
Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals





Abstract
1. Reducing Carbon Dioxide from the Air: The Challenge
2. Carbonic Anhydrases: Efficient Devices for CO2 Uptake
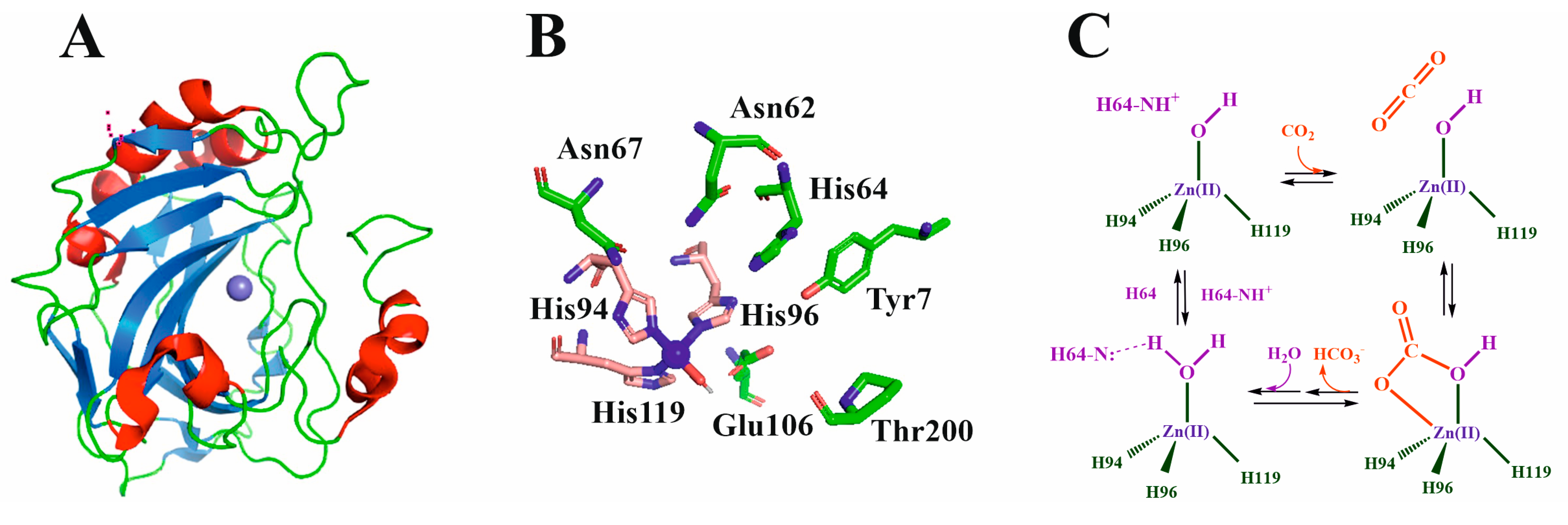
2.1. Classification and Structure of Carbonic Anhydrases
2.2. CA Mechanism of Action
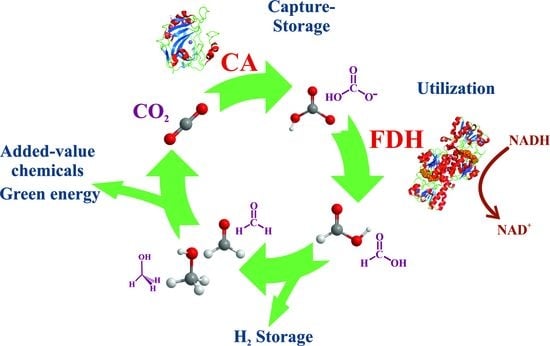
3. Formate Dehydrogenases: Natural Machines for Reducing CO2
3.1. Metal-Independent/NAD-Dependent FDHs
3.2. Metal-Dependent FDHs
4. Improving CA Performance: Enzyme Immobilization
4.1. Physical Adsorption
4.2. Entrapment and Encapsulation
4.3. Covalent Binding and Crosslinking
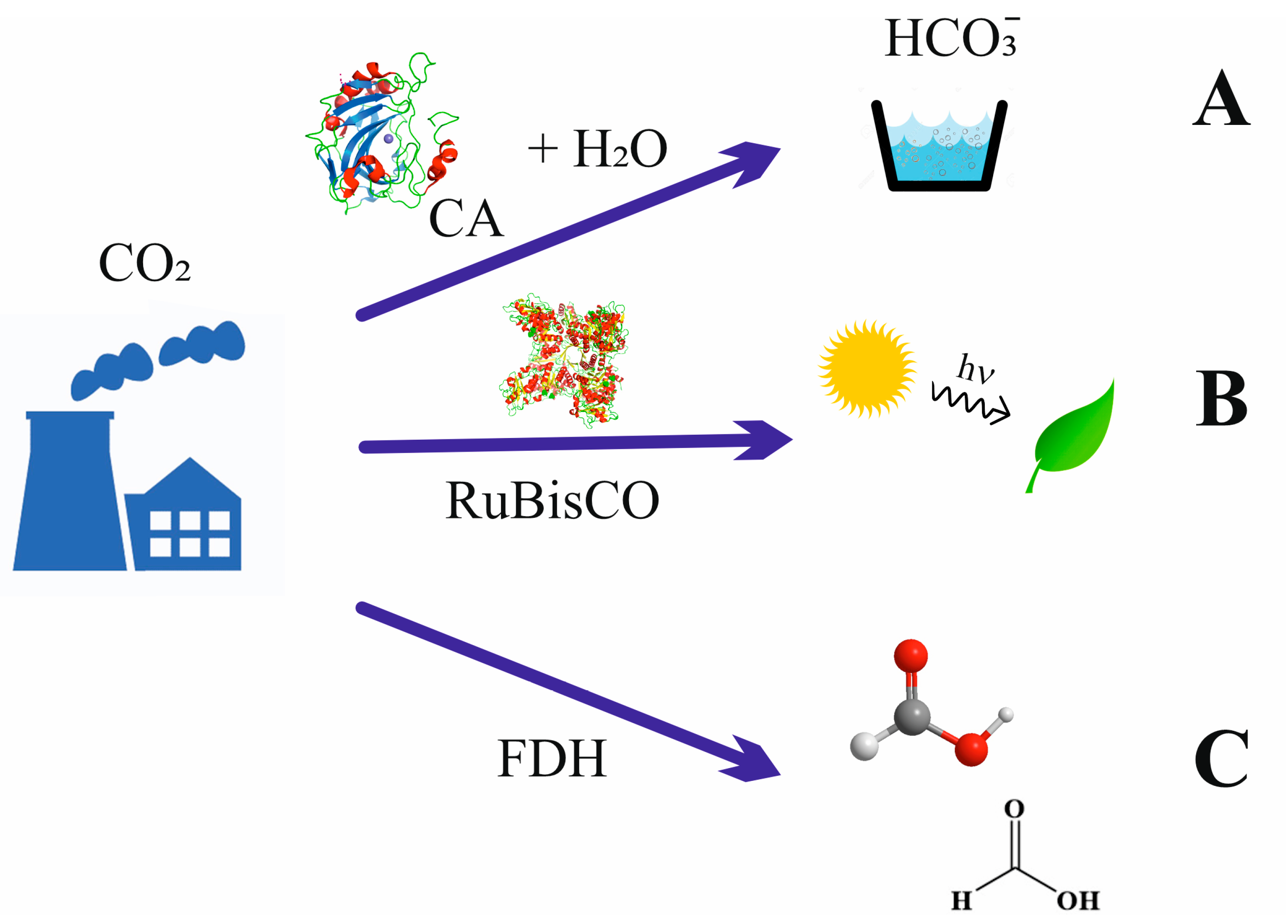
5. Carbon Capture Storage and Utilization: State-of-the-Art, Costs, and Perspectives
6. Carbonic Anhydrase in Carbon Capture Storage
6.1. Chemical Absorption
6.2. Chemical Carbonation
6.3. Mineralization
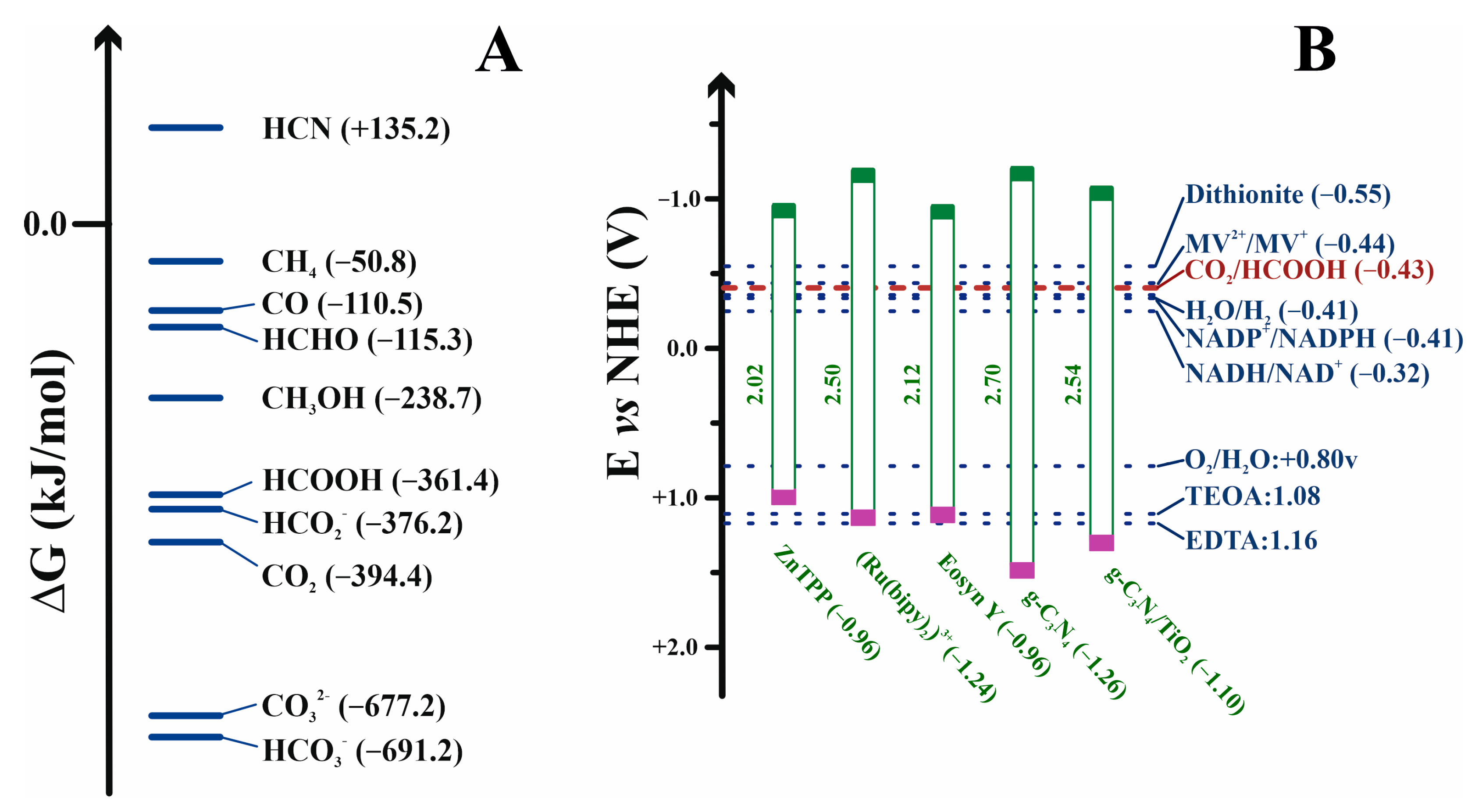
7. Biotechnological Aspects of CO2 Reduction
7.1. Hindrances to Biochemically Reducing CO2
7.2. Coupled reactions: Enzymatic Multicascades

7.3. Electrochemical Regeneration of NADH Cofactor
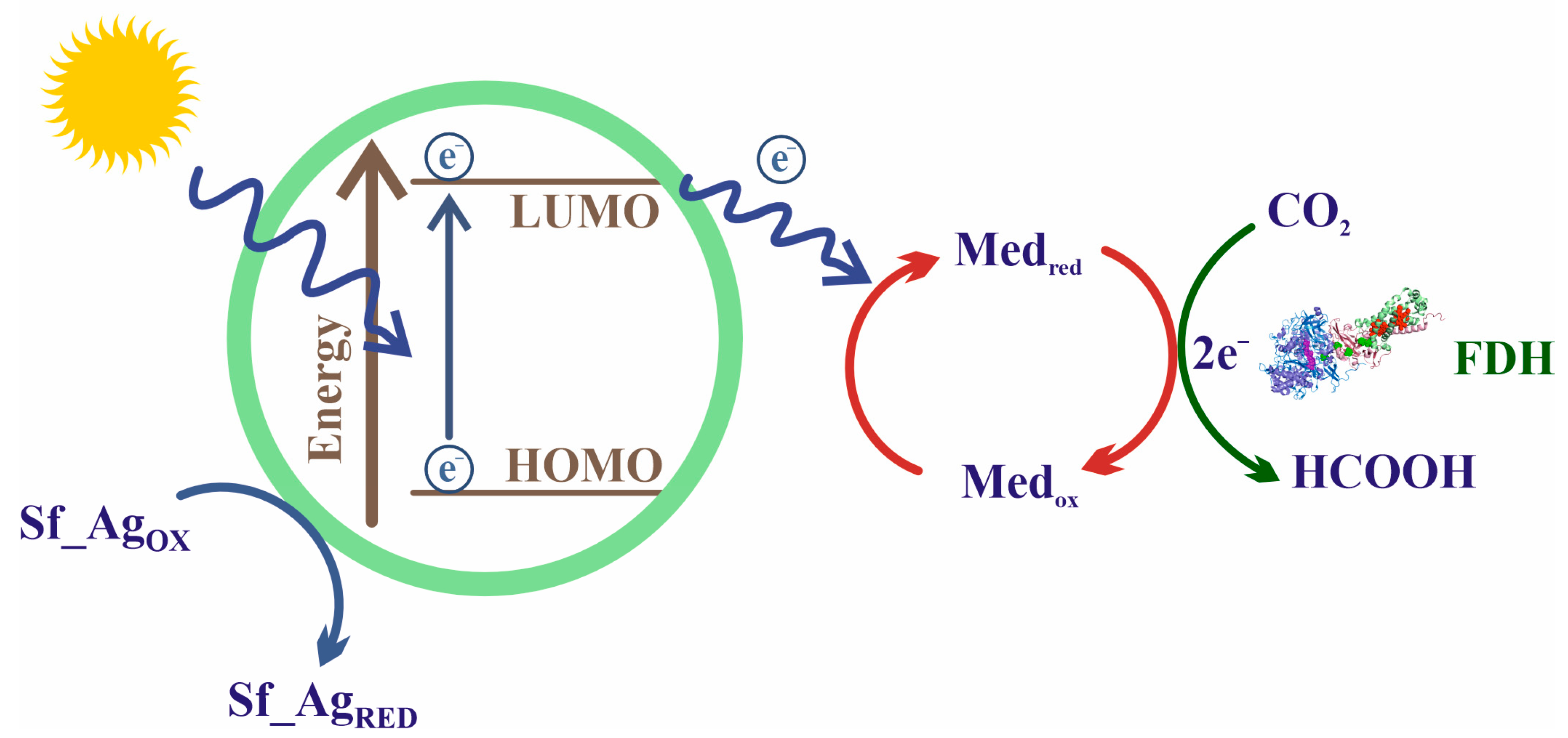
7.4. Photochemical NADH Regeneration
