
Privileged Scaffolds for Potent and Specific Inhibitors of Mono-ADP-Ribosylating PARPs



Abstract
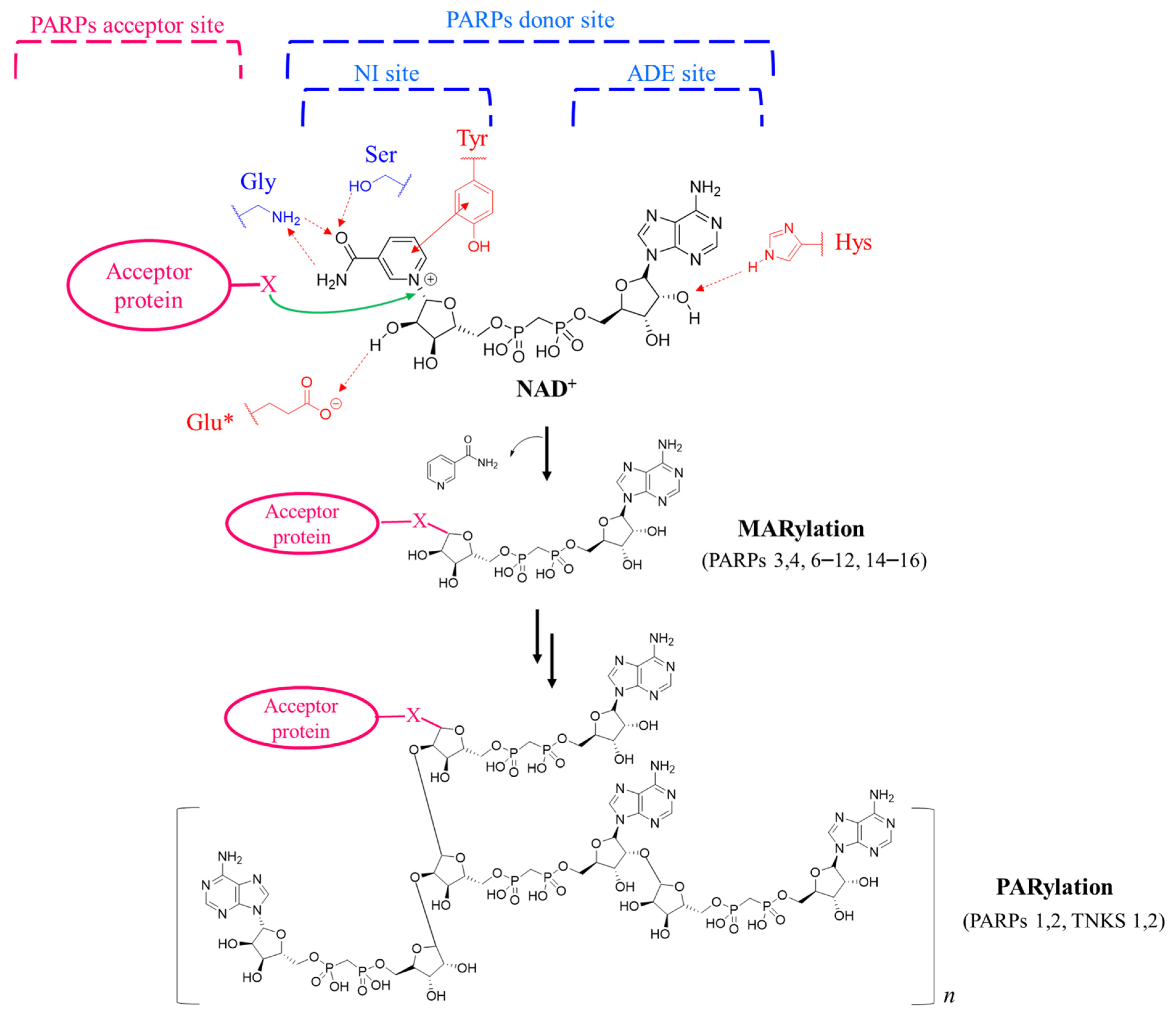
1. Introduction
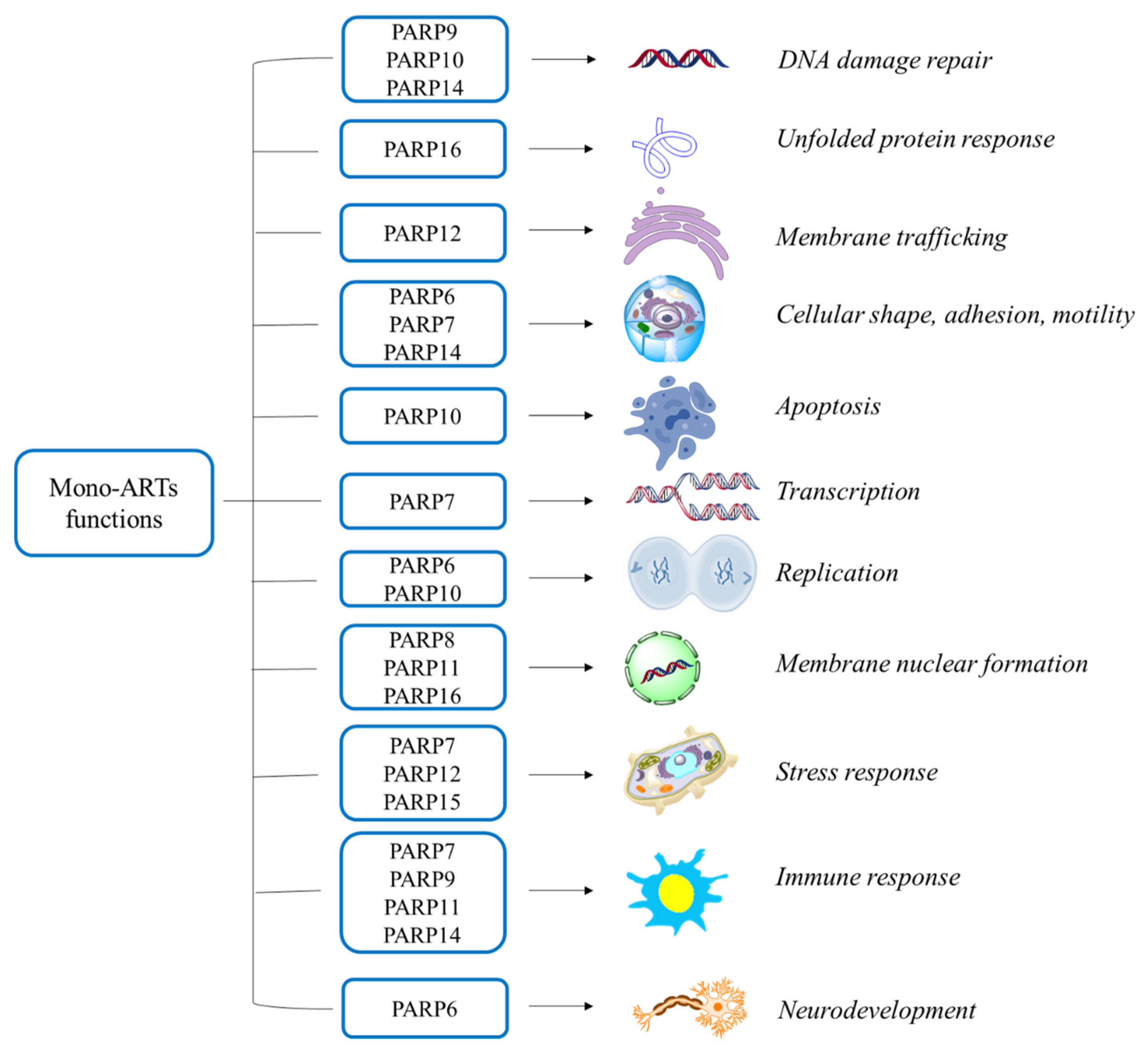
Mono-ARTs Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
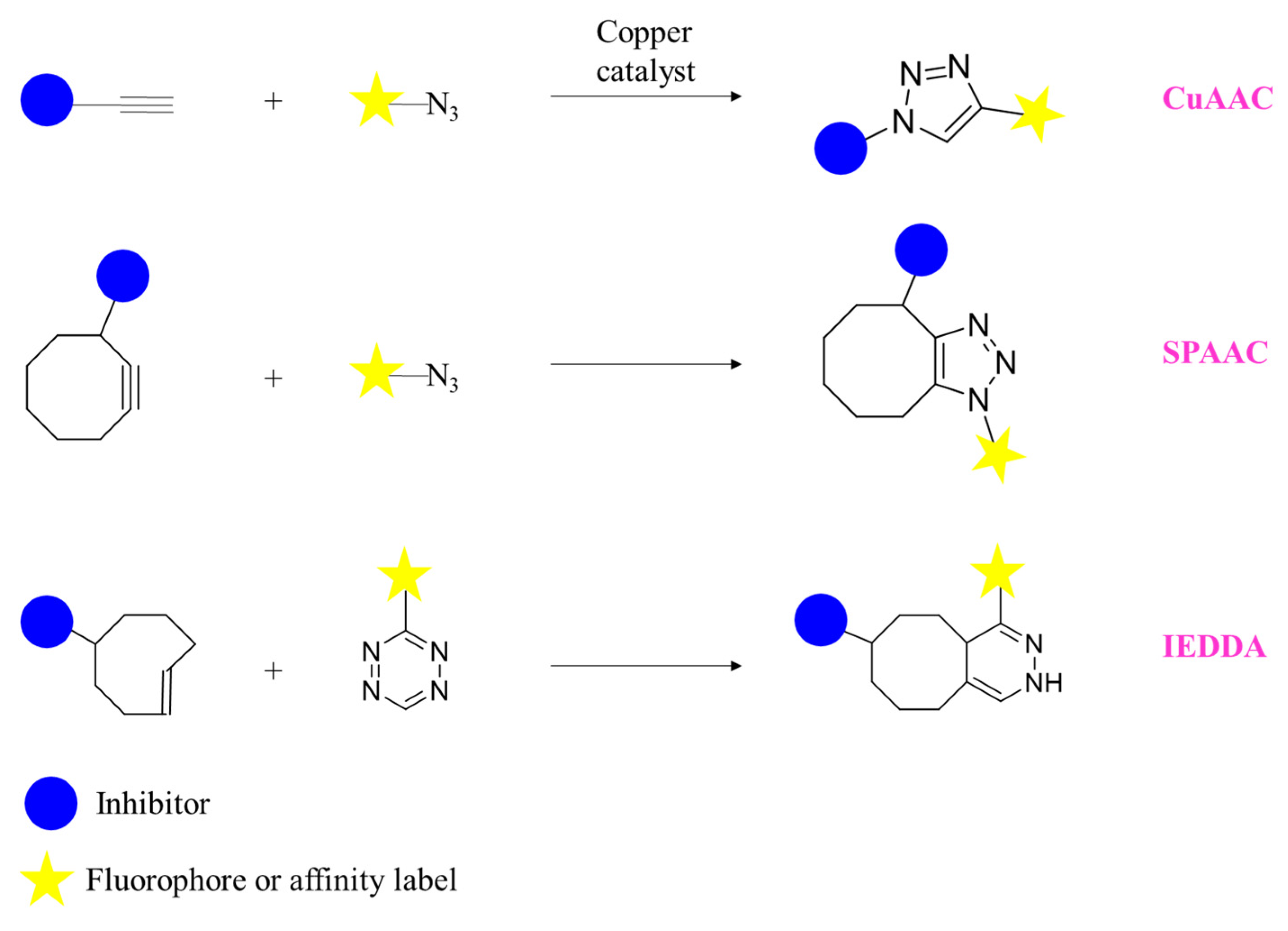{kind=link}
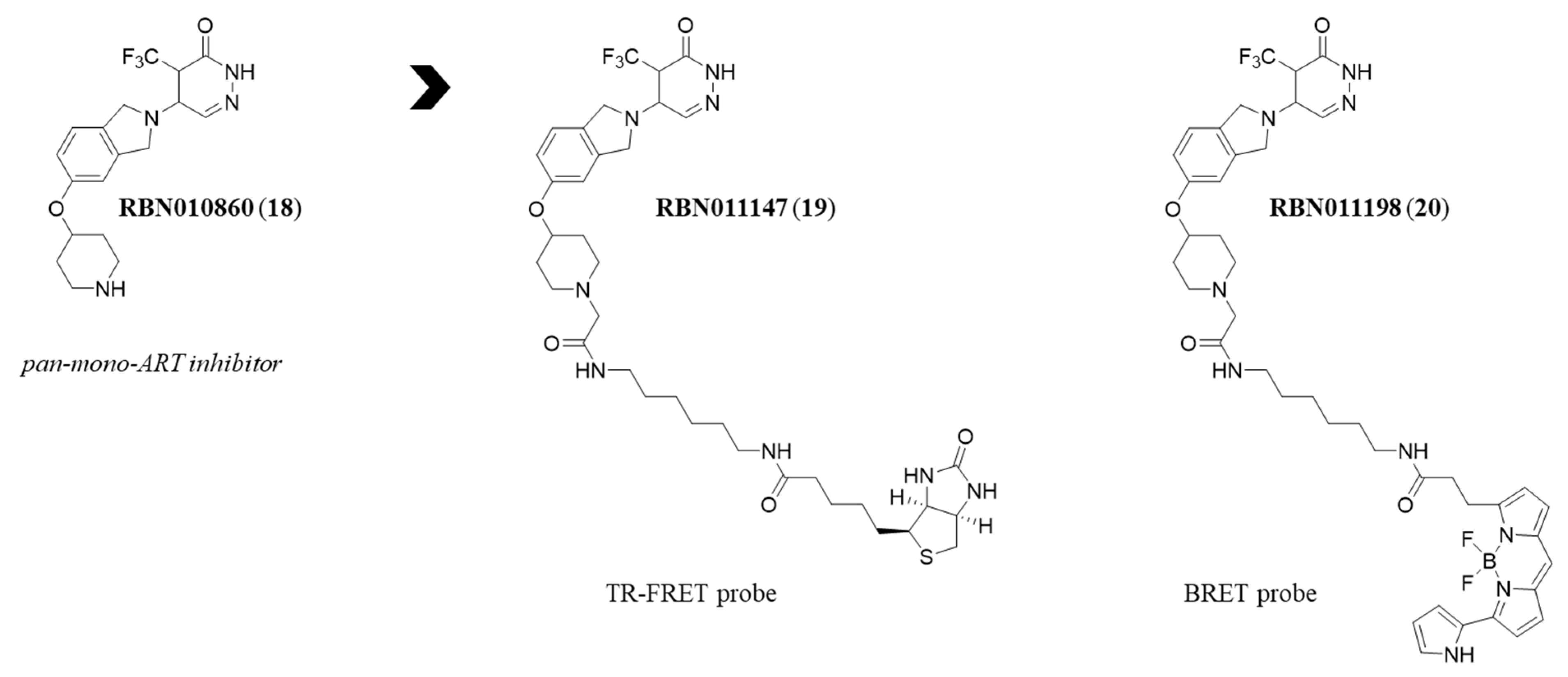{kind=link}
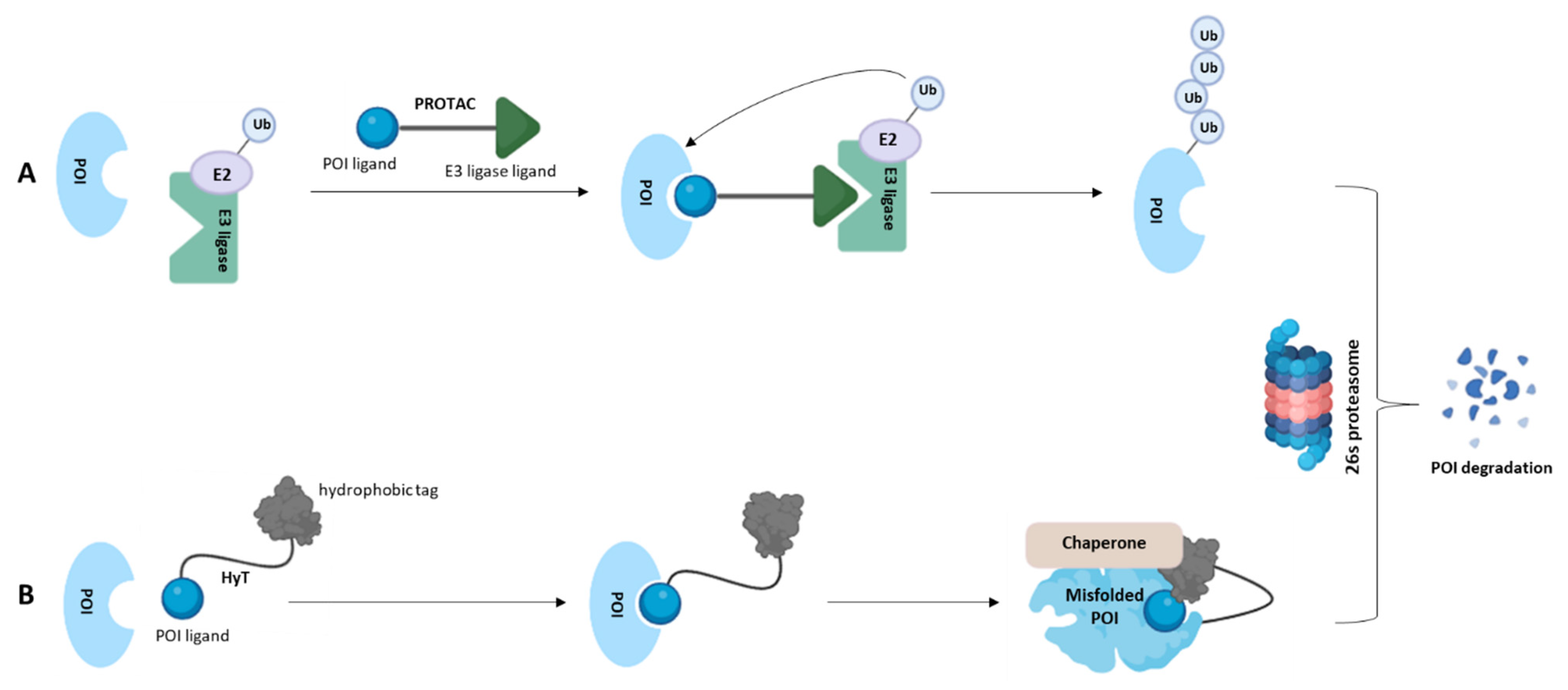{kind=link}
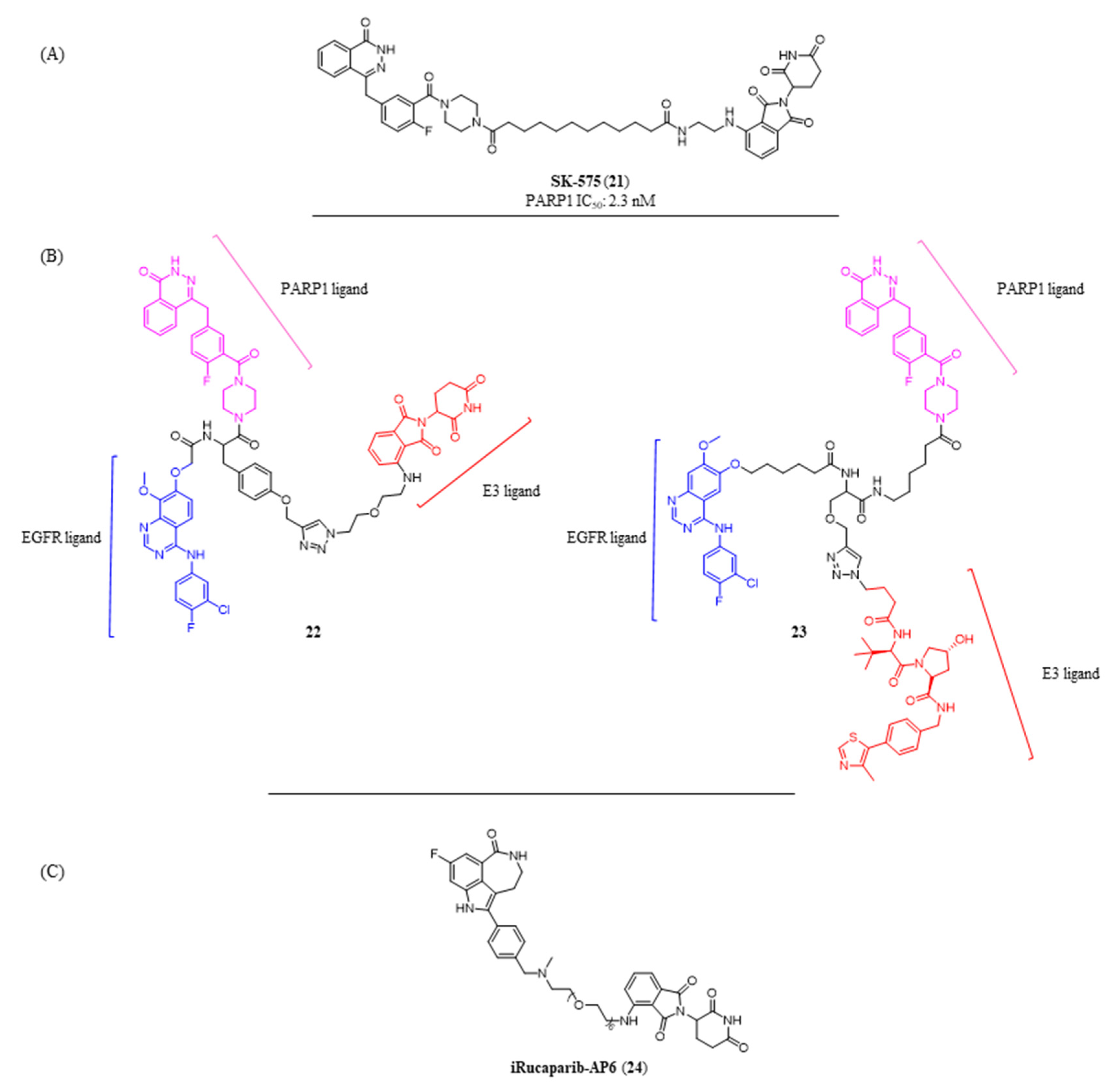{kind=link}
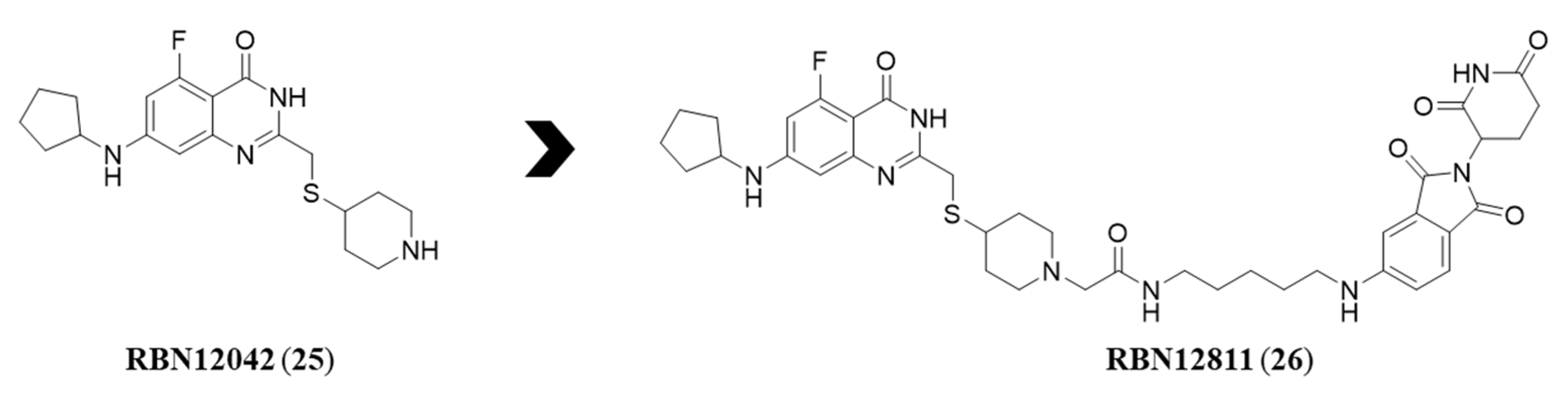{kind=link}
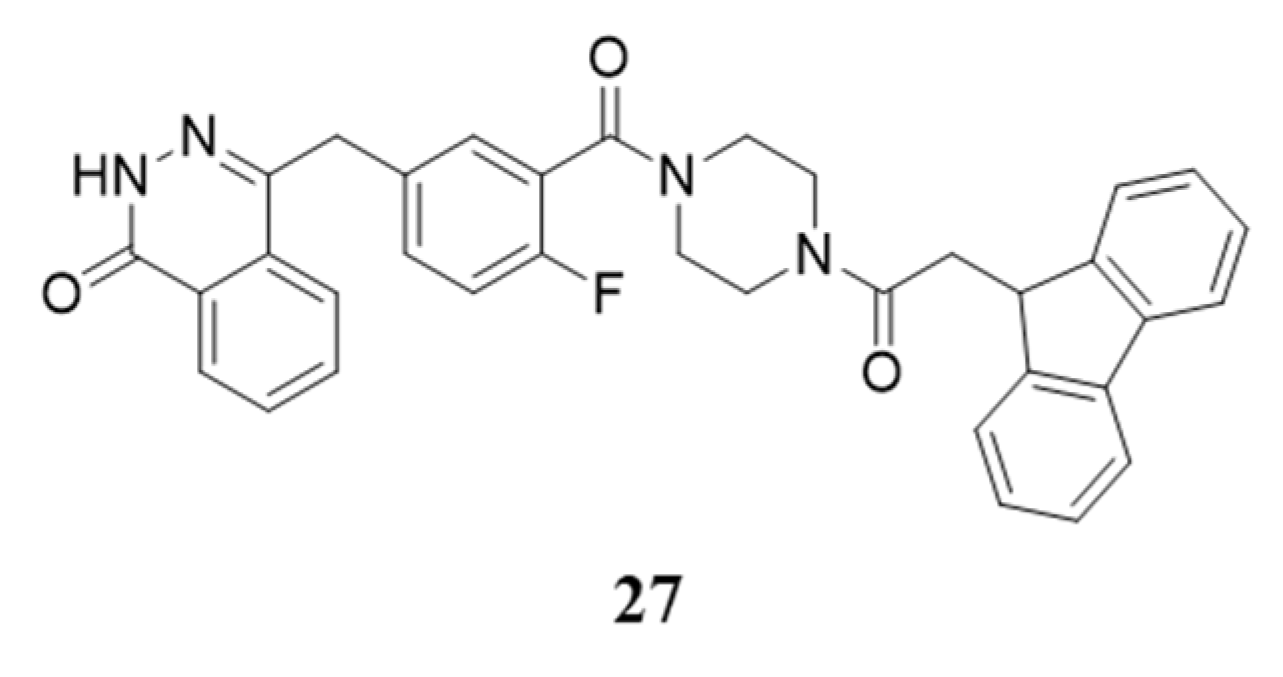{kind=link}
| Mono-ART | Cancer Type |
|---|---|
| PARP6 | MDA-MB468 and HCC1806 breast cancers a |
| PARP7 | Advanced metastatic solid tumours b |
| PARP10 | Ovarian cancer c Breast cancer d |
| PARP12 | MCF7 breast cancer e |
| PARP14 | Large B cell lymphoma f Multiple myeloma g Prostate cancer h Hepatocellular carcinoma i |
| PARP15 | B-aggressive lymphoma j Myeloid leukaemia k |
2. Specific Mono-ARTs Inhibitors
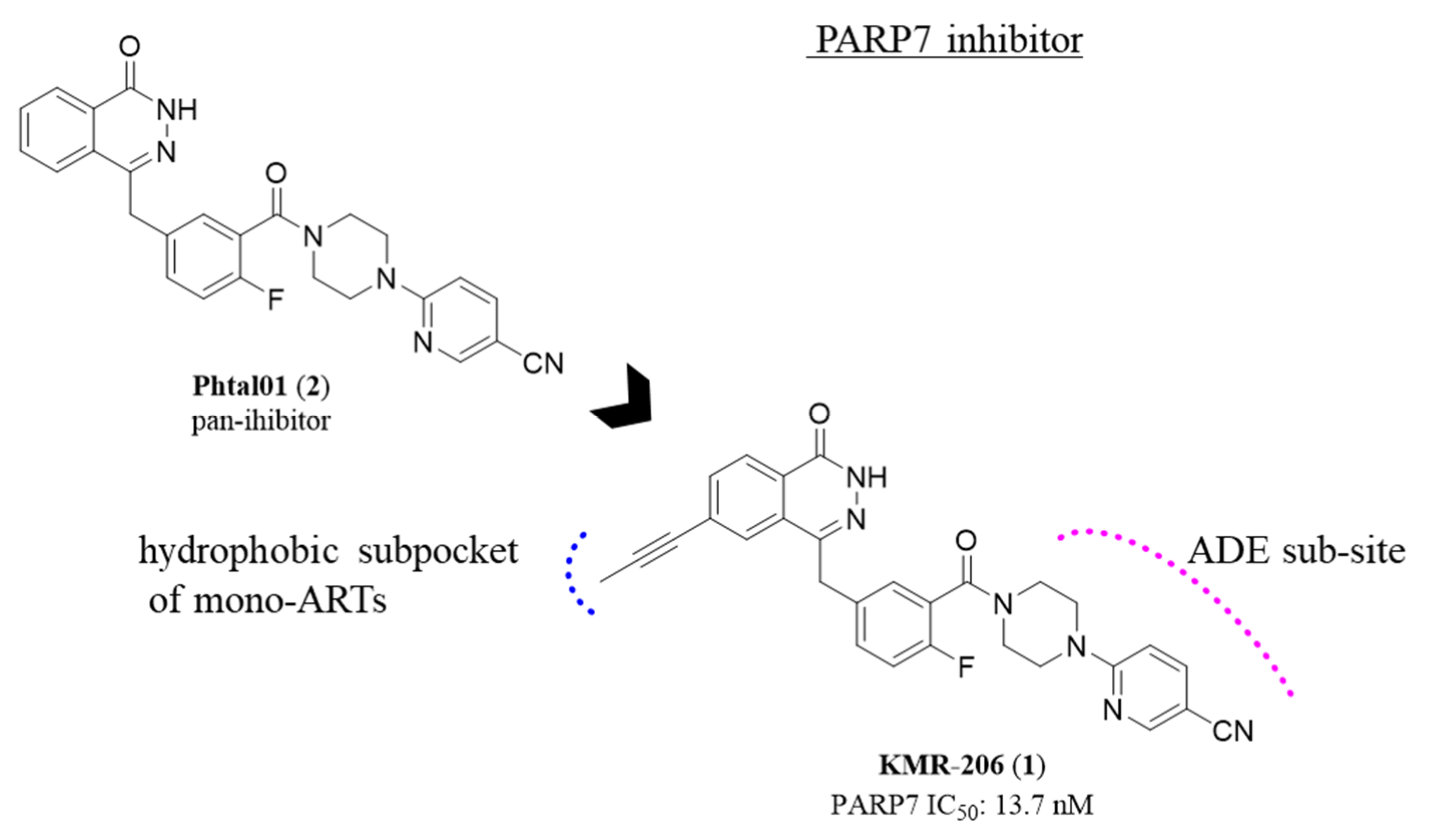
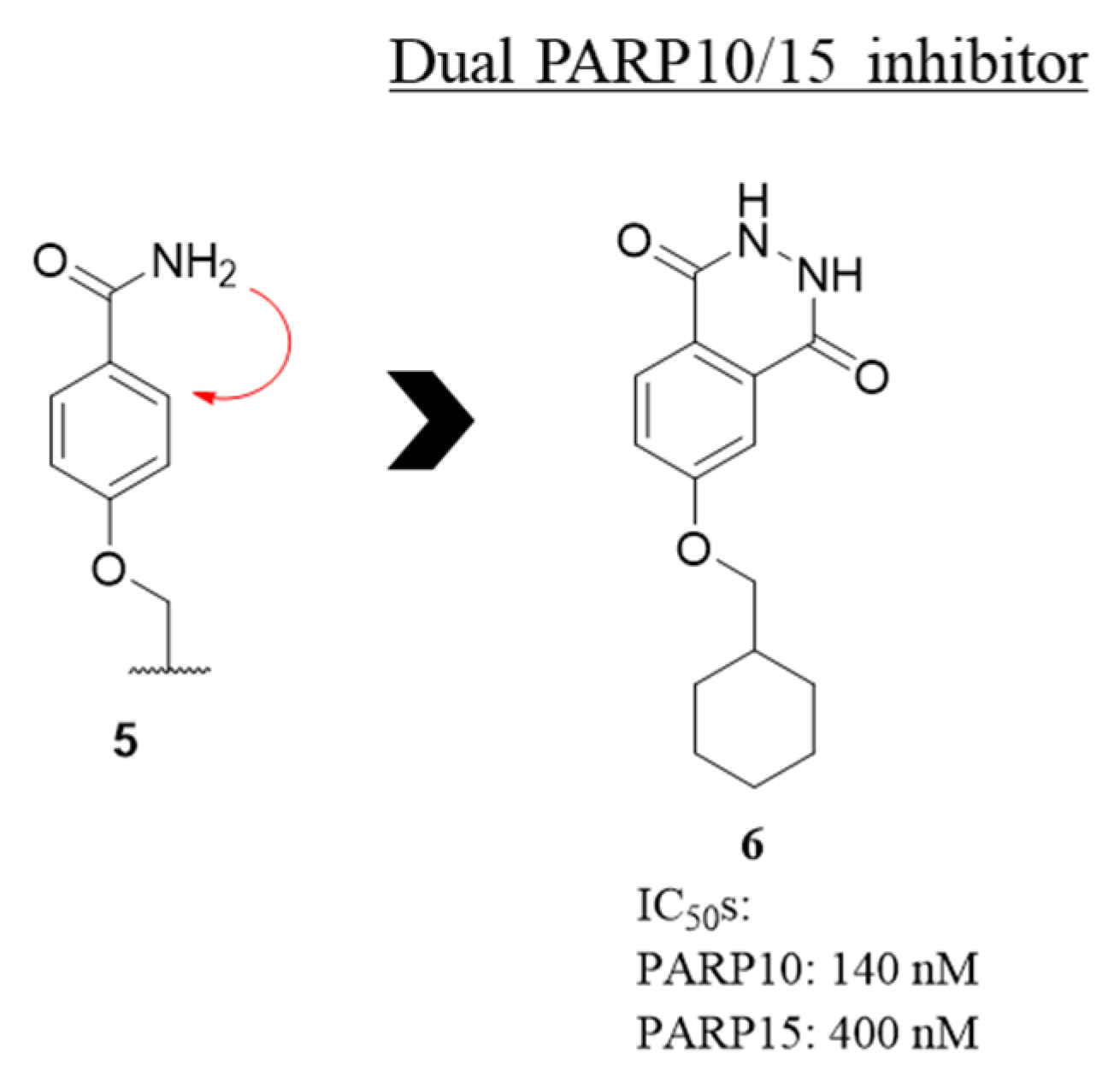
2.1. Phthalazin-1(2H)-one
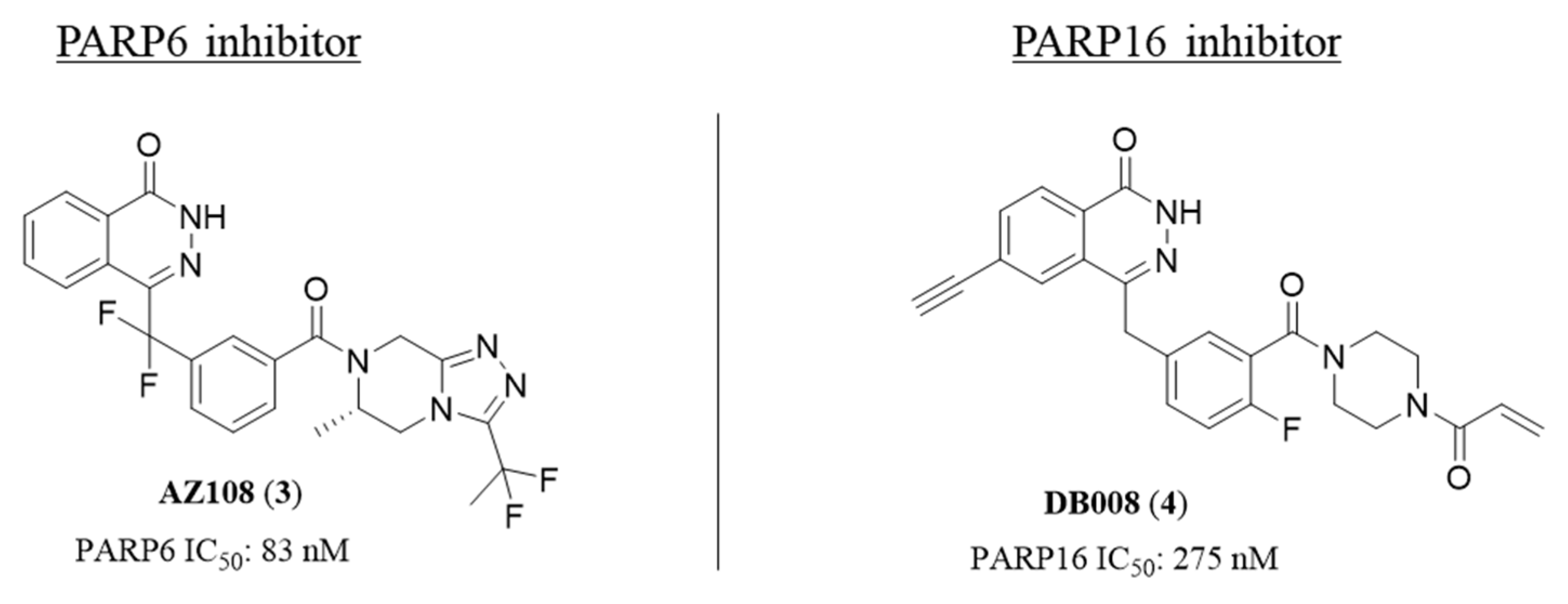
2.2. 4,5-Dihydropyridazin-3(2H)-one
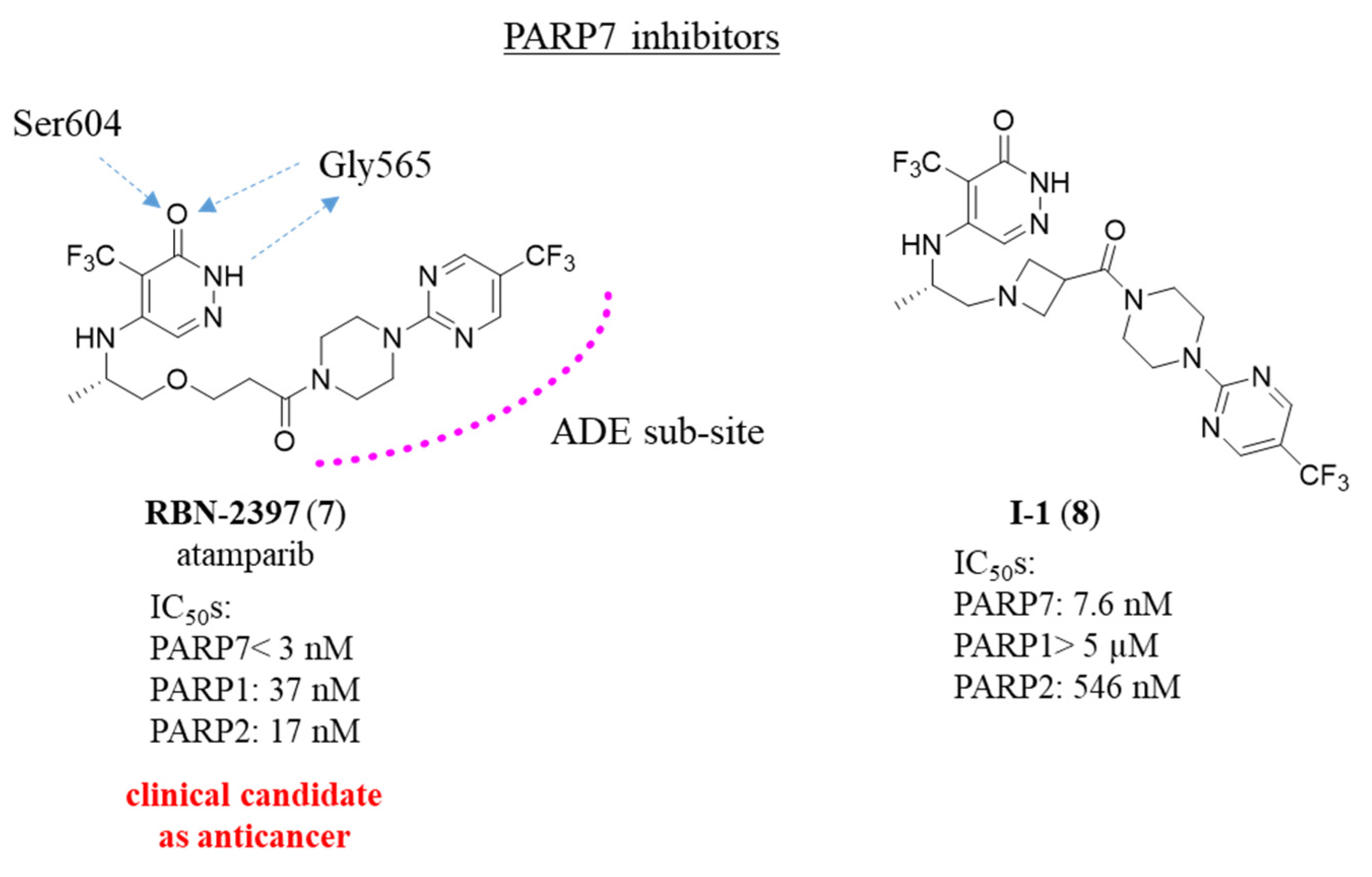
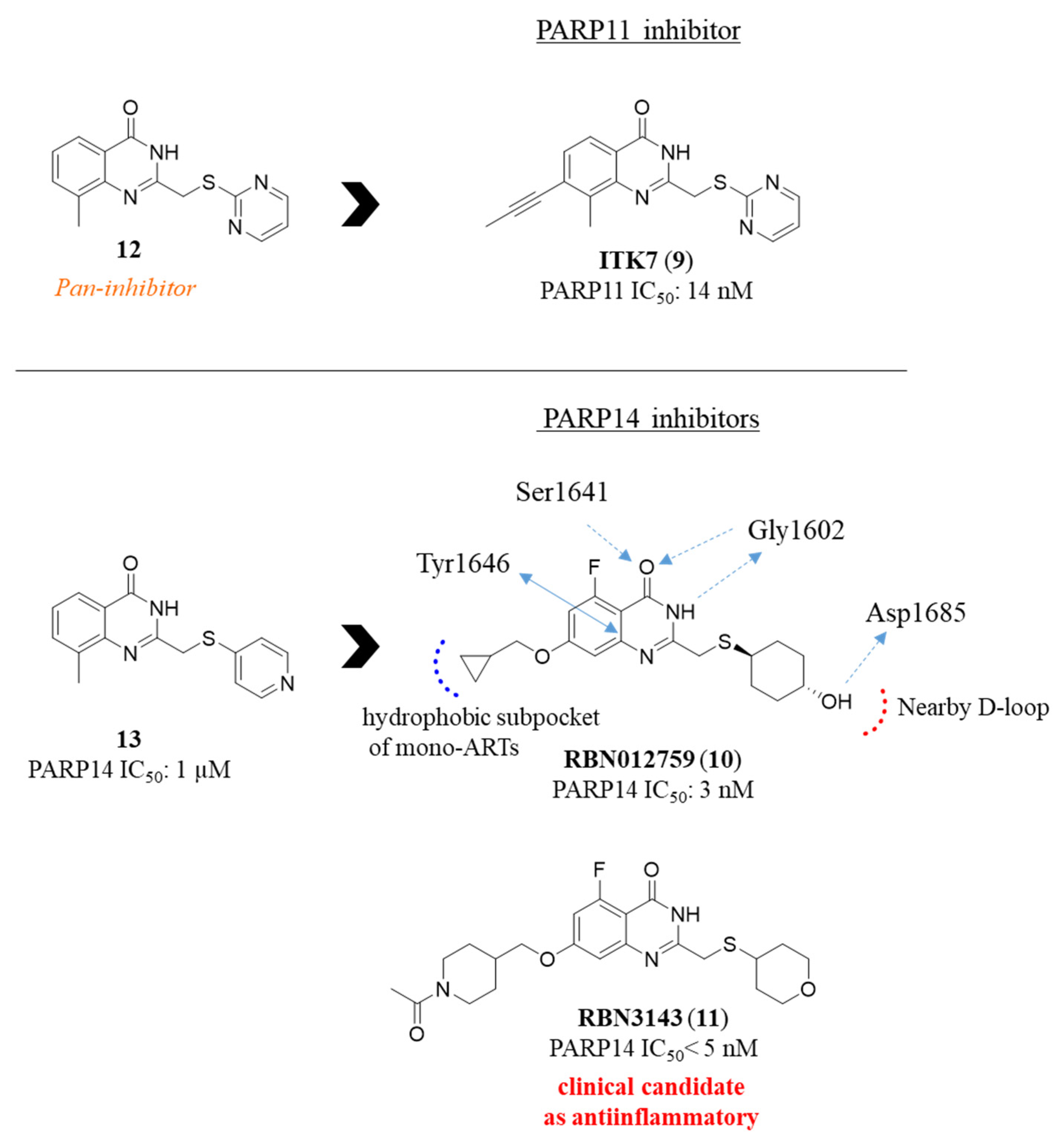
2.3. Quinazolin-4(3H)-one
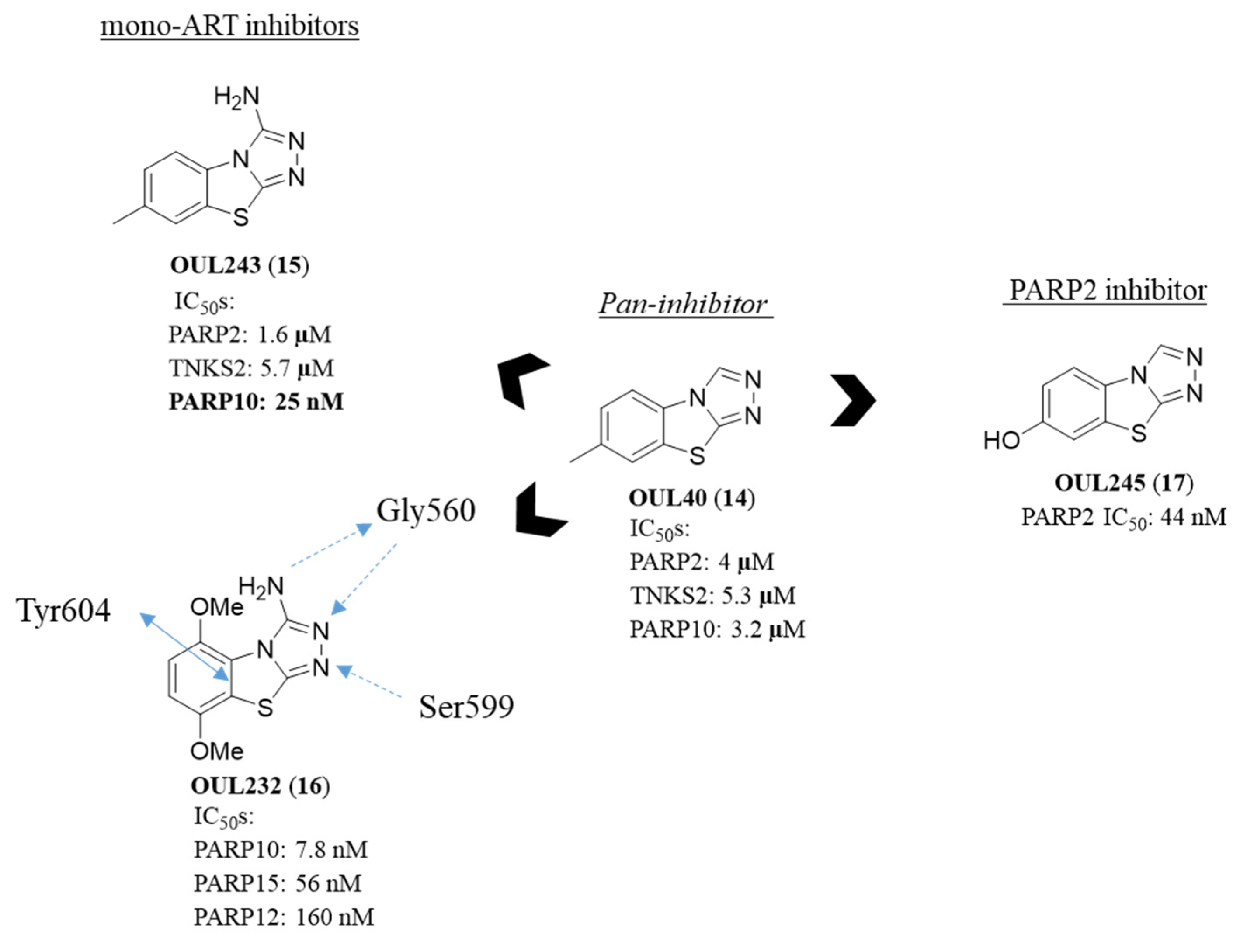
2.4. [1,2,4]Triazolo[3,4-b]benzothiazole
3. Applications of Specific ARTs Inhibitors
3.1. Specific Inhibitors as Precious Chemical Probes
3.2. Protein Degraders as Alternative to Catalytic Inhibitors
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| ADE | adenine ribose |
| ARTs | ADP-ribosyltransferase |
| ARTDs | diphtheria-toxin-like ADP-ribosyltransferases |
| BRCA | breast cancer gene |
| BRET | bioluminescence resonance energy transfer |
| CuAAC | copper assisted azide alkyne cycloaddition |
| ERα | estrogen receptor |
| HDAC | histone deacetylase |
| Hyt | hydrophobic tagging |
| HR | homologous recombination |
| IEDDA | inverse electron demand Dies-Alder reaction |
| NAD+ | β-nicotinamide adenine dinucleotide |
| NCE | new chemical entity |
| NHEJ | non-homologous end joining |
| NI | nicotinamide |
| PARPs | poly-ADP-ribose polymerases |
| PARPi | PARP inhibitors |
| PCNA | proliferating cell nuclear antigen |
| POI | protein of interest |
| PROTAC | proteolysis-targeting chimera |
| PTMs | post-translational modifications |
| SPAAC | strain promoted azide alkyne cycloaddition |
| SSBs | single strand breaks |
| TBT | [1,2,4]triazolo[3,4-b]benzothiazole |
| TNKS | tankyrase |
| TPD | targeted protein degradation |
| TR-FRET | time-resolved fluorescence resonance energy transfer |
| β-TrCP | β-transducin-repeat containing protein |
| UPR | unfolded protein response |
References
- Krishna, R.G.; Wold, F. Post-Translational Modifications of Proteins. In Methods in Protein Sequence Analysis; Springer: Boston, MA, USA, 1993; pp. 167–172. [Google Scholar] [CrossRef]
- Mikolčević, P.; Hloušek-Kasun, A.; Ahel, I.; Mikoč, A. ADP-Ribosylation Systems in Bacteria and Viruses. Comput. Struct. Biotechnol. J. 2021, 19, 2366–2383. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, I.J.; Moss, J. Structure and Function of Eukaryotic Mono-ADP-Ribosyltransferases. Rev. Physiol. Biochem. Pharmacol. 1996, 129, 51–104. [Google Scholar] [CrossRef] [PubMed]
- Gibson, B.A.; Kraus, W.L. New Insights into the Molecular and Cellular Functions of Poly(ADP-Ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Van Ness, B.G.; Howard, J.B.; Bodley, J.W. ADP-Ribosylation of Elongation Factor 2 by Diphtheria Toxin. Isolation and Properties of the Novel Ribosyl-Amino Acid and Its Hydrolysis Products. J. Biol. Chem. 1980, 255, 10717–10720. [Google Scholar] [CrossRef]
- Groslambert, J.; Prokhorova, E.; Ahel, I. ADP-Ribosylation of DNA and RNA. DNA Repair 2021, 105, 103144. [Google Scholar] [CrossRef]
- Munnur, D.; Bartlett, E.; Mikolčević, P.; Kirby, I.T.; Rack, J.G.M.; Mikoč, A.; Cohen, M.S.; Ahel, I. Reversible ADP-Ribosylation of RNA. Nucleic Acids Res. 2019, 47, 5658–5669. [Google Scholar] [CrossRef]
- Miwa, M.; Ishihara, M.; Takishima, S.; Takasuka, N.; Maeda, M.; Yamaizumi, Z.; Sugimura, T.; Yokoyama, S.; Miyazawa, T. The Branching and Linear Portions of Poly(Adenosine Diphosphate Ribose) Have the Same Alpha(1 Leads to 2) Ribose-Ribose Linkage. J. Biol. Chem. 1981, 256, 2916–2921. [Google Scholar] [CrossRef]
