
Contemporary Strategies for Immobilizing Metallophthalocyanines for Electrochemical Transformations of Carbon Dioxide



Abstract

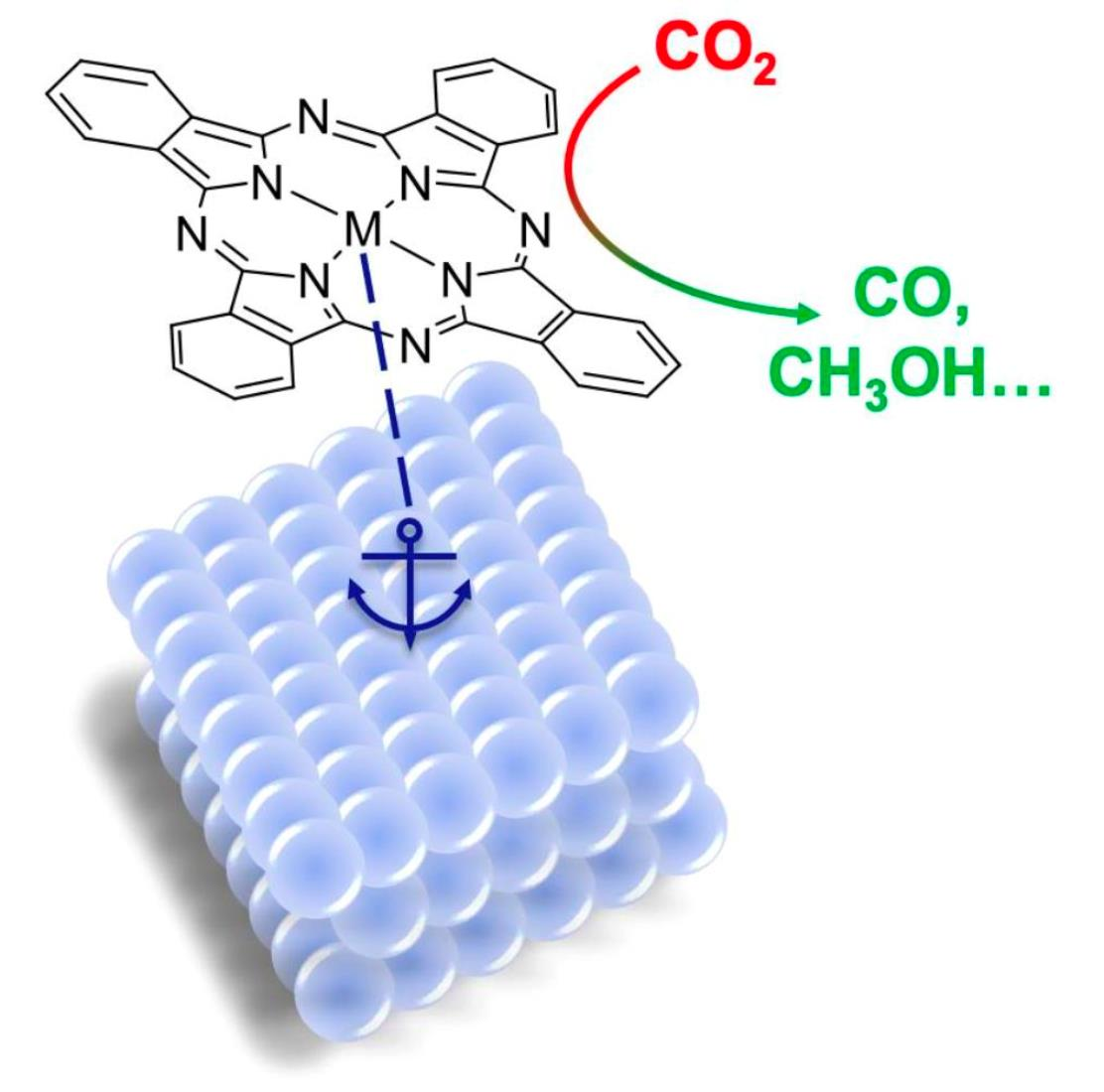{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
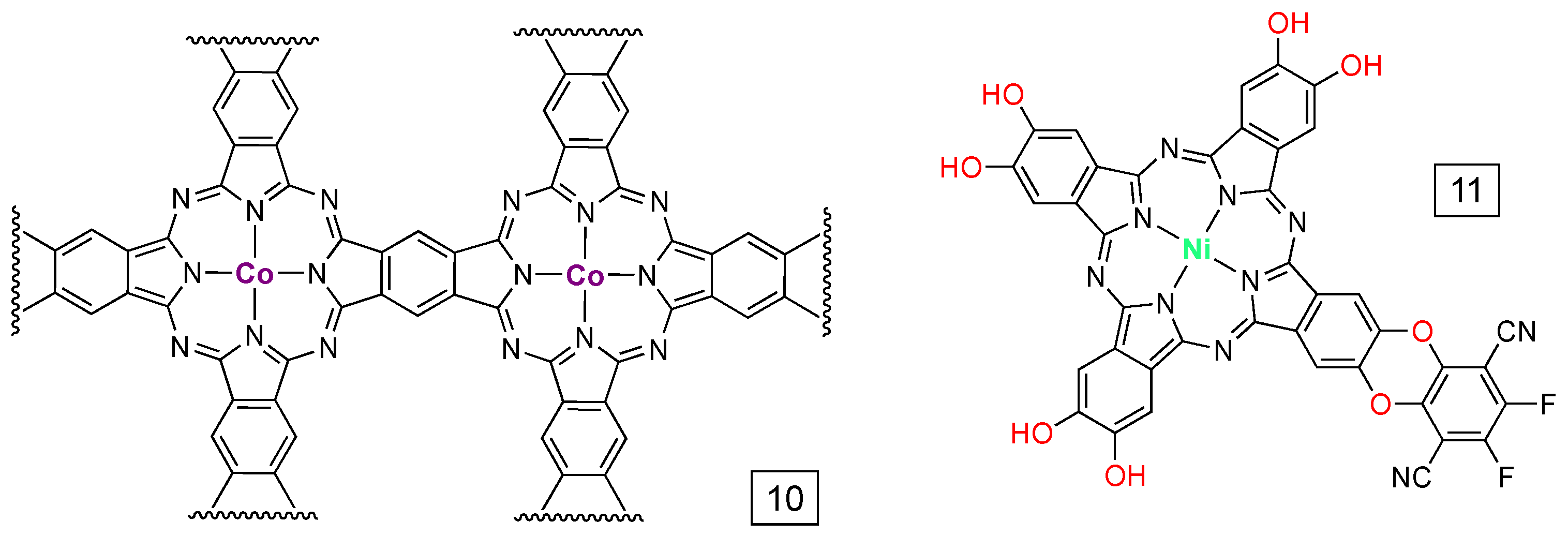{kind=link}
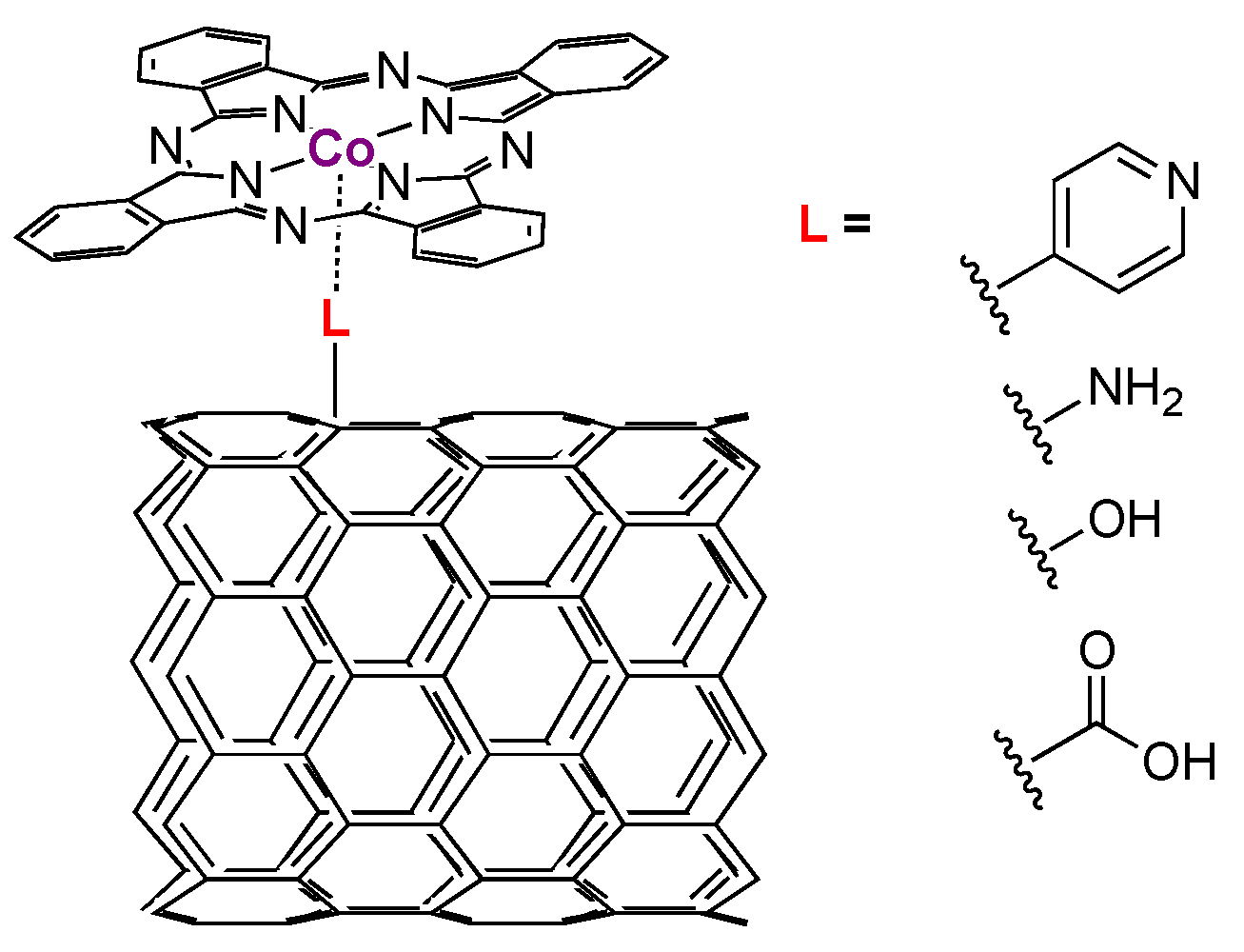{kind=link}
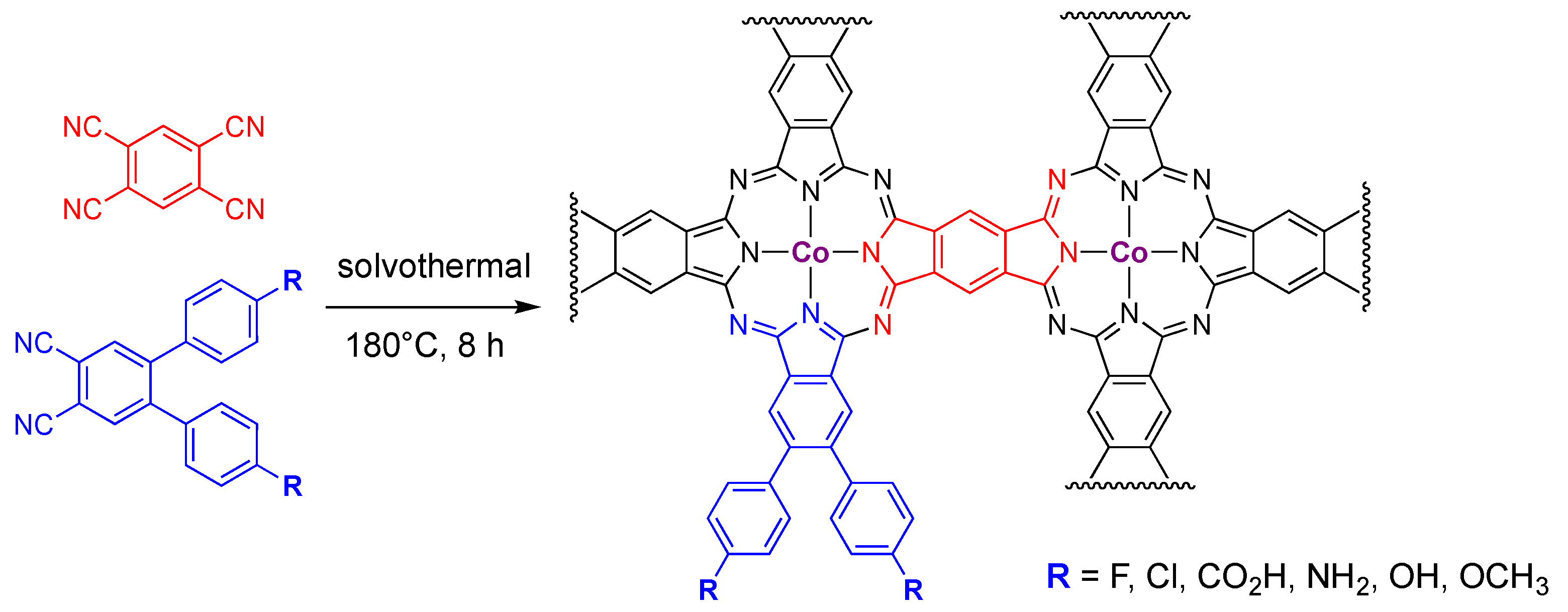{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Heterogeneous versus Homogeneous Catalysis
3. Selectivity and Mechanisms of CO2 Reduction
4. Direct Electrode Adsorption/Deposition and Catalyst Inks
5. PcM Materials Supported on Carbon Nanotubes
6. Network Materials Built with PcM
7. Polymer Incorporation
8. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Moser, F.H.; Thomas, A.L. Phthalocyanine Compounds. J. Chem. Educ. 1964, 41, 245. [Google Scholar] [CrossRef]
- Kasuga, K.; Tsutsuo, M. Some New Developments in the Chemistry of Metallophthalocyanines. Coord. Chem. Rev. 1980, 32, 67–95. [Google Scholar] [CrossRef]
- Gregory, P. Industrial Applications of Phthalocyanines. J. Porphyr. Phthalocya. 2000, 04, 432–437. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Vashistha, V.; Kumar Das, D. Recent Development on Metal Phthalocyanines Based Materials for Energy Conversion and Storage Applications. Coord. Chem. Rev. 2021, 431, 213678. [Google Scholar] [CrossRef]
- Yang, S.; Yu, Y.; Gao, X.; Zhang, Z.; Wang, F. Recent Advances in Electrocatalysis with Phthalocyanines. Chem. Soc. Rev. 2021, 50, 12985–13011. [Google Scholar] [CrossRef]
- Tsolekile, N.; Nelana, S.; Oluwafemi, O.S. Porphyrin as Diagnostic and Therapeutic Agent. Molecules 2019, 24, 2669. [Google Scholar] [CrossRef]
- Zheng, B.-D.; He, Q.-X.; Li, X.; Yoon, J.; Huang, J.-D. Phthalocyanines as Contrast Agents for Photothermal Therapy. Coord. Chem. Rev. 2021, 426, 213548. [Google Scholar] [CrossRef]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef]
- Domingo-Tafalla, B.; Chatterjee, T.; Palomares, E. Recent Advances in the Rational Designing of Metalloporphyrinoid-Based CO2 Reduction Catalysts: From Molecular Structural Tuning to the Application in Catalysis. J. Porphyr. Phthalocya. 2023, 27, 23–46. [Google Scholar] [CrossRef]
- Feng, Q.; Sun, Y.; Gu, X.; Dong, Z. Advances of Cobalt Phthalocyanine in Electrocatalytic CO2 Reduction to CO: A Mini Review. Electrocatalysis 2022, 13, 675–690. [Google Scholar] [CrossRef]
- Huang, S.; Chen, K.; Li, T.-T. Porphyrin and Phthalocyanine Based Covalent Organic Frameworks for Electrocatalysis. Coord. Chem. Rev. 2022, 464, 214563. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking Heterogeneous Electrocatalysts for the Oxygen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef]
- Matsubara, Y. Unified Benchmarking of Electrocatalysts in Noninnocent Second Coordination Spheres for CO2 Reduction. ACS Energy Lett. 2019, 4, 1999–2004. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Homogeneous Molecular Catalysis of Electrochemical Reactions: Catalyst Benchmarking and Optimization Strategies. J. Am. Chem. Soc. 2017, 139, 8245–8250. [Google Scholar] [CrossRef] [PubMed]