7.5. CO2 Reduction by Whole-Cell Bacteria
8. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- National Center for Environmental Information. Annual 2021 Global Climate Report. Available online: https://www.ncei.noaa.gov/access/monitoring/monthly-report/global/202113 (accessed on 24 February 2023).
- Intergovernmental Panel on Climate Change (IPCC). Climate Change 2022, Mitigation of Climate Change. Available online: https://www.ipcc.ch/report/ar6/wg3/downloads/report/IPCC_AR6_WGIII_FullReport.pdf (accessed on 24 February 2023).
- Fellowes, T.E.; Anggadi, F.; Byrne, M.; Vila-Concejo, A.; Bruce, E.; Baker, E. Stability of Coral Reef Islands and Associated Legal Maritime Zones in a Changing Ocean. Environ. Res. Lett. 2022, 17, 093003. [Google Scholar] [CrossRef]
- Borges, F.O.; Sampaio, E.; Santos, C.P.; Rosa, R. Impacts of Low Oxygen on Marine Life: Neglected, but a Crucial Priority for Research. Biol. Bull. 2022, 243, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Arrhenius, S.; Holden, E.S. On the Influence of Carbonic Acid in the Air Upon the Temperature of the Earth. Publ. Astron. Soc. Pac. 1897, 9, 14–24. [Google Scholar] [CrossRef]