- Yang, C.S.; Jividen, K.; Spencer, A.; Dworak, N.; Ni, L.; Oostdyk, L.T.; Chatterjee, M.; Kuśmider, B.; Reon, B.; Parlak, M.; et al. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017, 66, 503–516. [Google Scholar] [CrossRef]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-Wide Analysis of Poly(ADP-Ribose) Polymerase Activity. Nat. Commun. 2014, 5, 4426–4455. [Google Scholar] [CrossRef]
- Ruf, A.; Rolli, V.; De Murcia, G.; Schulz, G.E. The Mechanism of the Elongation and Branching Reaction of Poly(ADP-Ribose) Polymerase as Derived from Crystal Structures and Mutagenesis. J. Mol. Biol. 1998, 278, 57–65. [Google Scholar] [CrossRef]
- O’Sullivan, J.; Tedim Ferreira, M.; Gagné, J.P.; Sharma, A.K.; Hendzel, M.J.; Masson, J.Y.; Poirier, G.G. Emerging Roles of Eraser Enzymes in the Dynamic Control of Protein ADP-Ribosylation. Nat. Commun. 2019, 10, 1182. [Google Scholar] [CrossRef]
- Rack, J.G.M.; Palazzo, L.; Ahel, I. (ADP-Ribosyl)Hydrolases: Structure, Function, and Biology. Genes Dev. 2020, 34, 263–284. [Google Scholar] [CrossRef]
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP)–Function in Genome Maintenance and Relevance of Inhibitors for Anti-Cancer Therapy. Front. Mol. Biosci. 2020, 7, 191–212. [Google Scholar] [CrossRef]
- Fisher, A.E.O.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-Ribose) Polymerase 1 Accelerates Single-Strand Break Repair in Concert with Poly(ADP-Ribose) Glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell. Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Van Beek, L.; Mcclay, É.; Patel, S.; Schimpl, M.; Spagnolo, L.; Maia De Oliveira, T. PARP Power: A Structural Perspective on PARP1, PARP2, and PARP3 in DNA Damage Repair and Nucleosome Remodelling. Int. J. Mol. Sci. 2021, 22, 5112. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, H.; Lord, C.J.; Tutt, A.H.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Myers, S.H.; Ortega, J.A.; Cavalli, A. Synthetic Lethality through the Lens of Medicinal Chemistry. J. Med. Chem. 2020, 63, 14151–14183. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First Global Approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Dockery, L.; Gunderson, C.; Moore, K. Rucaparib: The Past, Present, and Future of a Newly Approved PARP Inhibitor for Ovarian Cancer. Onco. Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef]
- Heo, Y.A.; Duggan, S.T. Niraparib: A Review in Ovarian Cancer. Target. Oncol. 2018, 13, 533–539. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Talazoparib for GBRCAm HER2-Negative Locally Advanced or Metastatic Breast Cancer|FDA. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-gbrcam-her2-negative-locally-advanced-or-metastatic-breast-cancer (accessed on 7 January 2022).
- Ghisoni, E.; Giannone, G.; Tuninetti, V.; Genta, S.; Scotto, G.; Aglietta, M.; Sangiolo, D.; Mittica, G.; Valabrega, G. Veliparib: A New Therapeutic Option in Ovarian Cancer? Future Oncol. 2019, 15, 1975–1987. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Neuhausen, S.L.; et al. The Incidence of Pancreatic Cancer in BRCA1 and BRCA2 Mutation Carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- Oh, M.; Alkhushaym, N.; Fallatah, S.; Althagafi, A.; Aljadeed, R.; Alsowaida, Y.; Jeter, J.; Martin, J.R.; Babiker, H.M.; McBride, A.; et al. The Association of BRCA1 and BRCA2 Mutations with Prostate Cancer Risk, Frequency, and Mortality: A Meta-Analysis. Prostate 2019, 79, 880–895. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP Is Activated at Stalled Forks to Mediate Mre11-Dependent Replication Restart and Recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar]
- Riffell, J.L.; Lord, C.J.; Ashworth, A. Tankyrase-Targeted Therapeutics: Expanding Opportunities in the PARP Family. Nat. Rev. Drug Discov. 2012, 11, 923–936. [Google Scholar] [CrossRef]
- Haikarainen, T.; Krauss, S.; Lehtiö, L. Tankyrases: Structure, Function and Therapeutic Implications in Cancer. Curr. Pharm. Des. 2014, 20, 6472–6488. [Google Scholar] [CrossRef]
- Mehta, C.C.; Bhatt, H.G. Tankyrase Inhibitors as Antitumor Agents: A Patent Update (2013–2020). Expert Opin. Ther. Pat. 2021, 31, 645–661. [Google Scholar] [CrossRef]
