Antimicrobial Effect of FQH-2 in a Mouse Model of Topical Infection with S. aureus
3. Materials and Methods
3.1. Reagents
3.2. Instrumentation
3.3. Synthesis of Fluoroquinolone Analogs
Chemical Characterization of FQH 1–5
3.4. Antimicrobial Activity
3.5. In Vitro Antimicrobial Activity Assays for the Determination of the Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC)
3.6. Molecular Docking
3.7. Cytotoxicity Assays
3.8. Evaluation of the Antimicrobial Activity of FQH-2 in an In Vivo Model of Topical Infection
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis; World Health Organization: Geneva, Switzerland, 2022. [Google Scholar]
- World Health Organization. Antimicrobial Resistance. 2018. Available online: https://apps.who.int/iris/handle/10665/255204 (accessed on 14 November 2022).
- World Health Organization. Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- World Health Organization. WHO Publishes List of Bacteria for which New Antibiotics Are Urgently Needed; World Health Organization: Geneva, Switzerland, 2017; Available online: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 29 April 2019).
- Ikuta, K.S.; Swetschinski, L.R.; Aguilar, G.R.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Weaver, N.D.; Wool, E.E.; Han, C.; Hayoon, A.G.; et al. Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study. Lancet 2019, 400, 2221–2248. [Google Scholar]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Microbiol. 2020, 18, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Rani, P.; Atanasov, A.G.; Alzahrani, Q.; Gupta, R.; Kapoor, B.; Gulati, M.; Chawla, P. Discovery and Development of Antibacterial Agents: Fortuitous and Designed. Mini Rev. Med. Chem. 2022, 22, 984–1029. [Google Scholar] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Reguero, M.T.; Barreto, E.; Jiménez, F. Relación estructura química actividad biológica: Una revisión retrospectiva. Rev. Colomb. Cienc. Químico-Farm. 1989, 17, 81–84. [Google Scholar]
- Lin, H. The Computational Methods in Drug Targets Discovery. Curr. Drug Targets 2019, 20, 479–480. [Google Scholar] [CrossRef]
- Tutone, M.; Almerico, A.M. Computational Approaches: Drug Discovery and Design in Medicinal Chemistry and Bioinformatics. Molecules 2021, 26, 7500. [Google Scholar] [CrossRef]
- Fedorowicz, J.; Sączewski, J. Modifications of quinolones and fluoroquinolones: Hybrid compounds and dual-action molecules. Monatsh. Chem. 2018, 149, 1199–1245. [Google Scholar]
- Takahashi, H.; Hayakawa, I.; Akimoto, T. The history of the development and changes of quinolone antibacterial agents. Yakushigaku Zasshi 2003, 38, 161–179. [Google Scholar]
- Ball, P. Chapter 1—The Quinolones: History and Overview. In The Quinolones, 3rd ed.; Andriole, V.T., Ed.; Academic Press: San Diego, CA, USA, 2000; pp. 1–31. [Google Scholar]
- Pham, T.D.M.; Ziora, Z.M.; Blaskovich, M.A.T. Quinolone antibiotics. MedChemComm 2019, 10, 1719–1739. [Google Scholar] [CrossRef]
- Madurga, S.; Sánchez-Céspedes, J.; Belda, I.; Vila, J.; Giralt, E. Mechanism of binding of fluoroquinolones to the quinolone resistance-determining region of DNA gyrase: Towards an understanding of the molecular basis of quinolone resistance. Chembiochem 2008, 9, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, J.S.; Hooper, D.C. The fluoroquinolones: Structures, mechanisms of action and resistance, and spectra of activity in vitro. Antimicrob. Agents Chemother. 1985, 28, 581–586. [Google Scholar] [CrossRef]
- Peterson, L.R. Quinolone Molecular Structure-Activity Relationships: What We Have Learned about Improving Antimicrobial Activity. Clin. Infect. Dis. 2001, 33 (Suppl. 3), S180–S186. [Google Scholar] [CrossRef]