- Arrhenius, S. The Celestial Bodies, In Particular the Earth, as Abodes of Living Beings. In Worlds in the Making: The Evolution of the Universe; Harper: New York, NY, USA, 1908; pp. 39–63. [Google Scholar]
- Friedlingstein, P.; Andrew, R.M.; Rogelj, J.; Peters, G.P.; Canadell, J.G.; Knutti, R.; Luderer, G.; Raupach, M.R.; Schaeffer, M.; van Vuuren, D.P.; et al. Persistent Growth of CO2 Emissions and Implications for Reaching Climate Targets. Nat. Geosci. 2014, 7, 709–715. [Google Scholar] [CrossRef]
- Lindsey, R. Climate Change: Atmospheric Carbon Dioxide. Science and Information for a Climate-Smart Nation. Available online: https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide (accessed on 24 February 2023).
- IPCC. Climate Change 2007: Synthesis Report. Summary for Policymakers. Valencia (Spain) 2007. Available online: https://www.ctc-n.org/resources/ipcc-fourth-assessment-report-climate-change-2007-synthesis-report-summary-policymakers (accessed on 24 February 2023).
- EPA. United States Environmental Protection Agency. Overview of Greenhouse Gases. Available online: https://www.epa.gov/ghgemissions/overview-greenhouse-gases (accessed on 24 February 2023).