- Appel, A.M.; Helm, M.L. Determining the Overpotential for a Molecular Electrocatalyst. ACS Catal. 2014, 4, 630–633. [Google Scholar] [CrossRef]
- Jouny, M.; Luc, W.; Jiao, F. General Techno-Economic Analysis of CO2 Electrolysis Systems. Ind. Eng. Chem. Res. 2018, 57, 2165–2177. [Google Scholar] [CrossRef]
- Shin, H.; Hansen, K.U.; Jiao, F. Techno-Economic Assessment of Low-Temperature Carbon Dioxide Electrolysis. Nat. Sustain. 2021, 4, 911–919. [Google Scholar] [CrossRef]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Keith, D.W.; Holmes, G.; Angelo, D.S.; Heidel, K. A Process for Capturing CO2 from the Atmosphere. Joule 2018, 2, 1573–1594. [Google Scholar] [CrossRef]
- Kibria, M.G.; Edwards, J.P.; Gabardo, C.M.; Dinh, C.-T.; Seifitokaldani, A.; Sinton, D.; Sargent, E.H. Electrochemical CO2 Reduction into Chemical Feedstocks: From Mechanistic Electrocatalysis Models to System Design. Adv. Mater. 2019, 31, 1807166. [Google Scholar] [CrossRef] [PubMed]
- Masel, R.I.; Liu, Z.; Yang, H.; Kaczur, J.J.; Carrillo, D.; Ren, S.; Salvatore, D.; Berlinguette, C.P. An Industrial Perspective on Catalysts for Low-Temperature CO2 Electrolysis. Nat. Nanotechnol. 2021, 16, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Savéant, J.-M. Towards an Intelligent Design of Molecular Electrocatalysts. Nat. Rev. Chem. 2017, 1, 0087. [Google Scholar] [CrossRef]
- Barrett, J.A.; Miller, C.J.; Kubiak, C.P. Electrochemical Reduction of CO2 Using Group VII Metal Catalysts. Trends Chem. 2021, 3, 176–187. [Google Scholar] [CrossRef]
- Saha, P.; Amanullah, S.; Dey, A. Selectivity in Electrochemical CO2 Reduction. Acc. Chem. Res. 2022, 55, 134–144. [Google Scholar] [CrossRef]
- Meshitsuka, S.; Ichikawa, M.; Tamaru, K. Electrocatalysis by Metal Phthalocyanines in the Reduction of Carbon Dioxide. J. Chem. Soc. Chem. Commun. 1974, 5, 158–159. [Google Scholar] [CrossRef]
- Hiratsuka, K.; Takahashi, K.; Sasaki, H.; Toshima, S. Electrocatalytic Behavior of Tetrasulfonated Metal Phthalocyanines in the Reduction of Carbon Dioxide. Chem. Lett. 1977, 6, 1137–1140. [Google Scholar] [CrossRef]
- Lieber, C.M.; Lewis, N.S. Catalytic Reduction of Carbon Dioxide at Carbon Electrodes Modified with Cobalt Phthalocyanine. J. Am. Chem. Soc. 1984, 106, 5033–5034. [Google Scholar] [CrossRef]
- Abe, T.; Imaya, H.; Yoshida, T.; Tokita, S.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Electrochemical CO2 Reduction Catalysed by Cobalt Octacyanophthalocyanine and Its Mechanism. J. Porphyr. Phthalocya. 1997, 1, 315–321. [Google Scholar] [CrossRef]
- Furuya, N.; Matsui, K. Electroreduction of Carbon Dioxide on Gas-Diffusion Electrodes Modified by Metal Phthalocyanines. J. Electroanal. Chem. Interfacial Electrochem. 1989, 271, 181–191. [Google Scholar] [CrossRef]