- Yu, M.; Yang, Y.; Sykes, M.; Wang, S. Small-Molecule Inhibitors of Tankyrases as Prospective Therapeutics for Cancer. J. Med. Chem. 2022, 65, 5244–5273. [Google Scholar] [CrossRef]
- First-in-Human Dose-Escalation Study of STP1002 in Patients with Advanced-Stage Solid Tumors ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04505839 (accessed on 2 December 2021).
- Plummer, R.; Dua, D.; Cresti, N.; Drew, Y.; Stephens, P.; Foegh, M.; Knudsen, S.; Sachdev, P.; Mistry, B.M.; Dixit, V.; et al. First-in-Human Study of the PARP/Tankyrase Inhibitor E7449 in Patients with Advanced Solid Tumours and Evaluation of a Novel Drug-Response Predictor. Br. J. Cancer 2020, 123, 525–533. [Google Scholar] [CrossRef]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the Repurposing of PARP Inhibitors for the Therapy of Non-Oncological Diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Nizi, M.G.; Maksimainen, M.M.; Lehtiö, L.; Tabarrini, O. Medicinal Chemistry Perspective on Targeting Mono-ADP-Ribosylating PARPs with Small Molecules. J. Med. Chem. 2022, 65, 7532–7560. [Google Scholar] [CrossRef]
- Yu, M.; Schreek, S.; Cerni, C.; Schamberger, C.; Lesniewicz, K.; Poreba, E.; Vervoorts, J.; Walsemann, G.; Grötzinger, J.; Kremmer, E.; et al. PARP-10, a Novel Myc-Interacting Protein with Poly(ADP-Ribose) Polymerase Activity, Inhibits Transformation. Oncogene 2005, 24, 1982–1993. [Google Scholar] [CrossRef]
- Nicolae, C.; Aho, E.R.; Vlahos, A.H.; Choe, K.N.; De, S.; Karras, G.I.; Moldovan, G.L. The ADP-Ribosyltransferase PARP10/ARTD10 Interacts with Proliferating Cell Nuclear Antigen (PCNA) and Is Required for DNA Damage Tolerance. J. Biol. Chem. 2014, 289, 13627–13637. [Google Scholar] [CrossRef]
- Di Paola, S.; Matarese, M.; Barretta, M.L.; Dathan, N.; Colanzi, A.; Corda, D.; Grimaldi, G. PARP10 Mediates Mono-ADP-Ribosylation of Aurora-A Regulating G2/M Transition of the Cell Cycle. Cancers 2022, 14, 5210. [Google Scholar] [CrossRef]
- Kaufmann, M.; Feijs, K.L.H.; Lüscher, B. Function and Regulation of the Mono-ADP-Ribosyltransferase ARTD10. Curr. Top. Microbiol. Immunol. 2015, 384, 167–188. [Google Scholar] [CrossRef]
- Feijs, K.L.; Kleine, H.; Braczynski, A.; Forst, A.H.; Herzog, N.; Verheugd, P.; Linzen, U.; Kremmer, E.; Lüscher, B. ARTD10 Substrate Identification on Protein Microarrays: Regulation of GSK3β by Mono-ADP-Ribosylation. Cell Commun. Signal. 2013, 11, 5–16. [Google Scholar] [CrossRef]
- Verheugd, P.; Forst, A.H.; Milke, L.; Herzog, N.; Feijs, K.L.H.; Kremmer, E.; Kleine, H.; Lüscher, B. Regulation of NF-ΚB Signalling by the Mono-ADP-Ribosyltransferase ARTD10. Nat. Commun. 2013, 4, 1683. [Google Scholar] [CrossRef] [PubMed]
- Iwata, H.; Goettsch, C.; Sharma, A.; Ricchiuto, P.; Goh, W.W.B.; Halu, A.; Yamada, I.; Yoshida, H.; Hara, T.; Wei, M.; et al. PARP9 and PARP14 Cross-Regulate Macrophage Activation via STAT1 ADP-Ribosylation. Nat. Commun. 2016, 7, 12849. [Google Scholar] [CrossRef] [PubMed]
- Bilal Iqbal, M.; Johns, M.; Cao, J.; Liu, Y.; Yu, S.-C.; Hyde, G.D.; Laffan, M.A.; Marchese, F.P.; Cho, S.H.; Clark, A.R.; et al. PARP-14 Combines with Tristetraprolin in the Selective Posttranscriptional Control of Macrophage Tissue Factor Expression Key Points. Blood 2014, 124, 3646–3655. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, P.; Riley, J.P.; Patel, R.; Li, F.; Voss, L.; Goenka, S. PARP-14 Functions as a Transcriptional Switch for Stat6-Dependent Gene Activation. J. Biol. Chem. 2011, 286, 1767–1776. [Google Scholar] [CrossRef] [PubMed]
- Yao, N.; Chen, Q.; Shi, W.; Tang, L.; Fu, Y. PARP14 Promotes the Proliferation and Gemcitabine Chemoresistance of Pancreatic Cancer Cells through Activation of NF-ΚB Pathway. Mol. Carcinog. 2019, 58, 1291–1302. [Google Scholar] [CrossRef]
- Barbarulo, A.; Iansante, V.; Chaidos, A.; Naresh, K.; Rahemtulla, A.; Franzoso, G.; Karadimitris, A.; Haskard, D.O.; Papa, S.; Bubici, C. Poly(ADP-Ribose) Polymerase Family Member 14 (PARP14) Is a Novel Effector of the JNK2-Dependent pro-Survival Signal in Multiple Myeloma. Oncogene 2012, 32, 4231–4242. [Google Scholar] [CrossRef]