- Hoegh-Guldberg, O.; Jacob, D.; Taylor, M.; Bolanos, T.G.; Bindi, M.; Brown, S.; Camilloni, I.A.; Diedhiou, A.; Djalante, R.; Ebi, K.; et al. The Human Imperative of Stabilizing Global Climate Change at 1.5 Degrees C. Science 2019, 365, eaaw6974. [Google Scholar] [CrossRef]
- Rajabloo, T.; Valee, J.; Marenne, Y.; Coppens, L.; De Ceuninck, W. Carbon Capture and Utilization for Industrial Applications. Energy Rep. 2023, 9, 111–116. [Google Scholar] [CrossRef]
- Kamkeng, A.D.N.; Wang, M.; Hu, J.; Du, W.; Qian, F. Transformation technologies for CO2 utilisation: Current status, challenges and future prospects. Chem. Eng. J. 2021, 409, 128138. [Google Scholar] [CrossRef]
- Villa, R.; Porcar, R.; Nieto, S.; Donaire, A.; García-Verdugo, E.; Luis, S.V.; Lozano-Rodríguez, P. Sustainable chemo-enzymatic synthesis of glycerol carbonate (meth)acrylate from glycidol and carbon dioxide enabled by ionic liquid technologies. Green Chem. 2021, 11, 4191–4200. [Google Scholar] [CrossRef]
- Knoche, W. Chemical Reactions of CO2 in Water. In Biophysics and Physiology of Carbon Dioxide, Proceedings in Life Sciences; Bauer, C., Gros, G., Bartels, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1980. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshow, A. Chemistry of the Elements, 2nd ed.; Butterworth-Heinemann, Elsevier: Oxford, UK, 1997. [Google Scholar] [CrossRef]
- Talekar, S.; Jo, B.H.; Dordick, J.S.; Kim, J. Carbonic Anhydrase for CO2 Capture, Conversion and Utilization. Curr. Opin. Biotechnol. 2022, 74, 230–240. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and Mechanism of Carbonic Anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- De Oliveira Maciel, A.; Christakopoulos, P.; Rova, U.; Antonopoulou, I. Carbonic Anhydrase to Boost CO2 Sequestration: Improving Carbon Capture Utilization and Storage (CCUS). Chemosphere 2022, 299, 134419. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Walde, P. Immobilized Carbonic Anhydrase: Preparation, Characteristics and Biotechnological Applications. World J. Microbiol. Biotechnol. 2018, 34, 151. [Google Scholar] [CrossRef]
- Appel, A.M.; Bercaw, J.E.; Bocarsly, A.B.; Dobbek, H.; DuBois, D.L.; Dupuis, M.; Ferry, J.G.; Fujita, E.; Hille, R.; Kenis, P.J.A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113, 6621–6658. [Google Scholar] [CrossRef]
- Bierbaumer, S.; Nattermann, M.; Schulz, L.; Zschoche, R.; Erb, T.J.; Winkler, C.K.; Tinzl, M.; Glueck, S.M. Enzymatic Conversion of CO2: From Natural to Artificial Utilization. Chem. Rev. 2023, 123, 5702–5754. [Google Scholar] [CrossRef]
- Badger, M.R.; Sharwood, R.E. Rubisco, the Imperfect Winner: It’s All about the Base. J. Exp. Bot. 2023, 74, 562–580. [Google Scholar] [CrossRef]
- Bauwe, H. Photorespiration—Rubisco’s Repair Crew. J. Plant. Physiol. 2023, 280, 153899. [Google Scholar] [CrossRef]
- Alpdagtas, S.; Binay, B. Nadp+-Dependent Formate Dehydrogenase: A Review. Biocatal. Biotransform. 2021, 39, 260–268. [Google Scholar] [CrossRef]
- Alekseeva, A.A.; Savin, S.S.; Tishkov, V.I. NAD+-Dependent Formate Dehydrogenase from Plants. Acta Nat. 2011, 3, 38–54. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and Tungsten-Containing Formate Dehydrogenases: Aiming to Inspire a Catalyst for Carbon Dioxide Utilization. Inorganica Chim. Acta 2017, 455, 350–363. [Google Scholar] [CrossRef]
- Jormakka, M.; Byrne, B.; Iwata, S. Formate Dehydrogenase—A Versatile Enzyme in Changing Environments. Curr. Opin. Struct. Biol. 2003, 13, 418–423. [Google Scholar] [CrossRef]
- Yang, J.Y.; Kerr, T.A.; Wang, X.S.; Barlow, J.M. Reducing CO2 to HCO2- at Mild Potentials: Lessons from Formate Dehydrogenase. J. Am. Chem. Soc. 2020, 142, 19438–19445. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Zhao, Z.; Liu, W. Effect of Carbonic Anhydrase on Enzymatic Conversion of CO2 to Formic Acid and Optimization of Reaction Conditions. J. Mol. Catal. B Enzym. 2015, 116, 89–94. [Google Scholar] [CrossRef]
- Thauer, R.K.; Jungermann, K.; Decker, K. Energy-Conservation in Chemotropic Anaerobic Bacteria. Bacteriol. Rev. 1977, 41, 100–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Li, R.; Liu, J. Bioinspired NADH Regeneration Based on Conjugated Photocatalytic Systems. Solar RRL 2021, 5, 2000339. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, D.S.; Kuk, S.K.; Park, C.B. Photobiocatalysis: Activating Redox Enzymes by Direct or Indirect Transfer of Photoinduced Electrons. Angew. Chem. Int. Ed. 2018, 57, 7958–7985. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metabolites 2017, 7, 56. [Google Scholar] [CrossRef]
- DiMario, R.J.; Machingura, M.C.; Waldrop, G.L.; Moroney, J.V. The Many Types of Carbonic Anhydrases in Photosynthetic Organisms. Plant Sci. 2018, 268, 11–17. [Google Scholar] [CrossRef]
- Kupriyanova, E.; Pronina, N.; Los, D. Carbonic Anhydrase—A Universal Enzyme of the Carbon-Based Life. Photosynthetica 2017, 55, 3–19. [Google Scholar] [CrossRef]
- Zimmerman, S.A.; Ferry, J.G. The Beta and Gamma Classes of Carbonic Anhydrase. Curr. Pharm. Des. 2008, 14, 716–721. [Google Scholar] [CrossRef]