- Furuya, N.; Koide, S. Electroreduction of Carbon Dioxide by Metal Phthalocyanines. Electrochim. Acta 1991, 36, 1309–1313. [Google Scholar] [CrossRef]
- Corbin, N.; Zeng, J.; Williams, K.; Manthiram, K. Heterogeneous Molecular Catalysts for Electrocatalytic CO2 Reduction. Nano Res. 2019, 12, 2093–2125. [Google Scholar] [CrossRef]
- Lee, K.J.; Elgrishi, N.; Kandemir, B.; Dempsey, J.L. Electrochemical and Spectroscopic Methods for Evaluating Molecular Electrocatalysts. Nat. Rev. Chem. 2017, 1, 0039. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Multielectron, Multistep Molecular Catalysis of Electrochemical Reactions: Benchmarking of Homogeneous Catalysts. ChemElectroChem 2014, 1, 1226–1236. [Google Scholar] [CrossRef]
- Rountree, E.S.; McCarthy, B.D.; Eisenhart, T.T.; Dempsey, J.L. Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry. Inorg. Chem. 2014, 53, 9983–10002. [Google Scholar] [CrossRef]
- Costentin, C.; Dridi, H.; Savéant, J.-M. Molecular Catalysis of O2 Reduction by Iron Porphyrins in Water: Heterogeneous versus Homogeneous Pathways. J. Am. Chem. Soc. 2015, 137, 13535–13544. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Hu, G.; Rooney, C.L.; Brudvig, G.W.; Wang, H. Heterogeneous Nature of Electrocatalytic CO/CO2 Reduction by Cobalt Phthalocyanines. ChemSusChem 2020, 13, 6296–6299. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Aggarwal, A.; Bhupathiraju, N.V.S.D.K.; Arianna, G.; Tiwari, K.; Drain, C.M. Glycosylated Porphyrins, Phthalocyanines, and Other Porphyrinoids for Diagnostics and Therapeutics. Chem. Rev. 2015, 115, 10261–10306. [Google Scholar] [CrossRef]
- Urbani, M.; Ragoussi, M.-E.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines for Dye-Sensitized Solar Cells. Coord. Chem. Rev. 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Zhang, R.; Warren, J.J. Recent Developments in Metalloporphyrin Electrocatalysts for Reduction of Small Molecules: Strategies for Managing Electron and Proton Transfer Reactions. ChemSusChem 2021, 14, 293–302. [Google Scholar] [CrossRef]
- Wakerley, D.; Lamaison, S.; Wicks, J.; Clemens, A.; Feaster, J.; Corral, D.; Jaffer, S.A.; Sarkar, A.; Fontecave, M.; Duoss, E.B.; et al. Gas Diffusion Electrodes, Reactor Designs and Key Metrics of Low-Temperature CO2 Electrolysers. Nat. Energy 2022, 7, 130–143. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Z.; Lu, X.; Liang, Y.; Wang, H. Domino Electroreduction of CO2 to Methanol on a Molecular Catalyst. Nature 2019, 575, 639–642. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, J.; Chen, X.-J.; Yu, S.; Yu, L.; Si, R.; Wang, Y.; Wang, S.; Meng, X.; Wang, Y.; et al. Reaction Mechanisms of Well-Defined Metal–N4 Sites in Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 16339–16342. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Yoshida, T.; Tokita, S.; Taguchi, F.; Imaya, H.; Kaneko, M. Factors Affecting Selective Electrocatalytic CO2 Reduction with Cobalt Phthalocyanine Incorporated in a Polyvinylpyridine Membrane Coated on a Graphite Electrode. J. Electroanal. Chem. 1996, 412, 125–132. [Google Scholar] [CrossRef]