- Beermann, D.; Bergan, T.; Christ, W.; Craig, W.A.; Critchlow, S.E.; Dalhoff, A.; Everett, M.J.; Grohe, K.; Kuhlmann, J.; Lode, H.; et al. Mode of Action. In Quinolone Antibacterials; Kuhlmann, J., Dalhoff, A., Zeiler, H.-J., Eds.; Springer Science & Business Media: Berlin/Heidelberg, Germany, 1998; pp. 119–158. [Google Scholar]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Azam, M.A.; Thathan, J.; Jubie, S. Dual targeting DNA gyrase B (GyrB) and topoisomerse IV (ParE) inhibitors: A review. Bioorganic Chem. 2015, 62, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Higgins, P.G.; Fluit, A.C.; Schmitz, F.J. Fluoroquinolones: Structure and target sites. Curr. Drug Targets 2003, 4, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hernández-López, H.; Sánchez-Miranda, G.; Araujo-Huitrado, J.G.; Granados-López, A.J.; López, J.A.; Leyva-Ramos, S.; Chacón-García, L. Synthesis of Hybrid Fluoroquinolone-Boron Complexes and Their Evaluation in Cervical Cancer Cell Lines. J. Chem. 2019, 2019, 5608652. [Google Scholar] [CrossRef]
- Chu, D.T.; Fernandes, P.B. Fernandes, and chemotherapy, Structure-activity relationships of the fluoroquinolones. Antimicrob. Agents Chemother. 1989, 33, 131–135. [Google Scholar] [CrossRef]
- Wang, Y.N.; Bheemanaboina, R.R.Y.; Gao, W.W.; Kang, J.; Cai, G.X.; Zhou, C.H. Discovery of Benzimidazole-Quinolone Hybrids as New Cleaving Agents toward Drug-Resistant Pseudomonas aeruginosa DNA. ChemMedChem 2018, 13, 1004–1017. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [PubMed]
- Pintilie, L.; Stefaniu, A. Molecular Docking Studies of Some Novel Fluoroquinolone Derivatives. Preprints 2019. [Google Scholar] [CrossRef]
- Pintilie, L.U.C.I.A.; Stefaniu, A.M.A.L.I.A.; Nicu, A.I.; Maganu, M.; Caproiu, M.T. Design, Synthesis and Docking Studies of Some Novel Fluoroquinolone Compounds with Antibacterial Activity. Synthesis 2018, 665, 636w. [Google Scholar] [CrossRef]
- Allaka, T.R.; Katari, N.K.; Veeramreddy, V.; Anireddy, J.S. Molecular Modeling Studies of Novel Fluoroquinolone Molecules. Curr. Drug Discov. Technol. 2018, 15, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Millanao, A.R.; Mora, A.Y.; Villagra, N.A.; Bucarey, S.A.; Hidalgo, A.A. Biological Effects of Quinolones: A Family of Broad-Spectrum Antimicrobial Agents. Molecules 2021, 26, 7153. [Google Scholar] [CrossRef]
- Salahuddin; Shaharyar, M.; Mazumder, A. Benzimidazoles: A biologically active compounds. Arab. J. Chem. 2017, 10, S157–S173. [Google Scholar]
- Bansal, Y.; Kaur, M.; Bansal, G. Antimicrobial Potential of Benzimidazole Derived Molecules. Mini Rev. Med. Chem. 2019, 19, 624–646. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Yar, M.S.; Sharma, V.K.; Khan, A.A.; Ali, Z.; Haider, M.D.; Pathak, A. Recent Progress of Benzimidazole Hybrids for Anticancer Potential. Curr. Med. Chem. 2020, 27, 5970–6014. [Google Scholar] [PubMed]
- Pandey, V.; Shukla, A. Synthesis and biological activity of isoquinolinyl benzimidazoles. Indian J. Chem.-Sect. B 1999, 38, 1381–1383. [Google Scholar]
- Küçükbav, H.; Durmaz, R.; Gueven, M.; Guenal, S. Synthesis of some benzimidazole derivatives and their antibacterial and antifungal activities. Arzneimittelforschung 2001, 51, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Tülay Aşkin, Ç. (Ed.) Cytotoxic New Insights into Toxic Assessment; IntechOpen: Rijeka, Croatia, 2021; pp. 1–4. [Google Scholar]
- Azéma, J.; Guidetti, B.; Dewelle, J.; Le Calve, B.; Mijatovic, T.; Korolyov, A.; Vaysse, J.; Malet-Martino, M.; Martino, R.; Kiss, R. 7-((4-Substituted)piperazin-1-yl) derivatives of ciprofloxacin: Synthesis and in vitro biological evaluation as potential antitumor agents. Bioorg. Med. Chem. 2009, 17, 5396–5407. [Google Scholar] [CrossRef] [PubMed]