- McGinn, P.J.; Morel, F.M.M. Expression and Regulation of Carbonic Anhydrases in the Marine Diatom Thalassiosira pseudonana and in Natural Phytoplankton Assemblages from Great Bay, New Jersey. Physiol. Plant. 2008, 133, 78–91. [Google Scholar] [CrossRef]
- Roberts, S.B.; Lane, T.W.; Morel, F.M.M. Carbonic Anhydrase in the Marine Diatom Thalassiosira weissflogii (Bacillariophyceae). J. Phycol. 1997, 33, 845–850. [Google Scholar] [CrossRef]
- Beauchemin, M.; Morse, D. δ-Carbonic Anhydrases: Structure, Distribution, and Potential Roles. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Université de Montréal: Montréal, QC, Canada, 2015; pp. 337–349. [Google Scholar] [CrossRef]
- Dou, Z.; Heinhorst, S.; Williams, E.B.; Murin, C.D.; Shively, J.M.; Cannon, G.C. CO2 Fixation Kinetics of Halothiobacillus neapolitanus Mutant Carboxysomes Lacking Carbonic Anhydrase Suggest the Shell Acts as a Diffusional Barrier for CO2. J. Biol. Chem. 2008, 283, 10377–10384. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; Buonanno, M.; Esposito, D.; Nocentini, A.; Berrino, E.; Bua, S.; Polentarutti, M.; Supuran, C.T.; Monti, S.M.; et al. Zeta-Carbonic Anhydrases Show CS2 Hydrolase Activity: A New Metabolic Carbon Acquisition Pathway in Diatoms? Comput. Struct. Biotechnol. J. 2021, 19, 3427–3436. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; Fisher, G.M.; Andrews, K.T.; Poulsen, S.-A.; Capasso, C.; Supuran, C.T. Discovery of a New Family of Carbonic Anhydrases in the Malaria Pathogen Plasmodium falciparum—The η-Carbonic Anhydrases. Bioorg. Med. Chem. Lett. 2014, 24, 4389–4396. [Google Scholar] [CrossRef]
- Nawaly, H.; Tanaka, A.; Toyoshima, Y.; Tsuji, Y.; Matsuda, Y. Localization and Characterization Theta Carbonic Anhydrases in Thalassiosira pseudonana. Photosynth. Res. 2023, 156, 217–229. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T.; Capasso, C. An Overview on the Recently Discovered Iota-Carbonic Anhydrases. J. Enzym. Inhib. Med. Chem. 2021, 36, 1988–1995. [Google Scholar] [CrossRef]
- Del Prete, S.; Nocentini, A.; Supuran, C.T.; Capasso, C. Bacterial Iota-Carbonic Anhydrase: A New Active Class of Carbonic Anhydrase Identified in the Genome of the Gram-Negative Bacterium Burkholderia Territorii. J. Enzym. Inhib. Med. Chem. 2020, 35, 1060–1068. [Google Scholar] [CrossRef]
- Kikutani, S.; Nakajima, K.; Nagasato, C.; Tsuji, Y.; Miyatake, A.; Matsuda, Y. Thylakoid Luminal Theta-Carbonic Anhydrase Critical for Growth and Photosynthesis in the Marine Diatom Phaeodactylum Tricornutum. Proc. Natl. Acad. Sci. USA 2016, 113, 9828–9833. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; De Simone, G.; Monti, S.M. Cadmium-Containing Carbonic Anhydrase CDCA1 in Marine Diatom Thalassiosira weissflogii. Mar. Drugs 2015, 13, 1688–1697. [Google Scholar] [CrossRef]
- Angeli, A.; Buonanno, M.; Donald, W.A.; Monti, S.M.; Supuran, C.T. The Zinc–but Not Cadmium–Containing ζ-Carbonic from the Diatom Thalassiosira weissflogii Is Potently Activated by Amines and Amino Acids. Bioorg. Chem. 2018, 80, 261–265. [Google Scholar] [CrossRef]
- Nathan, V.K.; Ammini, P. Carbon Dioxide Sequestering Ability of Bacterial Carbonic Anhydrase in a Mangrove Soil Microcosm and Its Bio-Mineralization Properties. Water Air Soil Pollut. 2019, 230, 192. [Google Scholar] [CrossRef]
- Banci, L.; Bertini, I.; Luchinat, C.; Donaire, A.; Martinez, M.-J. The Factors Governing the Coordination Number in the Anion Derivatives of Carbonic Anhydrase. Comments Inorg. Chem. 1990, 9, 245–261. [Google Scholar] [CrossRef]
- Hirakawa, Y.; Senda, M.; Fukuda, K.; Yu, H.Y.; Ishida, M.; Taira, M.; Kinbara, K.; Senda, T. Characterization of a Novel Type of Carbonic Anhydrase That Acts without Metal Cofactors. BMC Biol. 2021, 19, 105. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.H.; Kim, I.G.; Seo, J.H.; Kang, D.G.; Cha, H.J. Engineered Escherichia coli with Periplasmic Carbonic Anhydrase as a Biocatalyst for CO2 Sequestration. Appl. Environ. Microbiol. 2013, 79, 6697–6705. [Google Scholar] [CrossRef]
- Ekinci, D.; Beydemir, S.; Alim, Z. Some Drugs Inhibit in vitro Hydratase and Esterase Activities of Human Carbonic Anhydrase-I and II. Pharmacol. Rep. 2007, 59, 580–587. [Google Scholar] [PubMed]
- Steger, F.; Reich, J.; Fuchs, W.; Rittmann, S.K.-M.R.; Gübitz, G.M.; Ribitsch, D.; Bochmann, G. Comparison of Carbonic Anhydrases for CO2 Sequestration. Int. J. Mol. Sci. 2022, 23, 957. [Google Scholar] [CrossRef]
- Faridi, S.; Satyanarayana, T. Novel Alkalistable α-Carbonic Anhydrase from the Polyextremophilic Bacterium Bacillus Halodurans: Characteristics and Applicability in Flue Gas CO2 Sequestration. Environ. Sci. Pollut. Res. 2016, 23, 15236–15249. [Google Scholar] [CrossRef]
- Capasso, C.; De Luca, V.; Carginale, V.; Cannio, R.; Rossi, M. Biochemical Properties of a Novel and Highly Thermostable Bacterial Alpha-Carbonic Anhydrase from Sulfurihydrogenibium yellowstonense YO3AOP1. J. Enzym. Inhib. Med. Chem. 2012, 27, 892–897. [Google Scholar] [CrossRef]