- Zhu, M.; Ye, R.; Jin, K.; Lazouski, N.; Manthiram, K. Elucidating the Reactivity and Mechanism of CO2 Electroreduction at Highly Dispersed Cobalt Phthalocyanine. ACS Energy Lett. 2018, 3, 1381–1386. [Google Scholar] [CrossRef]
- Han, N.; Wang, Y.; Ma, L.; Wen, J.; Li, J.; Zheng, H.; Nie, K.; Wang, X.; Zhao, F.; Li, Y.; et al. Supported Cobalt Polyphthalocyanine for High-Performance Electrocatalytic CO2 Reduction. Chem 2017, 3, 652–664. [Google Scholar] [CrossRef]
- Liu, Y.; McCrory, C.C.L. Modulating the Mechanism of Electrocatalytic CO2 Reduction by Cobalt Phthalocyanine through Polymer Coordination and Encapsulation. Nat. Commun. 2019, 10, 1683. [Google Scholar] [CrossRef]
- Abe, T.; Taguchi, F.; Yoshida, T.; Tokita, S.; Schnurpfeil, G.; Wöhrle, D.; Kaneko, M. Electrocatalytic CO2 Reduction by Cobalt Octabutoxyphthalocyanine Coated on Graphite Electrode. J. Mol. Catal. A-Chem. 1996, 112, 55–61. [Google Scholar] [CrossRef]
- Chang, Q.; Liu, Y.; Lee, J.-H.; Ologunagba, D.; Hwang, S.; Xie, Z.; Kattel, S.; Lee, J.H.; Chen, J.G. Metal-Coordinated Phthalocyanines as Platform Molecules for Understanding Isolated Metal Sites in the Electrochemical Reduction of CO2. J. Am. Chem. Soc. 2022, 144, 16131–16138. [Google Scholar] [CrossRef]
- Wang, X.; Cai, Z.-F.; Wang, Y.-Q.; Feng, Y.-C.; Yan, H.-J.; Wang, D.; Wan, L.-J. In Situ Scanning Tunneling Microscopy of Cobalt-Phthalocyanine-Catalyzed CO2 Reduction Reaction. Angew. Chem. Int. Ed. 2020, 59, 16098–16103. [Google Scholar] [CrossRef]
- Karapinar, D.; Zitolo, A.; Huan, T.N.; Zanna, S.; Taverna, D.; Galvão Tizei, L.H.; Giaume, D.; Marcus, P.; Mougel, V.; Fontecave, M. Carbon-Nanotube-Supported Copper Polyphthalocyanine for Efficient and Selective Electrocatalytic CO2 Reduction to CO. ChemSusChem 2020, 13, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Snow, A.W. Phthalocyanine Aggregation. In The Porphyrin Handbook; Elsevier: Amsterdam, The Netherlands, 2003; pp. 129–176. ISBN 978-0-08-092391-8. [Google Scholar]
- Kobayashi, N.; Ogata, H.; Nonaka, N.; Luk’yanets, E.A. Effect of Peripheral Substitution on the Electronic Absorption and Fluorescence Spectra of Metal-Free and Zinc Phthalocyanines. Chem. Eur. J. 2003, 9, 5123–5134. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Wagner, P.; Gambhir, S.; Jalili, R.; MacFarlane, D.R.; Wallace, G.G.; Officer, D.L. Steric Modification of a Cobalt Phthalocyanine/Graphene Catalyst To Give Enhanced and Stable Electrochemical CO2 Reduction to CO. ACS Energy Lett. 2019, 4, 666–672. [Google Scholar] [CrossRef]
- Ren, S.; Lees, E.W.; Hunt, C.; Jewlal, A.; Kim, Y.; Zhang, Z.; Mowbray, B.A.W.; Fink, A.G.; Melo, L.; Grant, E.R.; et al. Catalyst Aggregation Matters for Immobilized Molecular CO2RR Electrocatalysts. J. Am. Chem. Soc. 2023, 145, 4414–4420. [Google Scholar] [CrossRef]
