
Low-Energy Transformation Pathways between Naphthalene Isomers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. The DFTB Potential Energy
2.2. Threshold Method
2.3. Computational Details
3. Results and Discussion
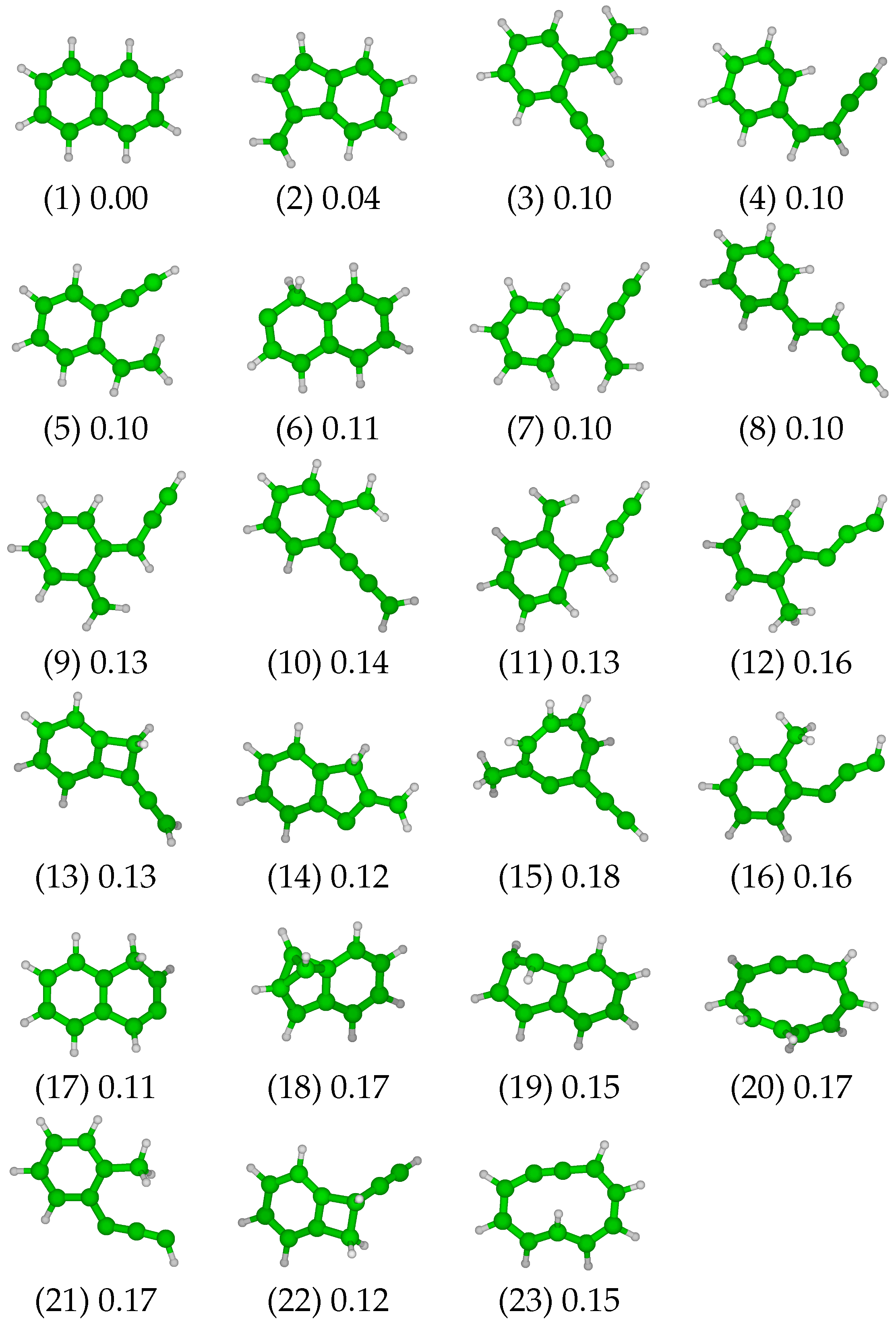
3.1. Low-Energy Naphthalene Isomers
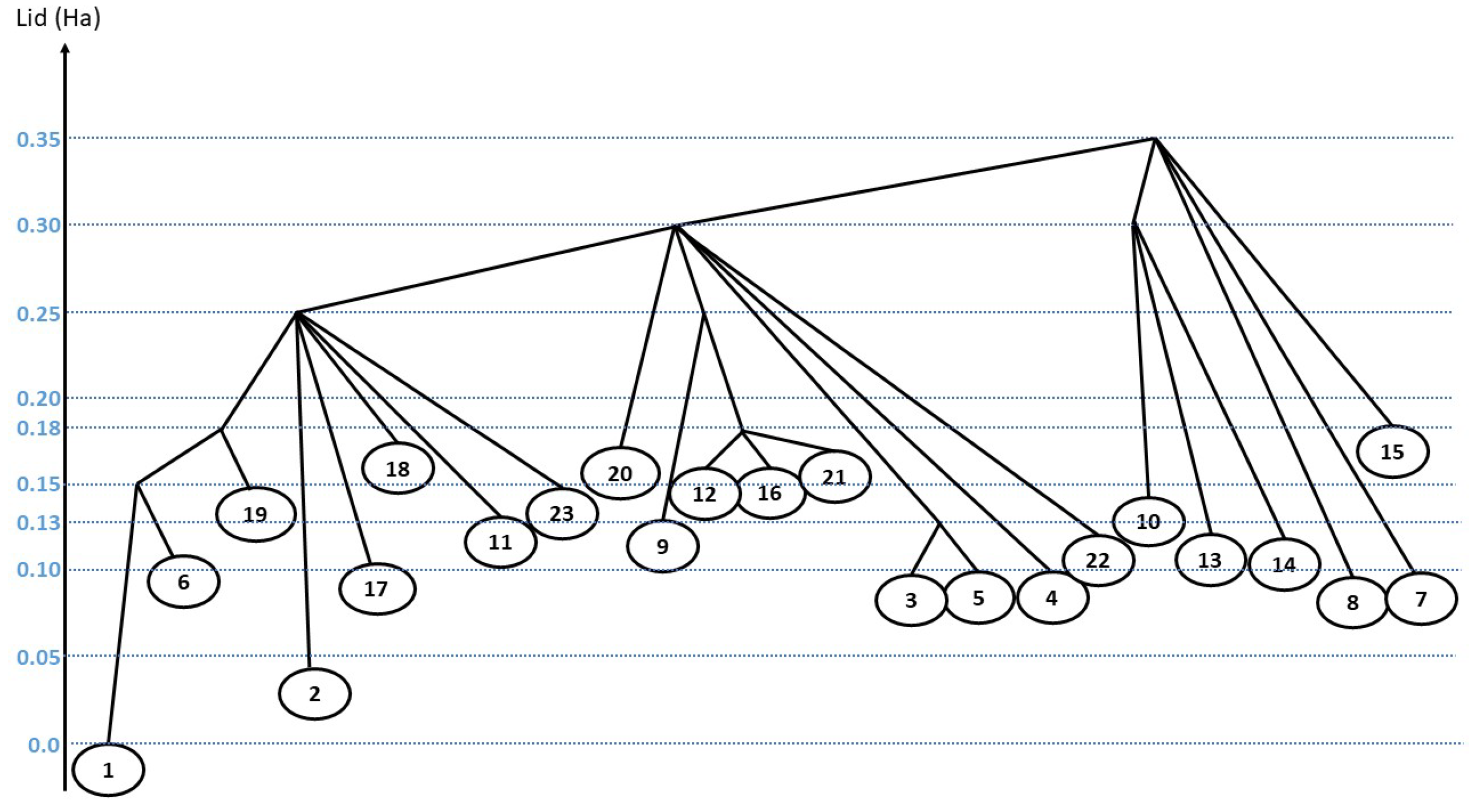
3.2. Disconnectivity Tree and Probability Flows
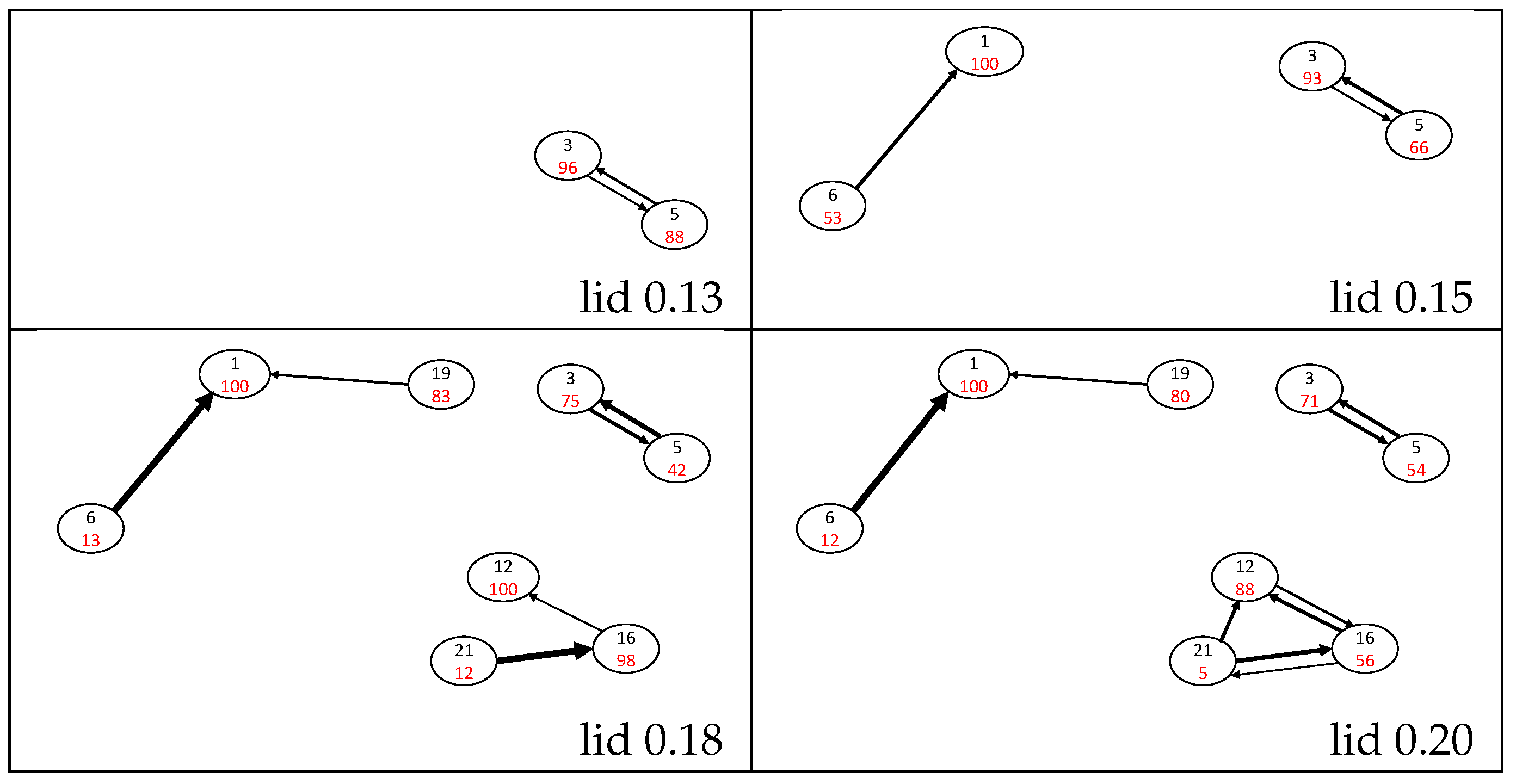
3.3. Return Probabilities and Transition Regions
3.4. Naphthalene Isomerization Pathways
3.5. Local Densities of States
4. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Allamandola, L.J.; Tielens, A.G.G.M.; Barker, J.R. Polycyclic aromatic hydrocarbons and the unidentified infrared emission bands: Auto exhaust along the milky way. Astrophys. J. Lett. 1985, 290, L25–L28. [Google Scholar] [CrossRef]
- Cernicharo, J.; Agúndez, M.; Cabezas, C.; Tercero, B.; Marcelino, N.; Pardo, J.R.; de Vicente, P. Pure hydrocarbon cycles in TMC-1: Discovery of ethynyl cyclopropenylidene, cyclopentadiene, and indene. Astron. Astrophys. 2021, 649, L15. [Google Scholar] [CrossRef]
- Micelotta, E.R.; Jones, A.P.; Tielens, A.G.G.M. Polycyclic aromatic hydrocarbon processing in interstellar shocks. Astron. Astrophys. 2010, 510, A36. [Google Scholar] [CrossRef]
- Micelotta, E.R.; Jones, A.P.; Tielens, A.G.G.M. Polycyclic aromatic hydrocarbon processing in a hot gas. Astron. Astrophys. 2010, 510, A37. [Google Scholar] [CrossRef]
- He, S.; Lin, Z.; Du, F.; Wang, X.; Liu, Y.; Tang, W. Simple unfused acceptors with optimal naphthalene isomerization enabling 10.72% as-cast organic solar cells. Chem. Eng. J. 2022, 441, 135973. [Google Scholar] [CrossRef]