- Ramanan, R.; Kannan, K.; Vinayagamoorthy, N.; Ramkumar, K.M.; Sivanesan, S.D.; Chakrabarti, T. Purification and Characterization of a Novel Plant-Type Carbonic Anhydrase from Bacillus subtilis. Biotechnol. Bioprocess Eng. 2009, 14, 32–37. [Google Scholar] [CrossRef]
- Kim, S.S.; Kim, N.J.; Hong, S.; Kim, S.S.; Sung, J.; Jin, M.S. The Structural Basis of the Low Catalytic Activities of the Two Minor Beta-Carbonic Anhydrases of the Filamentous Fungus Aspergillus fumigatus. J. Struct. Biol. 2019, 208, 61–68. [Google Scholar] [CrossRef]
- Sridharan, U.; Ragunathan, P.; Kuramitsu, S.; Yokoyama, S.; Kumarevel, T.; Ponnuraj, K. Structural and Functional Characterization of a Putative Carbonic Anhydrase from Geobacillus kaustophilus Reveals Its Cambialistic Function. Biochem. Biophys. Res. Commun. 2021, 547, 96–101. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, Y.; Wang, L.; Jing, Q.; Wang, X.; Xi, X.; Zhao, X.; Wang, H. Molecular Structure of Thermostable and Zinc-Ion-Binding Gamma-Class Carbonic Anhydrases. Biometals 2019, 32, 317–328. [Google Scholar] [CrossRef]
- Alber, B.E.; Ferry, J.G. A Carbonic Anhydrase from the Archaeon Methanosarcina thermophila. Proc. Natl. Acad. Sci. USA 1994, 91, 6909–6913. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; Di Fonzo, P.; Osman, S.M.; AlOthman, Z.; Donald, W.A.; Supuran, C.T.; Capasso, C. Sulfonamide Inhibition Profile of the Gamma-Carbonic Anhydrase Identified in the Genome of the Pathogenic Bacterium Burkholderia pseudomallei the Etiological Agent Responsible of Melioidosis. Bioorg. Med. Chem. Lett. 2017, 27, 490–495. [Google Scholar] [CrossRef]
- Del Prete, S.; Vullo, D.; De Luca, V.; Supuran, C.T.; Capasso, C. Biochemical Characterization of the Delta-Carbonic Anhydrase from the Marine Diatom Thalassiosira weissflogii, TweCA. J. Enzym. Inhib. Med. Chem. 2014, 29, 906–911. [Google Scholar] [CrossRef]
- Zhang, H.; Blanco-Ameijeiras, S.; Hopkinson, B.M.; Bernasconi, S.M.; Mejia, L.M.; Liu, C.; Stoll, H. An Isotope Label Method for Empirical Detection of Carbonic Anhydrase in the Calcification Pathway of the Coccolithophore Emiliania huxleyi. Geochim. Cosmochim. Acta 2021, 292, 78–93. [Google Scholar] [CrossRef]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M.M. Structure and Metal Exchange in the Cadmium Carbonic Anhydrase of Marine Diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. The η-Class Carbonic Anhydrases as Drug Targets for Antimalarial Agents. Expert Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef]
- Kitahara, M.; Fudo, S.; Yoneda, T.; Nukaga, M.; Hoshino, T. Anisotropic Distribution of Ammonium Sulfate Ions in Protein Crystallization. Cryst. Growth Des. 2019, 19, 6004–6010. [Google Scholar] [CrossRef]
- Woods, L.A.; Dolezal, O.; Ren, B.; Ryan, J.H.; Peat, T.S.; Poulsen, S.-A. Native State Mass Spectrometry, Surface Plasmon Resonance, and X-Ray Crystallography Correlate Strongly as a Fragment Screening Combination. J. Med. Chem. 2016, 59, 2192–2204. [Google Scholar] [CrossRef]
- Avvaru, B.S.; Busby, S.A.; Chalmers, M.J.; Griffin, P.R.; Venkatakrishnan, B.; Agbandje-McKenna, M.; Silverman, D.N.; McKenna, R. Apo-Human Carbonic Anhydrase II Revisited: Implications of the Loss of a Metal in Protein Structure, Stability, and Solvent Network. Biochemistry 2009, 48, 7365–7372. [Google Scholar] [CrossRef] [PubMed]
- Bergenhem, N.C.; Hallberg, M.; Wisén, S. Molecular Characterization of the Human Carbonic Anhydrase-Related Protein (HCA-RP VIII). Biochim. Biophys. Acta 1998, 1384, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.S.; Muniz, J.R.; Kramm, A.; Pilka, E.S.; Kochan, G.; Oppermann, U.; Yue, W.W. Crystal Structure of Human Carbonic Anhydrase-Related Protein VIII Reveals the Basis for Catalytic Silencing. Proteins 2009, 76, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Lakkis, M.M.; Bergenhem, N.C.; O’Shea, K.S.; Tashian, R.E. Expression of the Acatalytic Carbonic Anhydrase VIII Gene, Car8, during Mouse Embryonic Development. Histochem. J. 1997, 29, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Hirota, J.; Ando, H.; Hamada, K.; Mikoshiba, K. Carbonic Anhydrase-Related Protein is a Novel Binding Protein for Inositol 1,4,5-Trisphosphate Receptor Ttype 1. Biochem. J. 2003, 372, 435–441. [Google Scholar] [CrossRef]
- Borén, K.; Andersson, P.; Larsson, M.; Carlsson, U. Characterization of a Molten Globule State of Bovine Carbonic Anhydrase III: Loss of Asymmetrical Environment of the Aromatic Residues Has a Profound Effect on Both the Near- and Far-UV CD Spectrum. Biochim. Biophys. Acta 1999, 1430, 111–118. [Google Scholar] [CrossRef]
- Capasso, C.; Supuran, C.T. Inhibition of Bacterial Carbonic Anhydrases as a Novel Approach to Escape Drug Resistance. Curr. Top. Med. Chem. 2017, 17, 1237–1248. [Google Scholar] [CrossRef]
- Wilbur, K.M.; Anderson, N.G. Electrometric and Colorimetric Determination of Carbonic Anhydrase. J. Biol. Chem. 1948, 176, 147–154. [Google Scholar] [CrossRef]
- Fuchs, W.; Steger, F.; Reich, J.; Ribitsch, D.; Rittmann, S.K.-M.R.; Bochmann, G. A Simple and Straightforward Method for Activity Measurement of Carbonic Anhydrases. Catalysis 2021, 11, 819. [Google Scholar] [CrossRef]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase Activities of Human Carbonic Anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar] [CrossRef]