- Tajbakhsh, E.; McKearney, D.; Leznoff, D.B.; Warren, J.J. Heterogenous Preparations of Solution-Processable Cobalt Phthalocyanines for Carbon Dioxide Reduction Electrocatalysis. Inorganics 2023, 11, 43. [Google Scholar] [CrossRef]
- De Riccardis, A.; Lee, M.; Kazantsev, R.V.; Garza, A.J.; Zeng, G.; Larson, D.M.; Clark, E.L.; Lobaccaro, P.; Burroughs, P.W.W.; Bloise, E.; et al. Heterogenized Pyridine-Substituted Cobalt(II) Phthalocyanine Yields Reduction of CO2 by Tuning the Electron Affinity of the Co Center. ACS Appl. Mater. Interfaces 2020, 12, 5251–5258. [Google Scholar] [CrossRef]
- Berlinger, S.A.; Garg, S.; Weber, A.Z. Multicomponent, Multiphase Interactions in Fuel-Cell Inks. Curr. Opin. Electrochem. 2021, 29, 100744. [Google Scholar] [CrossRef]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulié, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Işci, U.; Berlinguette, C.P.; et al. CO2 Electrochemical Catalytic Reduction with a Highly Active Cobalt Phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Z.; Zhang, X.; Li, L.; Li, Y.; Xu, H.; Li, X.; Yu, X.; Zhang, Z.; Liang, Y.; et al. Highly Selective and Active CO2 Reduction Electrocatalysts Based on Cobalt Phthalocyanine/Carbon Nanotube Hybrid Structures. Nat. Commun. 2017, 8, 14675. [Google Scholar] [CrossRef]
- Jin, Y.; Zhan, X.; Zheng, Y.; Wang, H.; Liu, X.; Yu, B.; Ding, X.; Zheng, T.; Wang, K.; Qi, D.; et al. In-Situ Growing Nickel Phthalocyanine Supramolecular Structure on Carbon Nanotubes for Efficient Electrochemical CO2 Conversion. Appl. Cat. B-Environ. 2023, 327, 122446. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, J.; Guo, R.; Xu, J.; Fang, X.; Han, Y.-F. Cobalt Phthalocyanine Coordinated to Pyridine-Functionalized Carbon Nanotubes with Enhanced CO2 Electroreduction. Appl. Cat. B-Environ. 2019, 251, 112–118. [Google Scholar] [CrossRef]
- Li, H.; Pan, Y.; Wang, Z.; Yu, Y.; Xiong, J.; Du, H.; Lai, J.; Wang, L.; Feng, S. Coordination Engineering of Cobalt Phthalocyanine by Functionalized Carbon Nanotube for Efficient and Highly Stable Carbon Dioxide Reduction at High Current Density. Nano Res. 2022, 15, 3056–3064. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, M.; Li, J.; Xu, J.; Han, Y.-F. Structure–Activity Relationship of the Polymerized Cobalt Phthalocyanines for Electrocatalytic Carbon Dioxide Reduction. J. Phys. Chem. C 2020, 124, 16501–16507. [Google Scholar] [CrossRef]
- Geng, K.; He, T.; Liu, R.; Dalapati, S.; Tan, K.T.; Li, Z.; Tao, S.; Gong, Y.; Jiang, Q.; Jiang, D. Covalent Organic Frameworks: Design, Synthesis, and Functions. Chem. Rev. 2020, 120, 8814–8933. [Google Scholar] [CrossRef]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef]
- Diercks, C.S.; Lin, S.; Kornienko, N.; Kapustin, E.A.; Nichols, E.M.; Zhu, C.; Zhao, Y.; Chang, C.J.; Yaghi, O.M. Reticular Electronic Tuning of Porphyrin Active Sites in Covalent Organic Frameworks for Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2018, 140, 1116–1122. [Google Scholar] [CrossRef]