- Senkan, S.; Castaldi, M. Formation of polycyclic aromatic hydrocarbons (PAH) in methane combustion: Comparative new results from premixed flames. Combust. Flame 1996, 107, 141–150. [Google Scholar] [CrossRef]
- Sabbah, H.; Biennier, L.; Klippenstein, S.J.; Sims, I.R.; Rowe, B.R. Exploring the Role of PAHs in the Formation of Soot: Pyrene Dimerization. J. Phys. Chem. Lett. 2010, 1, 2962–2967. [Google Scholar] [CrossRef]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2016. [Google Scholar]
- Leboucher, H.; Simon, A.; Rapacioli, M. Structures and stabilities of PAH clusters solvated by water aggregates: The case of the pyrene dimer. J. Chem. Phys. 2023, 158, 114308. [Google Scholar] [CrossRef]
- Patel, A.B.; Shaikh, S.; Jain, K.R.; Desai, C.; Madamwar, D. Polycyclic Aromatic Hydrocarbons: Sources, Toxicity, and Remediation Approaches. Front. Microbiol. 2020, 11, 562813. [Google Scholar] [CrossRef]
- Pope, C.J.; Peters, W.A.; Howard, J.B. Thermodynamic driving forces for PAH isomerization and growth during thermal treatment of polluted soils. J. Hazard. Mater. 2000, 79, 189–208. [Google Scholar] [CrossRef]
- Castellanos, P.; Candian, A.; Zhen, J.; Linnartz, H.; Tielens, A.G.G.M. Photoinduced polycyclic aromatic hydrocarbon dehydrogenation The competition between H- and H2-loss. Astron. Astrophys. 2018, 616, A166. [Google Scholar] [CrossRef]
- Rodriguez Castillo, S.; Simon, A.; Joblin, C. Investigating the importance of edge-structure in the loss of H/H2 of PAH cations: The case of dibenzopyrene isomers. Int. J. Mass Spectrom. 