- Carter-O’connell, I.; Vermehren-Schmaedick, A.; Jin, H.; Morgan, R.K.; David, L.L.; Cohen, M.S. Combining Chemical Genetics with Proximity-Dependent Labeling Reveals Cellular Targets of Poly(ADP-Ribose) Polymerase 14 (PARP14). ACS Chem. Biol. 2018, 13, 2841–2848. [Google Scholar] [CrossRef]
- Qin, W.; Wu, H.-J.; Cao, L.-Q.; Li, H.-J.; He, C.-X.; Zhao, D.; Xing, L.; Li, P.-Q.; Jin, X.; Cao, H.-L. Research Progress on PARP14 as a Drug Target. Front. Pharmacol. 2019, 10, 172–184. [Google Scholar] [CrossRef]
- Camicia, R.; Bachmann, S.B.; Winkler, H.C.; Beer, M.; Tinguely, M.; Haralambieva, E.; Hassa, P.O. BAL1/ARTD9 Represses the Anti-Proliferative and pro-Apoptotic IFNγ-STAT1-IRF1-P53 Axis in Diffuse Large B-Cell Lymphoma. J. Cell Sci. 2013, 126, 1969–1980. [Google Scholar] [CrossRef]
- Bachmann, S.B.; Frommel, S.C.; Camicia, R.; Winkler, H.C.; Santoro, R.; Hassa, P.O. DTX3L and ARTD9 Inhibit IRF1 Expression and Mediate in Cooperation with ARTD8 Survival and Proliferation of Metastatic Prostate Cancer Cells. Mol. Cancer 2014, 13, 125–149. [Google Scholar] [CrossRef]
- Iansante, V.; Choy, P.M.; Fung, S.W.; Liu, Y.; Chai, J.-G.; Dyson, J.; Rio, A.D.; D’Santos, C.; Williams, R.; Chokshi, S.; et al. PARP14 Promotes the Warburg Effect in Hepatocellular Carcinoma by Inhibiting JNK1-Dependent PKM2 Phosphorylation and Activation. Nat. Commun. 2015, 6, 7882. [Google Scholar] [CrossRef]
- Riley, J.P.; Kulkarni, A.; Mehrotra, P.; Koh, B.; Perumal, N.B.; Kaplan, M.H.; Goenka, S. PARP-14 Binds Specific DNA Sequences to Promote Th2 Cell Gene Expression. PLoS ONE 2013, 8, 83127. [Google Scholar] [CrossRef]
- Macpherson, L.; Tamblyn, L.; Rajendra, S.; Bralha, F.; Mcpherson, J.P.; Matthews, J. Tetrachlorodibenzo-p-Dioxin Poly(ADP-Ribose) Polymerase (TiPARP, ARTD14) Is a Mono-ADP-Ribosyltransferase and Repressor of Aryl Hydrocarbon Receptor Transactivation. Nucleic Acids Res. 2013, 41, 1604–1621. [Google Scholar] [CrossRef]
- Bindesbøll, C.; Tan, S.; Bott, D.; Cho, T.; Tamblyn, L.; MacPherson, L.; Grønning-Wang, L.; Nebb, H.I.; Matthews, J. TCDD-Inducible Poly-ADP-Ribose Polymerase (TIPARP/PARP7) Mono-ADP-Ribosylates and Co-Activates Liver X Receptors. Biochem. J. 2016, 473, 899–910. [Google Scholar] [CrossRef]
- Yamada, T.; Horimoto, H.; Kameyama, T.; Hayakawa, S.; Yamato, H.; Dazai, M.; Takada, A.; Kida, H.; Bott, D.; Zhou, A.C.; et al. Constitutive Aryl Hydrocarbon Receptor Signaling Constrains Type I Interferon-Mediated Antiviral Innate Defense. Nat. Immunol. 2016, 17, 687–694. [Google Scholar] [CrossRef]
- Gan, Y.; Li, X.; Han, S.; Liang, Q.; Ma, X.; Rong, P.; Wang, W.; Li, W. The CGAS/STING Pathway: A Novel Target for Cancer Therapy. Front. Immunol. 2022, 12, 795401. [Google Scholar] [CrossRef]
- Zhu, H.; Tang, Y.D.; Zhan, G.; Su, C.; Zheng, C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front. Immunol. 2021, 12, 712556. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Vasbinder, M.M.; Abo, R.P.; Kunii, K.; Kuplast-Barr, K.G.; Gui, B.; Lu, A.Z.; Molina, J.R.; Minissale, E.; Swinger, K.K.; et al. PARP7 Negatively Regulates the Type I Interferon Response in Cancer Cells and Its Inhibition Triggers Antitumor Immunity. Cancer Cell 2021, 39, 1214–1226. [Google Scholar] [CrossRef]
- Lisi, L.; Cari, L.; Sorrentino, R.; Mini, E.; Fimognari, C.; Nocentini, G. Immune Modulation of Cancer: Mechanisms and Resistance. Pharmadvances 2021, 3, 550. [Google Scholar] [CrossRef]
- Palavalli Parsons, L.H.; Challa, S.; Gibson, B.A.; Nandu, T.; Stokes, M.S.; Huang, D.; Lea, J.S.; Lee Kraus, W. Identification of PARP-7 Substrates Reveals a Role for Marylation in Microtubule Control in Ovarian Cancer Cells. eLife 2021, 10, e60481. [Google Scholar] [CrossRef]
- Rasmussen, M.; Tan, S.; Somisetty, V.S.; Hutin, D.; Olafsen, N.E.; Moen, A.; Anonsen, J.H.; Grant, D.M.; Matthews, J. PARP7 and Mono-ADP-Ribosylation Negatively Regulate Estrogen Receptor α Signaling in Human Breast Cancer Cells. Cells 2021, 10, 623. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, H.; Liu, P.; Li, C.; Quanquin, N.; Ji, X.; Sun, N.; Du, P.; Qin, C.F.; Lu, N.; et al. PARP12 Suppresses Zika Virus Infection through PARP-Dependent Degradation of NS1 and NS3 Viral Proteins. Sci. Signal. 2018, 11, 9332. [Google Scholar] [CrossRef] [PubMed]