- Meng, Z.; Stolz, R.M.; Mirica, K.A. Two-Dimensional Chemiresistive Covalent Organic Framework with High Intrinsic Conductivity. J. Am. Chem. Soc. 2019, 141, 11929–11937. [Google Scholar] [CrossRef]
- Wang, M.; Ballabio, M.; Wang, M.; Lin, H.-H.; Biswal, B.P.; Han, X.; Paasch, S.; Brunner, E.; Liu, P.; Chen, M.; et al. Unveiling Electronic Properties in Metal–Phthalocyanine-Based Pyrazine-Linked Conjugated Two-Dimensional Covalent Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 16810–16816. [Google Scholar] [CrossRef]
- Huang, N.; Lee, K.H.; Yue, Y.; Xu, X.; Irle, S.; Jiang, Q.; Jiang, D. A Stable and Conductive Metallophthalocyanine Framework for Electrocatalytic Carbon Dioxide Reduction in Water. Angew. Chem. Int. Ed. 2020, 59, 16587–16593. [Google Scholar] [CrossRef]
- Han, B.; Ding, X.; Yu, B.; Wu, H.; Zhou, W.; Liu, W.; Wei, C.; Chen, B.; Qi, D.; Wang, H.; et al. Two-Dimensional Covalent Organic Frameworks with Cobalt(II)-Phthalocyanine Sites for Efficient Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2021, 143, 7104–7113. [Google Scholar] [CrossRef]
- Meng, Z.; Aykanat, A.; Mirica, K.A. Welding Metallophthalocyanines into Bimetallic Molecular Meshes for Ultrasensitive, Low-Power Chemiresistive Detection of Gases. J. Am. Chem. Soc. 2019, 141, 2046–2053. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Ghorbani-Asl, M.; Ly, K.H.; Zhang, J.; Ge, J.; Wang, M.; Liao, Z.; Makarov, D.; Zschech, E.; Brunner, E.; et al. Synergistic Electroreduction of Carbon Dioxide to Carbon Monoxide on Bimetallic Layered Conjugated Metal-Organic Frameworks. Nat. Commun. 2020, 11, 1409. [Google Scholar] [CrossRef]
- Yi, J.-D.; Si, D.-H.; Xie, R.; Yin, Q.; Zhang, M.-D.; Wu, Q.; Chai, G.-L.; Huang, Y.-B.; Cao, R. Conductive Two-Dimensional Phthalocyanine-Based Metal–Organic Framework Nanosheets for Efficient Electroreduction of CO2. Angew. Chem. Int. Ed. 2021, 60, 17108–17114. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Cai, P.; Xu, K.; Li, H.; Chen, H.; Zhou, H.-C.; Huang, N. Stable Bimetallic Polyphthalocyanine Covalent Organic Frameworks as Superior Electrocatalysts. J. Am. Chem. Soc. 2021, 143, 18052–18060. [Google Scholar] [CrossRef]
- Velický, M.; Toth, P.S.; Woods, C.R.; Novoselov, K.S.; Dryfe, R.A.W. Electrochemistry of the Basal Plane versus Edge Plane of Graphite Revisited. J. Phys. Chem. C 2019, 123, 11677–11685. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Salehi, M.; Le, Q.V.; Seifitokaldani, A.; Dinh, C.T. Fundamentals of Electrochemical CO2 Reduction on Single-Metal-Atom Catalysts. ACS Catal. 2020, 10, 10068–10095. [Google Scholar] [CrossRef]
- Cui, X.; An, W.; Liu, X.; Wang, H.; Men, Y.; Wang, J. C2N-Graphene Supported Single-Atom Catalysts for CO2 Electrochemical Reduction Reaction: Mechanistic Insight and Catalyst Screening. Nanoscale 2018, 10, 15262–15272. [Google Scholar] [CrossRef]