2018, 429, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Gatchell, M.; Stockett, M.H.; Delaunay, R.; Domaracka, A.; Micelotta, E.R.; Tielens, A.G.G.M.; Rousseau, P.; Adoui, L.; Huber, B.A.; et al. Formation of H2 from internally heated polycyclic aromatic hydrocarbons: Excitation energy dependence. J. Chem. Phys. 2015, 142, 144305. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Troe, J. Thermal isomerization of azulene to naphthalene in shock waves. Int. J. Chem. Kinet. 1988, 20, 379–386. [Google Scholar] [CrossRef]
- Jacovella, U.; Rossi, C.; Romanzin, C.; Alcaraz, C.; Thissen, R. Monitoring the Light-induced Isomerisation of the Prototypical Polycyclic Aromatic Hydrocarbons C10 through Ion-Molecule Reactions. ChemPhysChem 2023, 24, e202200474. [Google Scholar] [CrossRef]
- Lee, J.W.L.; Tikhonov, D.S.; Allum, F.; Boll, R.; Chopra, P.; Erk, B.; Gruet, S.; He, L.; Heathcote, D.; Kazemi, M.M.; et al. The kinetic energy of PAH dication and trication dissociation determined by recoil-frame covariance map imaging. Phys. Chem. Chem. Phys. 2022, 24, 23096–23105. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, A.; Joblin, C.; Mulas, G.; Mundlapati, V.R.; Bonnamy, A. Photodissociation of aliphatic PAH derivatives under relevant astrophysical conditions. Astron. Astrophys. 2021, 652, A42. [Google Scholar] [CrossRef]
- Panchagnula, S.; Bouwman, J.; Rap, D.B.; Castellanos, P.; Candian, A.; Mackie, C.; Banhatti, S.; Bruken, S.; Linnartz, H.; Tielens, A.G.G.M. Structural investigation of doubly-dehydrogenated pyrene cations. Phys. Chem. Chem. Phys. 2020, 22, 21651–21663. [Google Scholar] [CrossRef]