- Krieg, S.; Pott, F.; Potthoff, L.; Verheirstraeten, M.; Bütepage, M.; Golzmann, A.; Lippok, B.; Goffinet, C.; Lüscher, B.; Korn, P. Mono-ADP-Ribosylation by PARP10 Inhibits Chikungunya Virus NsP2 Proteolytic Activity and Viral Replication. Cell. Mol. Life Sci. 2023, 80, 72. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, Y.s.; Choi, Y.; Son, A.; Park, Y.; Lee, K.M.; Kim, J.; Kim, J.S.; Kim, V.N. The SARS-CoV-2 RNA Interactome. Mol. Cell 2021, 81, 2838–2850. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A Diverse Range of Gene Products Are Effectors of the Type i Interferon Antiviral Response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Leung, K.E.; Vyas, S.; Rood, J.E.; Bhutkar, A.; Sharp, P.A.; Chang, P. Poly(ADP-Ribose) Regulates Stress Responses and MicroRNA Activity in the Cytoplasm. Mol. Cell 2011, 42, 489–499. [Google Scholar] [CrossRef]
- Catara, G.; Grimaldi, G.; Schembri, L.; Spano, D.; Turacchio, G.; Lo Monte, M.; Beccari, A.R.; Valente, C.; Corda, D. PARP1-Produced Poly-ADP-Ribose Causes the PARP12 Translocation to Stress Granules and Impairment of Golgi Complex Functions. Sci. Rep. 2017, 7, 14035–14052. [Google Scholar] [CrossRef]
- Grimaldi, G.; Filograna, A.; Schembri, L.; Lo Monte, M.; Martino, R.D.; Pirozzi, M.; Spano, D.; Beccari, A.R.; Parashuraman, S.; Luini, A.; et al. PKD-Dependent PARP12-Catalyzed Mono-ADP-Ribosylation of Golgin-97 Is Required for E-Cadherin Transport from Golgi to Plasma Membrane. Proc. Natl. Acad. Sci. USA 2022, 119, e2026494119. [Google Scholar] [CrossRef]
- Shao, C.; Qiu, Y.; Liu, J.; Feng, H.; Shen, S.; Saiyin, H.; Yu, W.; Wei, Y.; Yu, L.; Su, W.; et al. PARP12 (ARTD12) Suppresses Hepatocellular Carcinoma Metastasis through Interacting with FHL2 and Regulating Its Stability. Cell Death Dis. 2018, 9, 856–870. [Google Scholar] [CrossRef]
- Gaston, J.; Cheradame, L.; Yvonnet, V.; Deas, O.; Poupon, M.F.; Judde, J.G.; Cairo, S.; Goffin, V. Intracellular STING Inactivation Sensitizes Breast Cancer Cells to Genotoxic Agents. Oncotarget 2016, 7, 77205–77224. [Google Scholar] [CrossRef]
- Johannes, J.W.; Almeida, L.; Daly, K.; Ferguson, A.D.; Grosskurth, N.E.; Guan, H.; Howard, T.; Ioannidis, S.; Kazmirski, S.; Lamb, M.L.; et al. Discovery of AZ0108, an Orally Bioavailable Phthalazinone PARP Inhibitor That Blocks Centrosome Clustering. Bioorg. Med. Chem. Lett. 2015, 25, 5743–5747. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A Systematic Analysis of the PARP Protein Family Identifies New Functions Critical for Cell Physiology. Nat. Commun. 2013, 4, 2240–2253. [Google Scholar] [CrossRef]
- Huang, J.Y.; Wang, K.; Vermehren-Schmaedick, A.; Adelman, J.P.; Cohen, M.S. PARP6 Is a Regulator of Hippocampal Dendritic Morphogenesis. Sci. Rep. 2016, 6, 18512–18522. [Google Scholar] [CrossRef]
- Guo, T.; Zuo, Y.; Qian, L.; Liu, J.; Yuan, Y.; Xu, K.; Miao, Y.; Feng, Q.; Chen, X.; Jin, L.; et al. ADP-Ribosyltransferase PARP11 Modulates the Interferon Antiviral Response by Mono-ADP-Ribosylating the Ubiquitin E3 Ligase β-TrCP. Nat. Microbiol. 2019, 4, 1872–1884. [Google Scholar] [CrossRef]
- Wang, Z.; Shaun, E.G.; Cheung, E.T.; Petteruti, P.; Zhang, J.; Wang, X.; Wang, W.; Gharahdaghi, F.; Wu, J.; Su, N.; et al. Pharmacological Inhibition of PARP6 Triggers Multipolar Spindle Formation and Elicits Therapeutic Effects in Breast Cancer. Cancer Res. 2018, 78, 6691–6702. [Google Scholar] [CrossRef]
- Aguiar, R.C.T.; Takeyama, K.; He, C.; Kreinbrink, K.; Shipp, M.A. B-Aggressive Lymphoma Family Proteins Have Unique Domains That Modulate Transcription and Exhibit Poly(ADP-Ribose) Polymerase Activity. J. Biol. Chem. 2005, 280, 33756–33765. [Google Scholar] [CrossRef]
- Lee, M.K.; Cheong, H.S.; Koh, Y.; Ahn, K.S.; Yoon, S.S.; Shin, H.D. Genetic Association of PARP15 Polymorphisms with Clinical Outcome of Acute Myeloid Leukemia in a Korean Population. Genet. Test. Mol. Biomark. 2016, 20, 696–701. [Google Scholar] [CrossRef]
- Challa, S.; Stokes, M.S.; Kraus, W.L. MARTs and MARylation in the Cytosol: Biological Functions, Mechanisms of Action, and Therapeutic Potential. Cells 2021, 10, 313. [Google Scholar] [CrossRef]
- Hopp, A.-K.; Hottiger, M.O. Uncovering the Invisible: Mono-ADP-Ribosylation Moved into the Spotlight. Cells 2021, 10, 680. [Google Scholar] [CrossRef]
