

Modulation of NRF2/KEAP1-Mediated Oxidative Stress for Cancer Treatment by Natural Products Using Pharmacophore-Based Screening, Molecular Docking, and Molecular Dynamics Studies
 , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
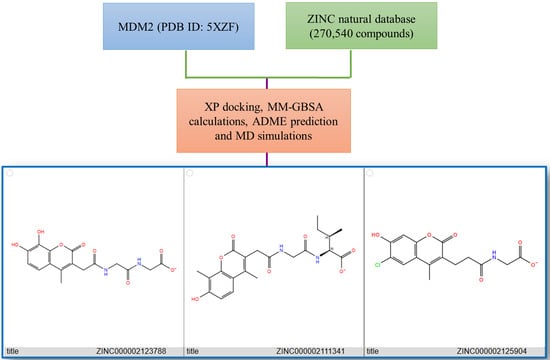
2.1. Pharmacophore Modeling and Virtual Screening
2.2. Molecular Docking and MM/GBSA Calculations
2.3. Ligand–Residue Interactions Analysis
2.4. ADMET Analysis
2.5. MD Simulations
3. Material and Methods
3.1. Protein and Ligands Preparation
3.2. Pharmacophore Generation and Virtual Screening
3.3. Docking and MM/GBSA Calculations
3.4. ADMET Prediction
3.5. MD Simulation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhang, S.; Duan, S.; Xie, Z.; Bao, W.; Xu, B.; Yang, W.; Zhou, L. Epigenetic Therapeutics Targeting NRF2/KEAP1 Signaling in Cancer Oxidative Stress. Front. Pharmacol. 2022, 13, 924817. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1–Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Song, M.Y.; Kim, E.H. Role of oxidative stress and nrf2/keap1 signaling in colorectal cancer: Mechanisms and therapeutic perspectives with phytochemicals. Antioxidants 2021, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Jiang, B.H. Interplay Between Reactive Oxygen Species and MicroRNAs in Cancer. Curr. Pharmacol. Rep. 2016, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The Tumorigenic Roles of the Cellular REDOX Regulatory Systems. Oxidative Med. Cell. Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R.R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Tsai, P.-Y.; Ka, S.-M.; Chang, J.-M.; Chen, H.-C.; Shui, H.-A.; Li, C.-Y.; Hua, K.-F.; Chang, W.-L.; Huang, J.-J.; Yang, S.-S.; et al. Epigallocatechin-3-gallate prevents lupus nephritis development in mice via enhancing the Nrf2 antioxidant pathway and inhibiting NLRP3 inflammasome activation. Free Radic. Biol. Med. 2011, 51, 744–754. [Google Scholar] [CrossRef]
- Cleasby, A.; Yon, J.; Day, P.J.; Richardson, C.; Tickle, I.J.; Williams, P.A.; Callahan, J.F.; Carr, R.; Concha, N.; Kerns, J.K.; et al. Structure of the BTB domain of Keap1 and its interaction with the triterpenoid antagonist CDDO. PLoS ONE 2014, 9, e98896. [Google Scholar] [CrossRef] [PubMed]
- Balstad, T.R.; Carlsen, H.; Myhrstad, M.C.W.; Kolberg, M.; Reiersen, H.; Gilen, L.; Ebihara, K.; Paur, I.; Blomhoff, R. Coffee, broccoli and spices are strong inducers of electrophile response element-dependent transcription in vitro and in vivo—Studies in electrophile response element transgenic mice. Mol. Nutr. Food Res. 2011, 55, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Das, L.; Vinayak, M. Long term effect of curcumin in restoration of tumour suppressor p53 and phase-II antioxidant enzymes via activation of Nrf2 signalling and modulation of inflammation in prevention of cancer. PLoS ONE 2015, 10, e0124000. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G. The Keap1—Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2016, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Sova, M.; Saso, L. Design and development of Nrf2 modulators for cancer chemoprevention and therapy: A review. Drug Des. Devel. Ther. 2018, 12, 3181–3197. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2—An update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.I.; Kobayashi, A.; Katsuoka, F.; Yamamoto, M. Two-site substrate recognition model for the Keap1-Nrf2 system: A hinge and latch mechanism. Biol. Chem. 2006, 387, 1311–1320. [Google Scholar] [CrossRef]
- Uetrecht, J. Handbook of Experimental Pharmacology: Preface; Springer: Berlin/Heidelberg, Germany, 2010; Volume 196, ISBN 9783642006623. [Google Scholar]
- Taguchi, K.; Yamamoto, M. The KeAP1—NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Free Radical Biology and Medicine Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef]