- Rouillé, G.; Steglich, M.; Hemberger, P.; Jäger, C.; Henning, T. Threshold Dissociation of the 1-ethynylpyrene Cation at Internal Energies Relevant to H i Regions. Astrophys. J. 2019, 885, 21. [Google Scholar] [CrossRef]
- Simon, A.; Champeaux, J.P.; Rapacioli, M.; Capelle, P.M.; Gadéa, F.X.; Sence, M. Dissociation of polycyclic aromatic hydrocarbons at high energy: MD/DFTB simulations versus collision experiments. Theor. Chem. Acc. 2018, 137, 106. [Google Scholar] [CrossRef]
- West, B.; Rodriguez Castillo, S.; Sit, A.; Mohamad, S.; Lowe, B.; Joblin, C.; Bodi, A.; Mayer, P.M. Unimolecular reaction energies for polycyclic aromatic hydrocarbon ions. Phys. Chem. Chem. Phys. 2018, 20, 7195–7205. [Google Scholar] [CrossRef] [PubMed]
- Parent, J.F.; Deslongchamps, P. High-Temperature Isomerization of Benzenoid Polycyclic Aromatic Hydrocarbons. Analysis through the Bent Bond and Antiperiplanar Hypothesis Orbital Model. J. Org. Chem. 2018, 83, 3299–3304. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Luo, Y. Formation of polyynes and ring-polyyne molecules following fragmentation of polycyclic aromatic hydrocarbons. Mon. Not. R. Astron. Soc. 2019, 486, 1875–1881. [Google Scholar] [CrossRef]
- Chen, T.; Luo, Y.; Li, A. Fragmentation and isomerization of polycyclic aromatic hydrocarbons in the interstellar medium: Coronene as a case study. Astron. Astrophys. 2020, 633, A103. [Google Scholar] [CrossRef]
- Simon, A.; Rapacioli, M.; Rouaut, G.; Trinquier, G.; Gadéa, F.X. Dissociation of polycyclic aromatic hydrocarbons: Molecular dynamics studies. Philos. Trans. R. Soc. A 2017, 375, 20160195. [Google Scholar] [CrossRef] [PubMed]