- Poltronieri, P.; Miwa, M.; Masutani, M. Adp-Ribosylation as Post-Translational Modification of Proteins: Use of Inhibitors in Cancer Control. Int. J. Mol. Sci. 2021, 22, 10829. [Google Scholar] [CrossRef]
- Poltronieri, P.; Celetti, A.; Palazzo, L. Mono(ADP-Ribosyl)Ation Enzymes and NAD+ Metabolism: A Focus on Diseases and Therapeutic Perspectives. Cells 2021, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Lei, Y.; Qi, J.; Liu, W.; Yao, K. Functional Roles of ADP-Ribosylation Writers, Readers and Erasers. Front. Cell Dev. Biol. 2022, 10, 941356. [Google Scholar] [CrossRef] [PubMed]
- Dhoonmoon, A.; Nicolae, C.M. Mono-ADP-Ribosylation by PARP10 and PARP14 in Genome Stability. NAR Cancer 2023, 5, zcad009. [Google Scholar] [CrossRef] [PubMed]
- Prime, M.E.; Courtney, S.M.; Brookfield, F.A.; Marston, R.W.; Walker, V.; Warne, J.; Boyd, A.E.; Kairies, N.A.; Von Der Saal, W.; Limberg, A.; et al. Phthalazinone Pyrazoles as Potent, Selective, and Orally Bioavailable Inhibitors of Aurora-A Kinase. J. Med. Chem. 2011, 54, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Zhao, Q.; Ma, X.; Lu, D.; Li, H.; Zeng, Y.; Tong, X.; Zeng, L.; Liu, J.; et al. Discovery of Phthalazinone Derivatives as Novel Hepatitis B Virus Capsid Inhibitors. J. Med. Chem. 2020, 63, 19. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, C.; Xie, C.; Jiang, J.; Gao, M.; Fu, L.; Li, Y.; Bao, X.; Fu, H.; Lou, L. Pharmacologic Characterization of Fluzoparib, a Novel Poly(ADP-ribose) Polymerase Inhibitor Undergoing Clinical Trials. Cancer Sci. 2019, 110, 1064–1075. [Google Scholar] [CrossRef]
- Sanderson, D.J.; Rodriguez, K.M.; Bejan, D.S.; Olafsen, N.E.; Bohn, I.D.; Kojic, A.; Sundalam, S.; Siordia, I.R.; Duell, A.K.; Deng, N.; et al. Structurally Distinct PARP7 Inhibitors Provide New Insights into the Function of PARP7 in Regulating Nucleic Acid-Sensing and IFN-β Signaling. Cell Chem. Biol. 2023, 30, 43–54. [Google Scholar] [CrossRef]
- Rodriguez, K.M.; Cohen, M.S. Chemical Genetic Methodologies for Identifying Protein Substrates of PARPs. Trends Biochem. Sci. 2022, 47, 390–402. [Google Scholar] [CrossRef]
- Kirby, I.T.; Kojic, A.; Arnold, M.R.; Thorsell, A.G.; Karlberg, T.; Vermehren-Schmaedick, A.; Sreenivasan, R.; Schultz, C.; Schüler, H.; Cohen, M.S. A Potent and Selective PARP11 Inhibitor Suggests Coupling between Cellular Localization and Catalytic Activity. Cell Chem. Biol. 2018, 25, 1547–1553. [Google Scholar] [CrossRef]
- Bejan, D.S.; Sundalam, S.; Jin, H.; Morgan, R.K.; Kirby, I.T.; Siordia, I.R.; Tivon, B.; London, N.; Cohen, M.S. Structure-Guided Design and Characterization of a Clickable, Covalent PARP16 Inhibitor. Chem. Sci. 2022, 13, 13898–13906. [Google Scholar] [CrossRef]
- Schenkel, L.B.; Molina, J.R.; Swinger, K.K.; Abo, R.; Blackwell, D.J.; Lu, A.Z.; Cheung, A.E.; Church, W.D.; Kunii, K.; Kuplast-Barr, K.G.; et al. A Potent and Selective PARP14 Inhibitor Decreases Protumor Macrophage Gene Expression and Elicits Inflammatory Responses in Tumor Explants. Cell Chem. Biol. 2021, 28, 1158–1168. [Google Scholar] [CrossRef]
- Nizi, M.G.; Maksimainen, M.M.; Murthy, S.; Massari, S.; Alaviuhkola, J.; Lippok, B.E.; Sowa, S.T.; Galera-Prat, A.; Prunskaite-Hyyryläinen, R.; Lüscher, B.; et al. Potent 2,3-Dihydrophthalazine-1,4-Dione Derivatives as Dual Inhibitors for Mono-ADP-Ribosyltransferases PARP10 and PARP15. Eur. J. Med. Chem. 2022, 237, 114362. [Google Scholar] [CrossRef]
- Murthy, S.; Desantis, J.; Verheugd, P.; Maksimainen, M.M.; Venkannagari, H.; Massari, S.; Ashok, Y.; Obaji, E.; Nkizinkinko, Y.; Lüscher, B.; et al. 4-(Phenoxy) and 4-(Benzyloxy)Benzamides as Potent and Selective Inhibitors of Mono-ADP-Ribosyltransferase PARP10/ARTD10. Eur. J. Med. Chem. 2018, 156, 93–102. [Google Scholar] [CrossRef]
- Abouzid, K.; Bekhit, S.A. Novel Anti-Inflammatory Agents Based on Pyridazinone Scaffold; Design, Synthesis and In Vivo Activity. Bioorg. Med. Chem. 2008, 16, 5547–5556. [Google Scholar] [CrossRef]
- Thyes, M.; Lehmann, H.D.; Gries, J.; Kónig, H.; Kretzschmar, R.; Kunze, J.; Lebkúcher, R.; Lenke, D. 6-Aryl-4,5-Dihydro-3(2H)-Pyridazinones. A New Class of Compounds with Platelet Aggregation Inhibiting and Hypotensive Activitiesf. J. Med. Chem. 1983, 26, 800–807. [Google Scholar] [CrossRef]