- Pandey, P.; Singh, A.K.; Singh, M.; Tewari, M.; Shukla, H.S.; Gambhir, I.S. The see-saw of Keap1-Nrf2 pathway in Cancer. Crit. Rev. Oncol. Hematol. 2017, 116, 89–98. [Google Scholar] [CrossRef]
- Schmoll, D.; Engel, C.K.; Glombik, H. The Keap1–Nrf2 protein–protein interaction: A suitable target for small molecules. Drug Discov. Today Technol. 2017, 24, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Harder, B.; Jiang, T.; Wu, T.; Tao, S.; Rojo de la Vega, M.; Tian, W.; Chapman, E.; Zhang, D.D. Molecular mechanisms of Nrf2 regulation and how these influence chemical modulation for disease intervention. Biochem. Soc. Trans. 2015, 43, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Tran, K.T.; Pallesen, J.S.; Solbak, S.M.; Narayanan, D.; Baig, A.; Zang, J.; Aguayo-Orozco, A.; Carmona, R.M.C.; Garcia, A.D.; Bach, A. A Comparative Assessment Study of Known Small # Molecule Keap1 # Nrf2 Protein # Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity A Comparative Assessment Study of Known Small-Molecule Keap1-Nrf2 Protein-Prot. J. Med. Chem. 2019, 62, 8028–8052. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.F.; Lefter, R.; Ali, A.; Abdellah, E.; Trus, C.; Ciobica, A.; Timofte, D. Review Article The Involvement of the Oxidative Stress Status in Cancer Pathology: A Double View on the Role of the Antioxidants. Oxidative Med. Cell. Longev. 2021, 2021, 9965916. [Google Scholar] [CrossRef] [PubMed]
- Crisman, E.; Duarte, P.; Dauden, E.; Cuadrado, A.; Rodríguez-Franco, M.I.; López, M.G.; León, R. KEAP1-NRF2 protein–protein interaction inhibitors: Design, pharmacological properties and therapeutic potential. Med. Res. Rev. 2023, 43, 237–287. [Google Scholar] [CrossRef] [PubMed]
- Rashighi, M.; Harris, J.E. 乳鼠心肌提取 (Suckling heart muscle extraction) HHS Public Access. Physiol. Behav. 2017, 176, 139–148. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jia, Z.; Pan, M.H.; Anandh Babu, P.V. Natural Products for the Prevention of Oxidative Stress-Related Diseases: Mechanisms and Strategies. Oxidative Med. Cell. Longev. 2016, 2016, 4628502. [Google Scholar] [CrossRef]
- Managing Cancer as a Chronic Illness. Available online: https://www.cancer.org/cancer/survivorship/long-term-health-concerns/cancer-as-a-chronic-illness.html (accessed on 9 August 2023).
- Vemula, D.; Jayasurya, P.; Sushmitha, V.; Kumar, Y.N.; Bhandari, V. CADD, AI and ML in drug discovery: A comprehensive review. Eur. J. Pharm. Sci. 2023, 181, 106324. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Rabbani, G.; Danishuddin, M.; Choi, I. Computer Aided Drug Design and its Application to the Development of Potential Drugs for Neurodegenerative Disorders. Curr. Neuropharmacol. 2018, 16, 740–748. [Google Scholar] [CrossRef]
- Lu, M.C.; Zhang, X.; Wu, F.; Tan, S.J.; Zhao, J.; You, Q.D.; Jiang, Z.Y. Discovery of a potent kelch-like ECH-associated protein 1-nuclear factor erythroid 2-related factor 2 (Keap1-Nrf2) protein-protein interaction inhibitor with natural proline structure as a cytoprotective agent against acetaminophen-induced hepatotoxicity. J. Med. Chem. 2019, 62, 6796–6813. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-S.; Hu, L.-B.; Zhang, H.; Shan, W.-X.; Wang, Y.; Li, X.; Liu, T.; Zhao, J.; You, Q.-D.; Jiang, Z.-Y. Design, Synthesis, and Structure-Activity Relationships of Indoline-Based Kelch-like ECH-Associated Protein 1-Nuclear Factor (Erythroid-Derived 2)-Like 2 (Keap1-Nrf2) Protein-Protein Interaction Inhibitors. J. Med. Chem. 2020, 63, 11149–11168. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Wu, Z.; Xing, C.; Miao, Z. Small molecules inhibiting Keap1-Nrf2 protein-protein interactions: A novel approach to activate Nrf2 function. Medchemcomm 2016, 8, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.G.; Jain, A.D.; Speltz, T.E.; Moore, T.W. Non-electrophilic modulators of the canonical Keap1/Nrf2 pathway. Bioorg. Med. Chem. Lett. 2015, 25, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhu, J.; Lin, H.; Gu, K.; Feng, F. Recent progress in the development of small molecule Nrf2 modulators: A patent review (2012–2016). Expert Opin. Ther. Pat. 2017, 27, 763–785. [Google Scholar] [CrossRef] [PubMed]