- Parneix, P.; Gamboa, A.; Falvo, C.; Bonnin, M.; Pino, T.; Calvo, F. Dehydrogenation effects on the stability of aromatic units in polycyclic aromatic hydrocarbons in the interstellar medium: A computational study at finite temperature. Mol. Astrophys. 2017, 7, 9–18. [Google Scholar] [CrossRef]
- Trinquier, G.; Simon, A.; Rapacioli, M.; Gadéa, F.X. PAH chemistry at eV internal energies. 1. H-shifted isomers. Mol. Astrophys. 2017, 7, 27–36. [Google Scholar] [CrossRef]
- Trinquier, G.; Simon, A.; Rapacioli, M.; Gadéa, F.X. PAH chemistry at eV internal energies. 2. Ring alteration and dissociation. Mol. Astrophys. 2017, 7, 37–59. [Google Scholar] [CrossRef]
- Dyakov, Y.A.; Ni, C.K.; Lin, S.H.; Lee, Y.T.; Mebel, A.M. Ab initio and RRKM study of photodissociation of azulene cation. Phys. Chem. Chem. Phys. 2006, 8, 1404–1415. [Google Scholar] [CrossRef]
- West, B.J.; Lesniak, L.; Mayer, P.M. Why Do Large Ionized Polycyclic Aromatic Hydrocarbons Not Lose C2H2? J. Phys. Chem. A 2019, 123, 3569–3574. [Google Scholar] [CrossRef]
- Solano, E.A.; Mayer, P.M. A complete map of the ion chemistry of the naphthalene radical cation? DFT and RRKM modeling of a complex potential energy surface. J. Chem. Phys. 2015, 143, 104305. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.; Fantuzzi, F.; Quitián-Lara, H.M.; Martins-Franco, Y.; Menéndez-Delmestre, K.; Boechat-Roberty, H.M.; Oliveira, R.R. Multiply charged naphthalene and its C10H8 isomers: Bonding, spectroscopy, and implications in AGN environments. Mon. Not. R. Astron. Soc. 2022, 512, 4669–4682. [Google Scholar] [CrossRef]
- Schön, J.C. 3.11—Energy landscapes in inorganic chemistry. In Comprehensive Inorganic Chemistry III, 3rd ed.; Reedijk, J., Poeppelmeier, K.R., Eds.; Elsevier: Oxford, UK, 2023; pp. 262–392. [Google Scholar] [CrossRef]