- Wang, M.; Kong, L.; Lu, X.; Wu, C.-M.L. First-Row Transition Metal Embedded Pyrazine-Based Graphynes as High-Performance Single Atom Catalysts for the CO2 Reduction Reaction. J. Mater. Chem. A 2022, 10, 9048–9058. [Google Scholar] [CrossRef]
- Yang, H.B.; Hung, S.-F.; Liu, S.; Yuan, K.; Miao, S.; Zhang, L.; Huang, X.; Wang, H.-Y.; Cai, W.; Chen, R.; et al. Atomically Dispersed Ni(I) as the Active Site for Electrochemical CO2 Reduction. Nat. Energy 2018, 3, 140–147. [Google Scholar] [CrossRef]
- Yang, D.; Wang, J.; Wang, Q.; Yuan, Z.; Dai, Y.; Zhou, C.; Wan, X.; Zhang, Q.; Yang, Y. Electrocatalytic CO2 Reduction over Atomically Precise Metal Nanoclusters Protected by Organic Ligands. ACS Nano 2022, 16, 15681–15704. [Google Scholar] [CrossRef]
- Muto, T.; Temma, T.; Kimura, M.; Hanabusa, K.; Shirai, H. A New Phthalocyanine Derivative Having Peripheral 2-Thienyl Substituents. Chem. Commun. 2000, 1649–1650. [Google Scholar] [CrossRef]
- Muto, T.; Temma, T.; Kimura, M.; Hanabusa, K.; Shirai, H. Elongation of the π-System of Phthalocyanines by Introduction of Thienyl Substituents at the Peripheral β Positions. Synthesis and Characterization. J. Org. Chem. 2001, 66, 6109–6115. [Google Scholar] [CrossRef] [PubMed]
- Obirai, J.; Rodrigues, N.P.; Bedioui, F.; Nyokong, T. Synthesis, Spectral and Electrochemical Properties of a New Family of Pyrrole Substituted Cobalt, Iron, Manganese, Nickel and Zinc Phthalocyanine Complexes. J. Porphyr. Phthalocya. 2003, 07, 508–520. [Google Scholar] [CrossRef]
- Luangchaiyaporn, J.; Wielend, D.; Solonenko, D.; Seelajaroen, H.; Gasiorowski, J.; Monecke, M.; Salvan, G.; Zahn, D.R.T.; Sariciftci, N.S.; Thamyongkit, P. High-Performance CoII-Phthalocyanine-Based Polymer for Practical Heterogeneous Electrochemical Reduction of Carbon Dioxide. Electrochim. Acta 2021, 367, 137506. [Google Scholar] [CrossRef]
- Chen, J.-M.; Xie, W.-J.; Yang, Z.-W.; He, L.-N. Cobalt Phthalocyanine Cross-Linked Polypyrrole for Efficient Electroreduction of Low Concentration CO2 To CO. ChemSusChem 2022, 15, e202201455. [Google Scholar] [CrossRef]
- Wang, Y.; Shi, Y.; Pan, L.; Ding, Y.; Zhao, Y.; Li, Y.; Shi, Y.; Yu, G. Dopant-Enabled Supramolecular Approach for Controlled Synthesis of Nanostructured Conductive Polymer Hydrogels. Nano Lett. 2015, 15, 7736–7741. [Google Scholar] [CrossRef]
- Yoshida, T.; Kamato, K.; Tsukamoto, M.; Iida, T.; Schlettwein, D.; Wöhrle, D.; Kaneko, M. Selective Electrocatalysis for CO2 Reduction in the Aqueous Phase Using Cobalt Phthalocyanine/Poly-4-Vinylpyridine Modified Electrodes. J. Electroanal. Chem. 1995, 385, 209–225. [Google Scholar] [CrossRef]
- Kramer, W.W.; McCrory, C.C.L. Polymer Coordination Promotes Selective CO2 Reduction by Cobalt Phthalocyanine. Chem. Sci. 2016, 7, 2506–2515. [Google Scholar] [CrossRef]
- Liu, Y.; Deb, A.; Leung, K.Y.; Nie, W.; Dean, W.S.; Penner-Hahn, J.E.; McCrory, C.C. Determining the Coordination Environment and Electronic Structure of Polymer-Encapsulated Cobalt Phthalocyanine under Electrocatalytic CO2 Reduction Conditions Using in Situ X-Ray Absorption Spectroscopy. Dalton Trans. 2020, 49, 16329–16339. [Google Scholar] [CrossRef]