- Imran, M. 2-Butyl-6-Phenyl-4,5-Dihydropyridazin-3(2H)-One: Synthesis, In Silico Studies and In Vitro Cyclooxygenase-2 Inhibitory Activity. Molbank 2020, 3, 1155. [Google Scholar] [CrossRef]
- Akhtar, W.; Shaquiquzzaman, M.; Akhter, M.; Verma, G.; Khan, M.F.; Alam, M.M. The Therapeutic Journey of Pyridazinone. Eur. J. Med. Chem. 2016, 123, 256–281. [Google Scholar] [CrossRef]
- Yang, C.; Wierbilowicz, K.; Dworak, N.M.; Bae, S.Y.; Tengse, S.B.; Abianeh, N.; Drake, J.M.; Abbas, T.; Ratan, A.; Wotton, D.; et al. Induction of PARP7 Creates a Vulnerability for Growth Inhibition by RBN2397 in Prostate Cancer Cells. Cancer Res. Commun. 2023, 3, 592–606. [Google Scholar] [CrossRef]
- Gu, H.; Yan, W.; Wang, Y.; Xu, W.; Huang, L.; Yang, J.; Zhai, B.; Wang, H.; Su, Y.; Zhu, Q.; et al. Discovery of the Potent and Highly Selective PARP7 Inhibitor as a Novel Immunotherapeutic Agent for Tumors. J. Med. Chem. 2022, 66, 490. [Google Scholar] [CrossRef]
- Khan, I.; Zaib, S.; Batool, S.; Abbas, N.; Ashraf, Z.; Iqbal, J.; Saeed, A. Quinazolines and Quinazolinones as Ubiquitous Structural Fragments in Medicinal Chemistry: An Update on the Development of Synthetic Methods and Pharmacological Diversification. Bioorg. Med. Chem. 2016, 24, 2361–2381. [Google Scholar] [CrossRef]
- Al-Salem, H.S.A.; Hegazy, G.H.; El-Taher, K.E.H.; El-Messery, S.M.; Al-Obaid, A.M.; El-Subbagh, H.I. Synthesis, Anticonvulsant Activity and Molecular Modeling Study of Some New Hydrazinecarbothioamide, Benzenesulfonohydrazide, and Phenacylacetohydrazide Analogues of 4(3H)-Quinazolinone. Bioorg. Med. Chem. Lett. 2015, 25, 1490–1499. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, W.; Jiang, L.; Wan, S.; Zhang, L.; Yu, R.; Jiang, T. 2-Pyridinyl-4(3H)-Quinazolinone: A Scaffold for Anti-Influenza A Virus Compounds. Chem. Biol. Drug Des. 2015, 86, 1221–1225. [Google Scholar] [CrossRef] [PubMed]
- Hemalatha, K.; Madhumitha, G.; Vasavi, C.S.; Munusami, P. 2,3-Dihydroquinazolin-4(1H)-Ones: Visible Light Mediated Synthesis, Solvatochromism and Biological Activity. J. Photochem. Photobiol. B Biol. 2015, 143, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Nkizinkiko, Y.; Desantis, J.; Koivunen, J.; Haikarainen, T.; Murthy, S.; Sancineto, L.; Massari, S.; Ianni, F.; Obaji, E.; Loza, M.I.; et al. 2-Phenylquinazolinones as Dual-Activity Tankyrase-Kinase Inhibitors. Sci. Rep. 2018, 8, 1680–1690. [Google Scholar] [CrossRef]
- Iwashita, A.; Hattori, K.; Yamamoto, H.; Ishida, J.; Kido, Y.; Kamijo, K.; Murano, K.; Miyake, H.; Kinoshita, T.; Warizaya, M.; et al. Discovery of Quinazolinone and Quinoxaline Derivatives as Potent and Selective Poly(ADP-Ribose) Polymerase-1/2 Inhibitors. FEBS Lett. 2005, 579, 1389–1393. [Google Scholar] [CrossRef]
- Ramadan, S.K.; Elrazaz, E.Z.; Abouzid, K.A.M.; El-Naggar, A.M. Design, Synthesis and in Silico Studies of New Quinazolinone Derivatives as Antitumor PARP-1 Inhibitors. RSC Adv. 2020, 10, 29475–29492. [Google Scholar] [CrossRef]
- Ghorab, W.M.; El-Sebaey, S.A.; Ghorab, M.M. Design, Synthesis and Molecular Modeling Study of Certain Quinazolinone Derivatives Targeting Poly (ADP-Ribose) Polymerase 1 (PARP-1) Enzyme as Anti-Breast Cancer and Radio-Sensitizers. J. Mol. Struct. 2023, 1273, 134358–134365. [Google Scholar] [CrossRef]
- RBN-3143–Reducing Th17 and Th2 Signaling to Treat Multiple Inflammatory Diseases through PARP14 Inhibition–Ribon Therapeutics. Available online: https://ribontx.com/rbn-3143/ (accessed on 24 November 2022).
- Eddie, A.M.; Chen, K.W.; Schenkel, L.B.; Swinger, K.K.; Molina, J.R.; Kunii, K.; Raybuck, A.L.; Keilhack, H.; Gibson-Corley, K.N.; Niepel, M.; et al. Selective Pharmaceutical Inhibition of PARP14 Mitigates Allergen-Induced IgE and Mucus Overproduction in a Mouse Model of Pulmonary Allergic Response. ImmunoHorizons 2022, 6, 432–446. [Google Scholar] [CrossRef]
- Ribon Therapeutics Initiates First-in-Human Phase 1 Clinical Study of RBN-3143 for the Treatment of Atopic Dermatitis–Ribon Therapeutics. Available online: https://ribontx.com/ribon-therapeutics-initiates-first-in-human-phase-1-clinical-study-of-rbn-3143-for-the-treatment-of-atopic-dermatitis/ (accessed on 29 March 2023).