- Schön, J.C. Studying the energy hypersurface of multi-minima systems—The threshold and the lid algorithm. Bunsenges. Phys. Chem. 1996, 100, 1388–1391. [Google Scholar] [CrossRef]
- Schön, J.C.; Putz, H.; Jansen, M. Studying the energy hypersurface of continuous systems—The threshold algorithm. J. Phys. Condens. Matter 1996, 8, 143–156. [Google Scholar] [CrossRef]
- Neelamraju, S.; Oligschleger, C.; Schön, J.C. The threshold algorithm: Description of the methodology and new developments. J. Chem. Phys. 2017, 147, 152713. [Google Scholar] [CrossRef]
- Porezag, D.; Frauenheim, T.; Köhler, T.; Seifert, G.; Kaschner, R. Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon. Phys. Rev. B 1995, 51, 12947–12957. [Google Scholar] [CrossRef]
- Seifert, G.; Porezag, D.; Frauenheim, T. Calculations of molecules, clusters, and solids with a simplified LCAO-DFT-LDA scheme. Int. J. Quant. Chem. 1996, 58, 185–192. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-Consistent-Charge Density-Functional Tight-Binding Method for Simulations of Complex Materials Properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Spiegelman, F.; Tarrat, N.; Cuny, J.; Dontot, L.; Posenitskiy, E.; Martí, C.; Simon, A.; Rapacioli, M. Density-functional tight-binding: Basic concepts and applications to molecules and clusters. Adv. Phys. X 2020, 5, 1710252. [Google Scholar] [CrossRef]
- Rapacioli, M.; Heine, T.; Dontot, L.; Buey, M.Y.; Tarrat, N.; Spiegelman, F.; Louisnard, F.; Cuny, J.; Morinière, M.; Dubosq, C.; et al. deMonNano Experiment. 2023. Available online: http://demon-nano.ups-tlse.fr/ (accessed on 26 July 2023).