- Duewelhenke, N.; Krut, O.; Eysel, P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob. Agents Chemother. 2007, 51, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Kloskowski, T.; Gurtowska, N.; Olkowska, J.; Nowak, J.M.; Adamowicz, J.; Tworkiewicz, J.; Dębski, R.; Grzanka, A.; Drewa, T. Ciprofloxacin is a potential topoisomerase II inhibitor for the treatment of NSCLC. Int. J. Oncol. 2012, 41, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Cheung, G.Y.C.; Bae, J.S.; Otto, M. Pathogenicity and virulence of Staphylococcus aureus. Virulence 2021, 12, 547–569. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K. Impetigo. Adv. Emerg. Nurs. J. 2020, 42, 262–269. [Google Scholar] [CrossRef]
- Tapia, A.G.P.; Vázquez, M.S.; Mata, D.C.B.; Charcas, R.M.; Morales, L.E.F.; del Río, L.T.V.; Romo, S.L. Prevalencia de infección de herida quirúrgica, causas y resistencia a los fármacos en el Hospital General de Zona núm. 2 del IMSS, San Luis Potosí. Rev. Espec. Médico-Quirúrgicas 2012, 17, 261–265. [Google Scholar]
- Kamel, C.; McGahan, L.; Mierzwinski-Urban, M.; Embil, J. Appendix 1. Classification of surgical wounds. In Preoperative Skin Antiseptic Preparations and Applications Techniques for Preventing Surgical Site Infections: A Systematic Review of the Clinical Evidence and Guidelines; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2011. [Google Scholar]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, 10–1128. [Google Scholar]
- Rasigade, J.P.; Dumitrescu, O.; Lina, G. New epidemiology of Staphylococcus aureus infections. Clin. Microbiol. Infect. 2014, 20, 587–588. [Google Scholar] [CrossRef]
- Wikler, M.A.J.C. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: Approved standard. Clsi (Nccls) 2006, 26, M7-A7. [Google Scholar]
- Clinical and Laboratory Standards Institute Wayne. M100, Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Agrawal, P.; Singh, H.; Srivastava, H.K.; Singh, S.; Kishore, G.; Raghava, G.P. Benchmarking of different molecular docking methods for protein-peptide docking. BMC Bioinform. 2019, 19 (Suppl. 13), 426. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Lim-Wilby, M. Molecular Docking. In Molecular Modeling of Proteins; Kukol, A., Ed.; Humana Press: Totowa, NJ, USA, 2008; pp. 365–382. [Google Scholar]
- Fährrolfes, R.; Bietz, S.; Flachsenberg, F.; Meyder, A.; Nittinger, E.; Otto, T.; Volkamer, A.; Rarey, M. ProteinsPlus: A web portal for structure analysis of macromolecules. Nucleic Acids Res. 2017, 45, W337–W343. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecula Graphics System; Version 2.0; Schrödinger, LLC.: Cambridge, MA, USA, 2019; Available online: https://pymol.org/2/ (accessed on 25 September 2019).
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Veselkov, D.A.; Laponogov, I.; Pan, X.S.; Selvarajah, J.; Skamrova, G.B.; Branstrom, A.; Narasimhan, J.; Prasad, J.V.V.; Fisher, L.M.; Sanderson, M.R. Structure of a quinolone-stabilized cleavage complex of topoisomerase IV from Klebsiella pneumoniae and comparison with a related Streptococcus pneumoniae complex. Acta Crystallogr. D Struct. Biol. 2016, 72 Pt 4, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, A.; Lotz, C.; Ortiz, J.; Lamour, V. Cryo-EM structure of the complete E. coli DNA gyrase nucleoprotein complex. Nat. Commun. 2019, 10, 4935. [Google Scholar]
- Grievink, H.W.; Luisman, T.; Kluft, C.; Moerland, M.; Malone, K.E. Comparison of Three Isolation Techniques for Human Peripheral Blood Mononuclear Cells: Cell Recovery and Viability, Population Composition, and Cell Functionality. Biopreserv. Biobank. 2016, 14, 410–415. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.C.; Ochoa-González, F.D.L.; Zapata-Zúñiga, M.; Mondragon-Marín, E.; Lara-Ramírez, E.E.; Ruíz-Carrillo, J.L.; DelaCruz-Flores, P.A.; Layseca-Espinosa, E.; Enciso-Moreno, J.A.; Castañeda-Delgado, J.E. GPR15 expressed in T lymphocytes from RA patients is involved in leukocyte chemotaxis to the synovium. J. Leukoc. Biol. 2022, 112, 1209–1221. [Google Scholar]
- National Research Council. Guide for the Care and Use of Laboratory Animals; National Research Council: Washington, DC, USA, 2010.