- Soucy, T.L.; Dean, W.S.; Zhou, J.; Rivera Cruz, K.E.; McCrory, C.C.L. Considering the Influence of Polymer–Catalyst Interactions on the Chemical Microenvironment of Electrocatalysts for the CO2 Reduction Reaction. Acc. Chem. Res. 2022, 55, 252–261. [Google Scholar] [CrossRef]
- Nichols, E.M.; Derrick, J.S.; Nistanaki, S.K.; Smith, P.T.; Chang, C.J. Positional Effects of Second-Sphere Amide Pendants on Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. Chem. Sci. 2018, 9, 2952–2960. [Google Scholar] [CrossRef]
- Derrick, J.S.; Loipersberger, M.; Nistanaki, S.K.; Rothweiler, A.V.; Head-Gordon, M.; Nichols, E.M.; Chang, C.J. Templating Bicarbonate in the Second Coordination Sphere Enhances Electrochemical CO2 Reduction Catalyzed by Iron Porphyrins. J. Am. Chem. Soc. 2022, 144, 11656–11663. [Google Scholar] [CrossRef]
- Amanullah, S.; Saha, P.; Dey, A. Activating the Fe(I) State of Iron Porphyrinoid with Second-Sphere Proton Transfer Residues for Selective Reduction of CO2 to HCOOH via Fe(III/II)–COOH Intermediate(s). J. Am. Chem. Soc. 2021, 143, 13579–13592. [Google Scholar] [CrossRef] [PubMed]
- Mondal, B.; Sen, P.; Rana, A.; Saha, D.; Das, P.; Dey, A. Reduction of CO2 to CO by an Iron Porphyrin Catalyst in the Presence of Oxygen. ACS Catal. 2019, 9, 3895–3899. [Google Scholar] [CrossRef]
- Hellman, A.N.; Haiges, R.; Marinescu, S.C. Rhenium Bipyridine Catalysts with Hydrogen Bonding Pendant Amines for CO2 Reduction. Dalton Trans. 2019, 48, 14251–14255. [Google Scholar] [CrossRef] [PubMed]
- Hellman, A.N.; Haiges, R.; Marinescu, S.C. Influence of Intermolecular Hydrogen Bonding Interactions on the Electrocatalytic Reduction of CO2 to CO by 6,6′-Amine Substituted Rhenium Bipyridine Complexes. ChemElectroChem 2021, 8, 1864–1872. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lawson, S.E.; Leznoff, D.B.; Warren, J.J. Contemporary Strategies for Immobilizing Metallophthalocyanines for Electrochemical Transformations of Carbon Dioxide. Molecules 2023, 28, 5878. https://doi.org/10.3390/molecules28155878
Lawson SE, Leznoff DB, Warren JJ. Contemporary Strategies for Immobilizing Metallophthalocyanines for Electrochemical Transformations of Carbon Dioxide. Molecules. 2023; 28(15):5878. https://doi.org/10.3390/molecules28155878
Chicago/Turabian StyleLawson, Scheryn E., Daniel B. Leznoff, and Jeffrey J. Warren. 2023. "Contemporary Strategies for Immobilizing Metallophthalocyanines for Electrochemical Transformations of Carbon Dioxide" Molecules 28, no. 15: 5878. https://doi.org/10.3390/molecules28155878
APA StyleLawson, S. E., Leznoff, D. B., & Warren, J. J. (2023). Contemporary Strategies for Immobilizing Metallophthalocyanines for Electrochemical Transformations of Carbon Dioxide. Molecules, 28(15), 5878. https://doi.org/10.3390/molecules28155878