- Rapacioli, M.; Schön, J.C.; Tarrat, N. Exploring energy landscapes at the DFTB quantum level using the threshold algorithm: The case of the anionic metal cluster . Theor. Chem. Acc. 2021, 140, 85. [Google Scholar] [CrossRef]
- Lee, J.W.L.; Stockett, M.H.; Ashworth, E.K.; Navarro Navarrete, J.E.; Gougoula, E.; Garg, D.; Ji, M.; Zhu, B.; Indrajith, S.; Zettergren, H.; et al. Cooling dynamics of energized naphthalene and azulene radical cations. J. Chem. Phys. 2023, 158, 174305. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C.; Koster, G.F. Simplified LCAO method for the periodic potential problem. Phys. Rev. 1954, 94, 1498–1524. [Google Scholar] [CrossRef]
- Rapacioli, M.; Tarrat, N. Periodic DFTB for Supported Clusters: Implementation and Application on Benzene Dimers Deposited on Graphene. Computation 2022, 10, 39. [Google Scholar] [CrossRef]
- Dontot, L.; Spiegelman, F.; Rapacioli, M. Structures and Energetics of Neutral and Cationic Pyrene Clusters. J. Phys. Chem. A 2019, 123, 9531–9543. [Google Scholar] [CrossRef] [PubMed]
- Tarrat, N.; Rapacioli, M.; Spiegelman, F. Au147 nanoparticles: Ordered or amorphous? J. Chem. Phys. 2018, 148, 204308. [Google Scholar] [CrossRef] [PubMed]
- Rapacioli, M.; Cazaux, S.; Foley, N.; Simon, A.; Hoekstra, R.; Schlathölter, T. Atomic hydrogen interactions with gas-phase coronene cations: Hydrogenation versus fragmentation. Phys. Chem. Chem. Phys. 2018, 20, 22427–22438. [Google Scholar] [CrossRef]
- Wevers, M.A.C.; Schön, J.C.; Jansen, M. Characteristic regions on the energy landscape of MgF2. J. Phys. A Math. Gen. 2001, 34, 4041. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Weber, T.A. Inherent structure in water. J. Phys. Chem. 1983, 87, 2833–2840. [Google Scholar] [CrossRef]
- Joswig, J.O.; Lorenz, T. Detecting and Quantifying Geometric Features in Large Series of Cluster Structures. Z. Phys. Chem. 2016, 230, 1057–1066. [Google Scholar] [CrossRef]
- Wevers, M.A.C.; Schön, J.C.; Jansen, M. Global aspects of the energy landscape of metastable crystal structures in ionic compounds. J. Phys. Condens. Matter 1999, 11, 6487. [Google Scholar] [CrossRef]
- Hoffmann, K.H.; Schön, J.C. Controlled dynamics on energy landscapes. Eur. Phys. J. B 2013, 86, 220. [Google Scholar] [CrossRef]
- Hoffmann, K.H.; Schön, J.C. Combining pressure and temperature control in dynamics on energy landscapes. Eur. Phys. J. B 2017, 90, 84. [Google Scholar] [CrossRef][Green Version]
- Hoffmann, K.H.; Fischer, A.; Schön, J.C. Chapter 10—Controlled dynamics and preferential trapping on energy landscapes. In Energy Landscapes of Nanoscale Systems; Wales, D.J., Ed.; Frontiers of Nanoscience; Elsevier: Amsterdam, The Netherlands, 2022; Volume 21, pp. 211–245. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomon, G.; Tarrat, N.; Schön, J.C.; Rapacioli, M. Low-Energy Transformation Pathways between Naphthalene Isomers. Molecules 2023, 28, 5778. https://doi.org/10.3390/molecules28155778
Salomon G, Tarrat N, Schön JC, Rapacioli M. Low-Energy Transformation Pathways between Naphthalene Isomers. Molecules. 2023; 28(15):5778. https://doi.org/10.3390/molecules28155778
Chicago/Turabian StyleSalomon, Grégoire, Nathalie Tarrat, J. Christian Schön, and Mathias Rapacioli. 2023. "Low-Energy Transformation Pathways between Naphthalene Isomers" Molecules 28, no. 15: 5778. https://doi.org/10.3390/molecules28155778
APA StyleSalomon, G., Tarrat, N., Schön, J. C., & Rapacioli, M. (2023). Low-Energy Transformation Pathways between Naphthalene Isomers. Molecules, 28(15), 5778. https://doi.org/10.3390/molecules28155778