- Boyenle, I.D.; Divine, U.C.; Adeyemi, R.; Ayinde, K.S.; Olaoba, O.T.; Apu, C.; Du, L.; Lu, Q.; Yin, X.; Adelusi, T.I. Direct Keap1-kelch inhibitors as potential drug candidates for oxidative stress-orchestrated diseases: A review on In silico perspective. Pharmacol. Res. 2021, 167, 105577. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S.; More, S.K.; Devadoss, D.; Zeng, B.; Kuhn, L.A.; Basson, M.D. Identification of potential small-molecule protein-protein inhibitors of cancer metastasis by 3D epitope-based computational screening. J. Physiol. Pharmacol. 2018, 69, 255–263. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertens 2003, 42, 1075–1081. [Google Scholar] [CrossRef]
- Van De Wier, B.; Koek, G.H.; Bast, A.; Haenen, G.R.M.M. The potential of flavonoids in the treatment of non-alcoholic fatty liver disease. Crit. Rev. Food Sci. Nutr. 2017, 57, 834–855. [Google Scholar] [CrossRef]
- Kwak, M.-K.; Kensler, T.W. Targeting NRF2 signaling for cancer chemoprevention. Toxicol. Appl. Pharmacol. 2010, 244, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, M.; Shamsara, J. An integrated structure- and pharmacophore-based MMP-12 virtual screening. Mol. Divers. 2018, 22, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Alzain, A.A.; Mukhtar, R.M.; Abdelmoniem, N.; Elbadwi, F.A.; Hussien, A.; Samman, W.A.; Ibrahim, S.R.M.; Mohamed, G.A.; Ashour, A. Computational Insights into Natural Antischistosomal Metabolites as SmHDAC8 Inhibitors: Molecular Docking, ADMET Profiling, and Molecular Dynamics Simulation. Metabolites 2023, 13, 658. [Google Scholar] [CrossRef] [PubMed]
- Alzain, A.A. Insights from computational studies on the potential of natural compounds as inhibitors against SARS-CoV-2 spike omicron variant. SAR QSAR Environ. Res. 2022, 33, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, D.; Tran, K.T.; Pallesen, J.S.; Solbak, S.M.; Qin, Y.; Mukminova, E.; Luchini, M.; Vasilyeva, K.O.; González Chichón, D.; Goutsiou, G.; et al. Development of Noncovalent Small-Molecule Keap1-Nrf2 Inhibitors by Fragment-Based Drug Discovery. J. Med. Chem. 2022, 65, 14481–14526. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, M.C.; Xu, L.L.; Yang, T.T.; Xi, M.Y.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; You, Q.D.; Sun, H.P. Discovery of potent Keap1-Nrf2 protein-protein interaction inhibitor based on molecular binding determinants analysis. J. Med. Chem. 2014, 57, 2736–2745. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-Y.; Xu, L.-L.; Lu, M.-C.; Pan, Y.; Huang, H.-Z.; Zhang, X.-J.; Sun, H.-P.; You, Q.-D. Investigation of the intermolecular recognition mechanism between the E3 ubiquitin ligase Keap1 and substrate based on multiple substrates analysis. J. Comput. Aided. Mol. Des. 2014, 28, 1233–1245. [Google Scholar] [CrossRef]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, Synthesis, and Evaluation of Triazole Derivatives That Induce Nrf2 Dependent Gene Products and Inhibit the Keap1-Nrf2 Protein-Protein Interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef]
- Zhuang, C.; Narayanapillai, S.; Zhang, W.; Sham, Y.Y.; Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 2014, 57, 1121–1126. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Score (kcal/mol) | MM/GBSA dG Bind (kcal/mol) |