- Effendi, S.S.W.; Ng, I.-S. The Prospective and Potential of Carbonic Anhydrase for Carbon Dioxide Sequestration: A Critical Review. Process Biochem. 2019, 87, 55–65. [Google Scholar] [CrossRef]
- Supuran, C.T.; De Simone, G. Carbonic Anhydrases: An Overview. In Carbonic Anhydrases as Biocatalysts: From Theory to Medical and Industrial Applications; Elsevier: Florence, Italy, 2015; pp. 3–13. [Google Scholar] [CrossRef]
- Niks, D.; Hille, R. Reductive Activation of CO2 by Formate Dehydrogenases. In Enzymes of Energy Technology; Methods in Enzymology; Armstrong, F., Ed.; Elsevier Acad. Press. Inc.: San Diego, CA, USA, 2018; Volume 613, pp. 277–295. [Google Scholar] [CrossRef]
- Nielsen, C.F.; Lange, L.; Meyer, A.S. Classification and Enzyme Kinetics of Formate Dehydrogenases for Biomanufacturing via CO2 Utilization. Biotechnol. Adv. 2019, 37, 107408. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, I.; Moura, J.J.G. Molybdenum and Tungsten-Containing Enzymes: An Overview. In Molibdenum and Tungsten Enzymes: Biochemistry; Hille, R., Schulzke, C., Kirk, M.L., Eds.; RSC Metallobiology Series; Royal Society of Chemistry: Cambridge, UK, 2017; Volume 5, pp. 1–80. [Google Scholar]
- Lemaire, O.N.; Jespersen, M.; Wagner, T. CO2-Fixation Strategies in Energy Extremophiles: What Can We Learn From Acetogens? Front. Microbiol. 2020, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.; Park, G.W.; Lee, J.; Lee, J.-S.; Min, K. Recent Progress in Formate Dehydrogenase (FDH) as a Non-Photosynthetic CO2 Utilizing Enzyme: A Short Review. J. CO2 Util. 2020, 42, 101353. [Google Scholar] [CrossRef]
- Alpdağtaş, S.; Turunen, O.; Valjakka, J.; Binay, B. The Challenges of Using NAD+-Dependent Formate Dehydrogenases for CO2 Conversion. Crit. Rev. Biotechnol. 2022, 42, 953–972. [Google Scholar] [CrossRef]
- Sakai, Y.; Murdanoto, A.P.; Konishi, T.; Iwamatsu, A.; Kato, N. Regulation of the Formate Dehydrogenase Gene, FDH1, in the Methylotrophic Yeast Candida boidinii and Growth Characteristics of an FDH1-Disrupted Strain on Methanol, Methylamine, and Choline. J. Bacteriol. 1997, 179, 4480–4485. [Google Scholar] [CrossRef]
- Tishkov, V.I.; Popov, V.O. Protein Engineering of Formate Dehydrogenase. Biomol. Eng. 2006, 23, 89–110. [Google Scholar] [CrossRef]
- Pagano, P.; Guo, Q.; Ranasinghe, C.; Schroeder, E.; Robben, K.; Hase, F.; Ye, H.; Wickersham, K.; Aspuru-Guzik, A.; Major, D.T.; et al. Oscillatory Active-Site Motions Correlate with Kinetic Isotope Effects in Formate Dehydrogenase. ACS Catal. 2019, 9, 11199–11206. [Google Scholar] [CrossRef]
- Yilmazer, B.; Isupov, M.N.; De Rose, S.A.; Bulut, H.; Benninghoff, J.C.; Binay, B.; Littlechild, J.A. Structural insights into the NAD+-dependent Formate Dehydrogenase Mechanism Revealed from the NADH Complex and the Formate NAD+-Ternary Complex of the Chaetomium thermophilum Enzyme J. Struct. Biol. 2020, 212, 107657. [Google Scholar] [CrossRef]
- Pala, U.; Yelmazer, B.; Corbacioglu, M.; Ruupunen, J.; Valjakka, J.; Turunen, O.; Binay, B. Functional Effects of Active Site Mutations in NAD+-Dependent Formate Dehydrogenases on Transformation of Hydrogen Carbonate to Formate. Protein Eng. Des. Sel. 2018, 31, 327–335. [Google Scholar] [CrossRef]
- Cakar, M.M.; Ruupunen, J.; Mangas-Sanchez, J.; Birmingham, W.R.; Yildirim, D.; Turunen, O.; Turner, N.J.; Valjakka, J.; Binay, B. Engineered Formate Dehydrogenase from Chaetomium Thermophilum, a Promising Enzymatic Solution for Biotechnical CO2 Fixation. Biotechnol. Lett. 2020, 42, 2251–2262. [Google Scholar] [CrossRef]
- Guo, Q.; Gakhar, L.; Wickersham, K.; Francis, K.; Vardi-Kilshtain, A.; Major, D.T.; Cheatum, C.M.; Kohen, A. Structural and Kinetic Studies of Formate Dehydrogenase from Candida boidinii. Biochemistry 2016, 55, 2760–2771. [Google Scholar] [CrossRef]
- Sato, R.; Amao, Y. Studies on the Catalytic Mechanism of Formate Dehydrogenase from Candida boidinii Using Isotope-Labelled Substrate and Co-Enzyme. Catal. Today 2023, 411–412, 113796. [Google Scholar] [CrossRef]
- Jiang, W.; Lin, P.; Yang, R.; Fang, B. Identification of Catalysis, Substrate, and Coenzyme Binding Sites and Improvement Catalytic Efficiency of Formate Dehydrogenase from Candida boidinii. Appl. Microbiol. Biotechnol. 2016, 100, 8425–8437. [Google Scholar] [CrossRef]
- Altas, N.; Aslan, A.S.; Karatas, E.; Chronopoulou, E.; Labrou, N.E.; Binay, B. Heterologous Production of Extreme Alkaline Thermostable NAD+-Dependent Formate Dehydrogenase with Wide-Range pH Activity from Myceliophthora thermophila. Process Biochem. 2017, 61, 110–118. [Google Scholar] [CrossRef]
- Choe, H.; Joo, J.C.; Cho, D.H.; Kim, M.H.; Lee, S.H.; Jung, K.D.; Kim, Y.H. Efficient CO2-Reducing Activity of NAD-Dependent Formate Dehydrogenase from Thiobacillus sp. KNK65MA for Formate Production from CO2 Gas. PLoS ONE 2014, 9, e103111. [Google Scholar] [CrossRef]