- Murthy, S.; Nizi, M.G.; Maksimainen, M.M.; Massari, S.; Alaviuhkola, J.; Lippok, B.E.; Vagaggini, C.; Sowa, S.T.; Galera-Prat, A.; Ashok, Y.; et al. Triazolo[3,4-b]Benzothiazole Scaffold as Versatile Nicotinamide Mimic Allowing Nanomolar Inhibition of Different PARP Enzymes. J. Med. Chem. 2022, 66, 1301–1320. [Google Scholar] [CrossRef]
- Deng, X.-Q.; Song, M.-X.; Wei, C.-X.; Li, F.-N.; Quan, Z.-S. Synthesis and Anticonvulsant Activity of 7-Alkoxy-Triazolo-[3, 4-b] Benzo[d]Thiazoles. Med. Chem. 2010, 6, 313–320. [Google Scholar] [CrossRef]
- Mamatha, S.V.; Belagali, S.L.; Bhat, M. Synthesis and SAR Evaluation of Mercaptotriazolo Derivatives as Anti-Inflammatory Agents. Int. J. ChemTech Res. 2020, 13, 187–198. [Google Scholar] [CrossRef]
- Aggarwal, R.; Sumran, G. An Insight on Medicinal Attributes of 1,2,4-Triazoles. Eur. J. Med. Chem. 2020, 205, 112,652–112,648. [Google Scholar] [CrossRef]
- Felicetti, T.; Pismataro, M.C.; Cecchetti, V.; Tabarrini, O.; Massari, S. Triazolopyrimidine Nuclei: Privileged Scaffolds for Developing Antiviral Agents with a Proper Pharmacokinetic Profile. Curr. Med. Chem. 2021, 29, 1379–1407. [Google Scholar] [CrossRef]
- Massari, S.; Bertagnin, C.; Pismataro, M.C.; Donnadio, A.; Nannetti, G.; Felicetti, T.; Di Bona, S.; Nizi, M.G.; Tensi, L.; Manfroni, G.; et al. Synthesis and Characterization of 1,2,4-Triazolo[1,5-a]Pyrimidine-2-Carboxamide-Based Compounds Targeting the PA-PB1 Interface of Influenza A Virus Polymerase. Eur. J. Med. Chem. 2021, 209, 112944–112963. [Google Scholar] [CrossRef]
- Gao, F.; Wang, T.; Xiao, J.; Huang, G. Antibacterial Activity Study of 1,2,4-Triazole Derivatives. Eur. J. Med. Chem. 2019, 173, 274–281. [Google Scholar] [CrossRef]
- Li, S.; Li, X.y.; Zhang, T.j.; Kamara, M.O.; Liang, J.w.; Zhu, J.; Meng, F.h. Design, Synthesis and Biological Evaluation of Homoerythrina Alkaloid Derivatives Bearing a Triazole Moiety as PARP-1 Inhibitors and as Potential Antitumor Drugs. Bioorg. Chem. 2020, 94, 103385. [Google Scholar] [CrossRef]
- Abu-Hashem, A.A.; Abu-Zied, K.M.; AbdelSalam Zaki, M.E.; El-Shehry, M.F.; Awad, H.M.; Khedr, M.A. Design, Synthesis, and Anticancer Potential of the Enzyme (PARP-1) Inhibitor with Computational Studies of New Triazole, Thiazolidinone, -Thieno [2, 3-d] Pyrimidinones. Lett. Drug Des. Discov. 2020, 17, 799–817. [Google Scholar] [CrossRef]
- Leenders, R.G.G.; Brinch, S.A.; Sowa, S.T.; Amundsen-Isaksen, E.; Galera-Prat, A.; Murthy, S.; Aertssen, S.; Smits, J.N.; Nieczypor, P.; Damen, E.; et al. Development of a 1,2,4-Triazole-Based Lead Tankyrase Inhibitor: Part II. J. Med. Chem. 2021, 64, 17936–17949. [Google Scholar] [CrossRef]
- Gan, Y.; Sha, H.; Zou, R.; Xu, M.; Zhang, Y.; Feng, J.; Wu, J. Research Progress on Mono-ADP-Ribosyltransferases in Human Cell Biology. Front. Cell Dev. Biol. 2022, 10, 815. [Google Scholar] [CrossRef]
- Dasovich, M.; Leung, A.K.L. PARPs and ADP-Ribosylation: Deciphering the Complexity with Molecular Tools. Mol. Cell 2023, 83, 1552–1572. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Reed, A. Pipeline Impact of Radiolabeled Compounds in Drug Discovery and Development. ACS Med. Chem. Lett. 2022, 13, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Carney, B.; Kossatz, S.; Reiner, T. Molecular Imaging of PARP. J. Nucl. Med. 2017, 58, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Li, K.; Chen, K.; He, S.; Zhang, J.; Gu, B.; Xu, X.; Song, S. PET Imaging of PARP Expression Using 68Ga-Labelled Inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 2606–2620. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.Y.; Chen, Z.; Destro, G.; Veal, M.; Lau, D.; O’Neill, E.; Dias, G.; Mosley, M.; Kersemans, V.; Guibbal, F.; et al. Imaging PARP with [18F]Rucaparib in Pancreatic Cancer Models. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3668–3678. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, J. Current Status and Progress in Using Radiolabelled PARP-1 Inhibitors for Imaging PARP-1 Expression in Tumours. Eur. J. Med. Chem. 2022, 242, 114690. [Google Scholar] [CrossRef]
- Howard, R.T.; Hemsley, P.; Petteruti, P.; Saunders, C.N.; Bermejo, J.A.M.; Scott, J.S.; Johannes, J.W.; Tate, E.W. Structure-Guided Design and In-Cell Target Profiling of a Cell-Active Target Engagement Probe for PARP Inhibitors. ACS Chem. Biol. 2020, 15, 333. [Google Scholar] [CrossRef]
- Rutkowska, A.; Thomson, D.W.; Vappiani, J.; Werner, T.; Mueller, K.M.; Dittus, L.; Krause, J.; Muelbaier, M.; Bergamini, G.; Bantscheff, M. A Modular Probe Strategy for Drug Localization, Target Identification and Target Occupancy Measurement on Single Cell Level. ACS Chem. Biol. 2016, 11, 2541–2550. [Google Scholar] [CrossRef]
- Voorneveld, J.; Florea, B.I.; Bakkum, T.; Mendowicz, R.J.; van der Veer, M.S.; Gagestein, B.; van Kasteren, S.I.; van der Stelt, M.; Overkleeft, H.S.; Filippov, D.V. Olaparib-Based Photoaffinity Probes for PARP-1 Detection in Living Cells. ChemBioChem 2020, 21, 2431–2434. [Google Scholar] [CrossRef]
- Bejan, D.S.; Cohen, M.S. Methods for Profiling the Target and Off-Target Landscape of PARP Inhibitors. Curr. Res. Chem. Biol. 2022, 2, 100027. [Google Scholar] [CrossRef]
- Wigle, T.J.; Blackwell, D.J.; Schenkel, L.B.; Ren, Y.; Church, W.D.; Desai, H.J.; Swinger, K.K.; Santospago, A.G.; Majer, C.R.; Lu, A.Z.; et al. In Vitro and Cellular Probes to Study PARP Enzyme Target Engagement. Cell Chem. Biol. 2020, 27, 877–887. [Google Scholar] [CrossRef]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Crews, C.M. Proteolysis Targeting Chimeras (PROTACs) Come of Age: Entering the Third Decade of Targeted Protein Degradation. RSC Chem. Biol. 2021, 2, 725. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein Degraders Enter the Clinic—A New Approach to Cancer Therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Koroleva, O.A.; Dutikova, Y.V.; Trubnikov, A.V.; Zenov, F.A.; Manasova, E.V.; Shtil, A.A.; Kurkin, A.V. PROTAC: Targeted Drug Strategy. Principles and Limitations. Russ. Chem. Bull. 2022, 71, 2310–2334. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, X.; Rao, Y. PROTAC Technology: Opportunities and Challenges. ACS Med. Chem. Lett. 2020, 11, 237–240. [Google Scholar] [CrossRef]
- Zhao, Q.; Lan, T.; Su, S.; Rao, Y. Induction of Apoptosis in MDA-MB-231 Breast Cancer Cells by a PARP1-Targeting PROTAC Small Molecule. Chem. Commun. 2019, 55, 369–372. [Google Scholar] [CrossRef]
- Cao, C.; Yang, J.; Chen, Y.; Zhou, P.; Wang, Y.; Du, W.; Zhao, L.; Chen, Y. Discovery of SK-575 as a Highly Potent and Efficacious Proteolysis-Targeting Chimera Degrader of PARP1 for Treating Cancers. J. Med. Chem. 2020, 63, 11012–11033. [Google Scholar] [CrossRef]
- Zheng, M.; Huo, J.; Gu, X.; Wang, Y.; Wu, C.; Zhang, Q.; Wang, W.; Liu, Y.; Liu, Y.; Zhou, X.; et al. Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 2021, 64, 7839–7852. [Google Scholar] [CrossRef]
- Kim, C.; Chen, C.; Yu, Y. Avoid the Trap: Targeting PARP1 beyond Human Malignancy. Cell Chem. Biol. 2021, 28, 456–462. [Google Scholar] [CrossRef]
- Pieper, A.A.; Walles, T.; Wei, G.; Clements, E.E.; Verma, A.; Snyder, S.H.; Zweier, J.L. Myocardial Postischemic Injury Is Reduced by PolyADPripose Polymerase-1 Gene Disruption. Mol. Med. 2000, 6, 271. [Google Scholar] [CrossRef]
- Wang, S.; Han, L.; Han, J.; Li, P.; Ding, Q.; Zhang, Q.J.; Liu, Z.P.; Chen, C.; Yu, Y. Uncoupling of PARP1 Trapping and Inhibition Using Selective PARP1 Degradation. Nat. Chem. Biol. 2019, 15, 1223–1231. [Google Scholar] [CrossRef]
- Wigle, T.J.; Ren, Y.; Molina, J.R.; Blackwell, D.J.; Schenkel, L.B.; Swinger, K.K.; Kuplast-Barr, K.; Majer, C.R.; Church, W.D.; Lu, A.Z.; et al. Targeted Degradation of PARP14 Using a Heterobifunctional Small Molecule. ChemBioChem 2021, 22, 2107–2110. [Google Scholar] [CrossRef]
- Zhong, Y.; Chi, F.; Wu, H.; Liu, Y.; Xie, Z.; Huang, W.; Shi, W.; Qian, H. Emerging Targeted Protein Degradation Tools for Innovative Drug Discovery: From Classical PROTACs to the Novel and Beyond. Eur. J. Med. Chem. 2022, 231, 114142. [Google Scholar] [CrossRef]
- Li, H.; Dong, J.; Cai, M.; Xu, Z.; Cheng, X.D.; Qin, J.J. Protein Degradation Technology: A Strategic Paradigm Shift in Drug Discovery. J. Hematol. Oncol. 2021, 14, 138. [Google Scholar] [CrossRef]
- Go, A.; Jang, J.W.; Lee, W.; Ha, J.D.; Kim, H.J.; Nam, H.J. Augmentation of the Antitumor Effects of PARP Inhibitors in Triple-Negative Breast Cancer via Degradation by Hydrophobic Tagging Modulation. Eur. J. Med. Chem. 2020, 204, 112635. [Google Scholar] [CrossRef]
- Kleine, H.; Poreba, E.; Lesniewicz, K.; Hassa, P.O.; Hottiger, M.O.; Litchfield, D.W.; Shilton, B.H.; Lüscher, B. Substrate-Assisted Catalysis by PARP10 Limits Its Activity to Mono-ADP-Ribosylation. Mol. Cell 2008, 32, 57–69. [Google Scholar] [CrossRef]
- Holechek, J.; Lease, R.; Thorsell, A.G.; Karlberg, T.; McCadden, C.; Grant, R.; Keen, A.; Callahan, E.; Schüler, H.; Ferraris, D. Design, Synthesis and Evaluation of Potent and Selective Inhibitors of Mono-(ADP-Ribosyl)Transferases PARP10 and PARP14. Bioorg. Med. Chem. Lett. 2018, 28, 2050–2054. [Google Scholar] [CrossRef]
- Upton, K.; Meyers, M.; Thorsell, A.G.; Karlberg, T.; Holechek, J.; Lease, R.; Schey, G.; Wolf, E.; Lucente, A.; Schüler, H.; et al. Design and Synthesis of Potent Inhibitors of the Mono(ADP-Ribosyl)Transferase, PARP14. Bioorg. Med. Chem. Lett. 2017, 27, 2907–2911. [Google Scholar] [CrossRef]
- Ekblad, T.; Lindgren, A.E.G.; Andersson, C.D.; Caraballo, R.; Thorsell, A.G.; Karlberg, T.; Spjut, S.; Linusson, A.; Schüler, H.; Elofsson, M. Towards Small Molecule Inhibitors of Mono-ADP-Ribosyltransferases. Eur. J. Med. Chem. 2015, 95, 546–551. [Google Scholar] [CrossRef]
- Andersson, C.D.; Karlberg, T.; Ekblad, T.; Lindgren, A.E.G.; Thorsell, A.-G.; Spjut, S.; Uciechowska, U.; Niemiec, M.S.; Wittung-Stafshede, P.; Weigelt, J.; et al. Discovery of Ligands for ADP-Ribosyltransferases via Docking-Based Virtual Screening. J. Med. Chem. 2012, 55, 7706–7718. [Google Scholar] [CrossRef]
- Schuller, M.; Riedel, K.; Gibbs-Seymour, I.; Uth, K.; Sieg, C.; Gehring, A.P.; Ahel, I.; Bracher, F.; Kessler, B.M.; Elkins, J.M.; et al. Discovery of a Selective Allosteric Inhibitor Targeting Macrodomain 2 of Polyadenosine-Diphosphate-Ribose Polymerase 14. ACS Chem. Biol. 2017, 12, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, T.; Verheugd, P.; Lindgren, A.E.; Nyman, T.; Elofsson, M.; Schüler, H. Identification of Poly(ADP-Ribose) Polymerase Macrodomain Inhibitors Using an AlphaScreen Protocol. SLAS Discov. 2018, 23, 353–362. [Google Scholar] [CrossRef] [PubMed]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nizi, M.G.; Sarnari, C.; Tabarrini, O. Privileged Scaffolds for Potent and Specific Inhibitors of Mono-ADP-Ribosylating PARPs. Molecules 2023, 28, 5849. https://doi.org/10.3390/molecules28155849
Nizi MG, Sarnari C, Tabarrini O. Privileged Scaffolds for Potent and Specific Inhibitors of Mono-ADP-Ribosylating PARPs. Molecules. 2023; 28(15):5849. https://doi.org/10.3390/molecules28155849
Chicago/Turabian StyleNizi, Maria Giulia, Chiara Sarnari, and Oriana Tabarrini. 2023. "Privileged Scaffolds for Potent and Specific Inhibitors of Mono-ADP-Ribosylating PARPs" Molecules 28, no. 15: 5849. https://doi.org/10.3390/molecules28155849
APA StyleNizi, M. G., Sarnari, C., & Tabarrini, O. (2023). Privileged Scaffolds for Potent and Specific Inhibitors of Mono-ADP-Ribosylating PARPs. Molecules, 28(15), 5849. https://doi.org/10.3390/molecules28155849