- Bele, A.A.; Jadhav, V.M.; Nikam, S.R.; Kadam, V.J. Antibacterial potential of herbal formulation. Res. J. Microbiol. 2009, 4, 164–167. [Google Scholar] [CrossRef]
- Rittenhouse, S.; Singley, C.; Hoover, J.; Page, R.; Payne, D. Use of the surgical wound infection model to determine the efficacious dosing regimen of retapamulin, a novel topical antibiotic. Antimicrob. Agents Chemother. 2006, 50, 3886–3888. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, J.D.; Polk, H.C., Jr. Murine Thigh Suture Model. In Handbook of Animals Models of Infeection: Experimental Models in Antimicrobial Chemotherapy; Zak, O., Sande, M.A., Eds.; Academic Press: Cambridge, MA, USA, 1999. [Google Scholar]
- Morton, D.B.; Griffiths, P.H. Guidelines on the recognition of pain, distress and discomfort in experimental animals and an hypothesis for assessment. Vet. Rec. 1985, 116, 431–436. [Google Scholar] [CrossRef] [PubMed]
- McRipley, R.J.; Whitney, R.R. Characterization and quantitation of experimental surgical-wound infections used to evaluate topical antibacterial agents. Antimicrob. Agents Chemother. 1976, 10, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Soma, L.R. Anesthetic and analgesic considerations in the experimental animal. Ann. N. Y. Acad. Sci. 1983, 406, 32–47. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label | Structure | S. aureus (ATCC 25923) | E. faecalis (ATCC 29212) | E. coli (ATCC 25922) | K. pneumoniae * | ||||
|---|---|---|---|---|---|---|---|---|---|
| MIC (mg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | MIC (µg/mL) | MBC (µg/mL) | ||
| CPX | CLSI Values C17H18FN3O3 experimental values | 0.125–0.5 | 0.250 | 0.250–2 | 0.250 | 0.016–0.004 | 0.008 | <1 | 1 |
| 0.250 | 0.250 | 0.250 | 0.250 | 0.013 ± 0.005 a | 0.033 ± 0.002 a | 0.5 | 0.5 | ||
| FQH-1 |  C24H21FN2O3 | 32 | >128 b,c | 128 b | >128 b,c | 1.667 ± 0.577 a | 4 | 64 | 64 |
| FQH-2 |  C19H14FN3O3 | 0.5 | 0.5 | 4 | 4 | 1.333 ± 0.577 a | 2 | 16 | 32 |
| FQH-3 |  C16H12FN3O5 | 128 | 128 | 128 | >128 b,c | 1.667 ± 0.577 a | 4 | 128 | 128 |
| FQH-4 |  C27H21FN3O5 | 128 | >128 c | 128 | >128 b,c | 2 | 4 | 32 | 32 |
| FQH-5 |  C21H22FN2O7 | 128 | >128 b,c | 128 | >128 b | 4 | 4 | 64 | 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Medellín-Luna, M.F.; Hernández-López, H.; Castañeda-Delgado, J.E.; Martinez-Gutierrez, F.; Lara-Ramírez, E.; Espinoza-Rodríguez, J.J.; García-Cruz, S.; Portales-Pérez, D.P.; Cervantes-Villagrana, A.R. Fluoroquinolone Analogs, SAR Analysis, and the Antimicrobial Evaluation of 7-Benzimidazol-1-yl-fluoroquinolone in In Vitro, In Silico, and In Vivo Models. Molecules 2023, 28, 6018. https://doi.org/10.3390/molecules28166018
Medellín-Luna MF, Hernández-López H, Castañeda-Delgado JE, Martinez-Gutierrez F, Lara-Ramírez E, Espinoza-Rodríguez JJ, García-Cruz S, Portales-Pérez DP, Cervantes-Villagrana AR. Fluoroquinolone Analogs, SAR Analysis, and the Antimicrobial Evaluation of 7-Benzimidazol-1-yl-fluoroquinolone in In Vitro, In Silico, and In Vivo Models. Molecules. 2023; 28(16):6018. https://doi.org/10.3390/molecules28166018
Chicago/Turabian StyleMedellín-Luna, Mitzzy Fátima, Hiram Hernández-López, Julio Enrique Castañeda-Delgado, Fidel Martinez-Gutierrez, Edgar Lara-Ramírez, Joan Jair Espinoza-Rodríguez, Salvador García-Cruz, Diana Patricia Portales-Pérez, and Alberto Rafael Cervantes-Villagrana. 2023. "Fluoroquinolone Analogs, SAR Analysis, and the Antimicrobial Evaluation of 7-Benzimidazol-1-yl-fluoroquinolone in In Vitro, In Silico, and In Vivo Models" Molecules 28, no. 16: 6018. https://doi.org/10.3390/molecules28166018
APA StyleMedellín-Luna, M. F., Hernández-López, H., Castañeda-Delgado, J. E., Martinez-Gutierrez, F., Lara-Ramírez, E., Espinoza-Rodríguez, J. J., García-Cruz, S., Portales-Pérez, D. P., & Cervantes-Villagrana, A. R. (2023). Fluoroquinolone Analogs, SAR Analysis, and the Antimicrobial Evaluation of 7-Benzimidazol-1-yl-fluoroquinolone in In Vitro, In Silico, and In Vivo Models. Molecules, 28(16), 6018. https://doi.org/10.3390/molecules28166018