|---|---|---|
| ZINC000002123788 | −9.728 | −72.29 |
| ZINC000002111341 | −8.259 | −70.75 |
| ZINC000002125904 | −7.718 | −63.23 |
| ZINC000096111705 | −7.612 | −56.53 |
| ZINC000012530057 | −7.564 | −59.29 |
| ZINC000002108268 | −7.458 | −60.97 |
| ZINC000002101617 | −7.130 | −57.75 |
| ZINC000000488978 | −6.847 | −57.77 |
| ZINC000002126056 | −6.792 | −58.51 |
| ZINC000002152231 | −6.653 | −60.81 |
| 7OFE-ligand | −6.633 | −56.36 |
| Compound | Pi-Cation | Pi-Pi Stacking | Salt Bridge | H-Bond | Hydrophobic Interactions | Other Interactions | Number of Interactions Made in Each Sub-Pocket |
|---|---|---|---|---|---|---|---|
| ZINC000002123788 | ARG415 | - | ARG415 | SER363, ASN414, ARG415, SER508, GLN530, SER555 | TYR334, TYR525, ALA556, TYR572, PHE577 | Charged positive: ARG415 Polar interaction: ASN382, SER363, ASN414, SER508, GLN530, SER555, SER602 | P1(2), P2(3), P3(3), P4(1), P5(3) |
| ZINC000002111341 | ARG415 | TYR334 | ARG415 | SER363, ARG415, GLN530, SER555 | TYR334, ILE461, PHE478, TYR525, ALA556, TYR572, PHE577 | Charged positive: ARG415, ARG483 Polar interaction: SER363, ASN382, ASN414, SER508, GLN530, SER555, SER602 | P1(5), P2(4), P3(3), P4(2), P5(3) |
| ZINC000002125904 | - | - | ARG415 | ARG415, SER508, GLN530, SER555 | TYR334, TYR525, ALA556, TYR572, PHE577 | Charged positive: ARG415 Polar interaction: SER363, ASN382, ASN414, SER508, GLN530, SER555, SER602 | P1(2), P2(3), P3(4), P4(2), P5(3) |
| Compound | QPlogPo/w a | QPlogS b | QPlogHERG c | QPlogBB d | QPPCaco e nm/sec | % Human Oral Absorption f | Rule of Five g |
|---|---|---|---|---|---|---|---|
| ZINC000002123788 | −1.267 | −0.752 | −0.258 | −3.565 | 0.206 | 7.256 | 0 |
| ZINC000002111341 | 1.438 | −2.786 | −0.233 | −2.099 | 9.546 | 52.902 | 0 |
| ZINC000002125904 | 0.905 | −2.373 | −1.215 | −1.994 | 5.175 | 45.022 | 0 |
| Recommended values | −2.0–6.5 | −6.5–0.5 | Below −5 | −3–1.2 | <25 poor >500 great | <25 poor >80% high | 0–4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alzain, A.A.; Mukhtar, R.M.; Abdelmoniem, N.; Shoaib, T.H.; Osman, W.; Alsulaimany, M.; Aljohani, A.K.B.; Almadani, S.A.; Alsaadi, B.H.; Althubyani, M.M.; et al. Modulation of NRF2/KEAP1-Mediated Oxidative Stress for Cancer Treatment by Natural Products Using Pharmacophore-Based Screening, Molecular Docking, and Molecular Dynamics Studies. Molecules 2023, 28, 6003. https://doi.org/10.3390/molecules28166003
Alzain AA, Mukhtar RM, Abdelmoniem N, Shoaib TH, Osman W, Alsulaimany M, Aljohani AKB, Almadani SA, Alsaadi BH, Althubyani MM, et al. Modulation of NRF2/KEAP1-Mediated Oxidative Stress for Cancer Treatment by Natural Products Using Pharmacophore-Based Screening, Molecular Docking, and Molecular Dynamics Studies. Molecules. 2023; 28(16):6003. https://doi.org/10.3390/molecules28166003
Chicago/Turabian StyleAlzain, Abdulrahim A., Rua M. Mukhtar, Nihal Abdelmoniem, Tagyedeen H. Shoaib, Wadah Osman, Marwa Alsulaimany, Ahmed K. B. Aljohani, Sara A. Almadani, Baiaan H. Alsaadi, Maryam M. Althubyani, and et al. 2023. "Modulation of NRF2/KEAP1-Mediated Oxidative Stress for Cancer Treatment by Natural Products Using Pharmacophore-Based Screening, Molecular Docking, and Molecular Dynamics Studies" Molecules 28, no. 16: 6003. https://doi.org/10.3390/molecules28166003
APA StyleAlzain, A. A., Mukhtar, R. M., Abdelmoniem, N., Shoaib, T. H., Osman, W., Alsulaimany, M., Aljohani, A. K. B., Almadani, S. A., Alsaadi, B. H., Althubyani, M. M., Mohamed, S. G. A., Mohamed, G. A., & Ibrahim, S. R. M. (2023). Modulation of NRF2/KEAP1-Mediated Oxidative Stress for Cancer Treatment by Natural Products Using Pharmacophore-Based Screening, Molecular Docking, and Molecular Dynamics Studies. Molecules, 28(16), 6003. https://doi.org/10.3390/molecules28166003