- Aslan, A.S.; Valjakka, J.; Ruupunen, J.; Yildirim, D.; Turner, N.J.; Turunen, O.; Binay, B. Chaetomium thermophilum Formate Dehydrogenase Has High Activity in the Reduction of Hydrogen Carbonate (HCO3−) to Formate. Protein Eng. Des. Sel. 2017, 30, 47–55. [Google Scholar] [CrossRef]
- de Bok, F.A.M.; Hagedoorn, P.L.; Silva, P.J.; Hagen, W.R.; Schiltz, E.; Fritsche, K.; Stams, A.J.M. Two W-Containing Formate Dehydrogenases (CO2-Reductases) Involved in Syntrophic Propionate Oxidation by Syntrophobacter fumaroxidans. Eur. J. Biochem. 2003, 270, 2476–2485. [Google Scholar] [CrossRef]
- Maia, L.B.; Fonseca, L.; Moura, I.; Moura, J.J.G. Reduction of Carbon Dioxide by a Molybdenum-Containing Formate Dehydrogenase: A Kinetic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 8834–8846. [Google Scholar] [CrossRef]
- Bassegoda, A.; Madden, C.; Wakerley, D.W.; Reisner, E.; Hirst, J. Reversible Interconversion of CO2 and Formate by a Molybdenum-Containing Formate Dehydrogenase. J. Am. Chem. Soc. 2014, 136, 15473–15476. [Google Scholar] [CrossRef]
- Axley, M.J.; Grahame, D.A.; Stadtman, T.C. Escherichia coli Formate-Hydrogen Lyase. Purification and Properties of the Selenium-Dependent Formate Dehydrogenase Component. J. Biol. Chem. 1990, 265, 18213–18218. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.R.; Mota, C.; Mourato, C.; Domingos, R.M.; Santos, M.F.A.; Gesto, D.; Guigliarelli, B.; Santos-Silva, T.; Romao, M.J.; Cardoso Pereira, I.A. Toward the Mechanistic Understanding of Enzymatic CO2 Reduction. ACS Catal. 2020, 10, 3844–3856. [Google Scholar] [CrossRef]
- da Silva, S.M.; Pimentel, C.; Valente, F.M.A.; Rodrigues-Pousada, C.; Pereira, I.A.C. Tungsten and Molybdenum Regulation of Formate Dehydrogenase Expression in Desulfovibrio Vulgaris Hildenborough. J. Bacteriol. 2011, 193, 2909–2916. [Google Scholar] [CrossRef] [PubMed]
- Schuchmann, K.; Mueller, V. Direct and Reversible Hydrogenation of CO2 to Formate by a Bacterial Carbon Dioxide Reductase. Science 2013, 342, 1382–1385. [Google Scholar] [CrossRef]
- Yu, X.; Niks, D.; Mulchandani, A.; Hille, R. Efficient Reduction of CO2 by the Molybdenum-Containing Formate Dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J. Biol. Chem. 2017, 292, 16872–16879. [Google Scholar] [CrossRef]
- Radon, C.; Mittelstadt, G.; Duffus, B.R.; Burger, J.; Hartmann, T.; Mielke, T.; Teutloff, C.; Leimkuhler, S.; Wendler, P. Cryo-EM Structures Reveal Intricate Fe-S Cluster Arrangement and Charging in Rhodobacter capsulatus Formate Dehydrogenase. Nat. Commun. 2020, 11, 1912. [Google Scholar] [CrossRef]
- Hartmann, T.; Leimkuehler, S. The Oxygen-Tolerant and NAD+-Dependent Formate Dehydrogenase from Rhodobacter capsulatus Is Able to Catalyze the Reduction of CO2 to Formate. FEBS J. 2013, 280, 6083–6096. [Google Scholar] [CrossRef]
- Ruschig, U.; Muller, U.; Willnow, P.; Hopner, T. CO2 Recution to Formate by NADH Catalyzed by Formate Dehydrogenase from Pseudomonas oxalaticus. Eur. J. Biochem. 1976, 70, 325–330. [Google Scholar] [CrossRef]
- Müller, U.; Willnow, P.; Ruschig, U.; Höpner, T. Formate Dehydrogenase from Pseudomonas oxalaticus. Eur. J. Biochem. 1978, 83, 485–498. [Google Scholar] [CrossRef]
- Cakar, M.M.; Mangas-Sanchez, J.; Birmingham, W.R.; Turner, N.J.; Binay, B. Discovery of a New Metal and NAD+-Dependent Formate Dehydrogenase from Clostridium ljungdahlii. Prep. Biochem. Biotechnol. 2018, 48, 327–334. [Google Scholar] [CrossRef]
- Min, K.; Moon, M.; Park, G.W.; Lee, J.-P.; Kim, S.J.; Lee, J.-S. Newly Explored Formate Dehydrogenases from Clostridium Species Catalyze Carbon Dioxide to Formate. Bioresour. Technol. 2022, 348, 126832. [Google Scholar] [CrossRef]
- Almendra, M.J.; Brondino, C.D.; Gavel, O.; Pereira, A.S.; Tavares, P.; Bursakov, S.; Duarte, R.; Caldeira, J.; Moura, J.J.G.; Moura, I. Purification and Characterization of a Tungsten-Containing Formate Dehydrogenase from Desulfovibrio gigas. Biochemistry 1999, 38, 16366–16372. [Google Scholar] [CrossRef]
- Yamamoto, I.; Saiki, T.; Liu, S.M.; Ljundgdahl, L.G. Purification and Properties of NADP-Dependent Formate Dehydrogenase from Clostridium thermoaceticum, a Tungsten Selenium Iron Protein. J. Biol. Chem. 1983, 258, 1826–1832. [Google Scholar] [CrossRef]
- Cordas, C.M.; Moura, J.J.G. Molybdenum and Tungsten Enzymes Redox Properties—A Brief Overview. Coord. Chem. Rev. 2019, 394, 53–64. [Google Scholar] [CrossRef]
- Hartmann, T.; Schwanhold, N.; Leimkuehler, S. Assembly and Catalysis of Molybdenum or Tungsten-Containing Formate Dehydrogenases from Bacteria. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 1090–1100. [Google Scholar] [CrossRef]
- Kirk, M.L.; Hille, R. Spectroscopic Studies of Mononuclear Molybdenum Enzyme Centers. Molecules 2022, 27, 4802. [Google Scholar] [CrossRef]
- Jormakka, M.; Tornroth, S.; Byrne, B.; Iwata, S. Molecular Basis of Proton Motive Force Generation: Structure of Formate Dehydrogenase-N. Science 2002, 295, 1863–1868. [Google Scholar] [CrossRef]
- Niks, D.; Hille, R. Molybdenum- and Tungsten-Containing Formate Dehydrogenases and Formylmethanofuran Dehydrogenases: Structure, Mechanism, and Cofactor Insertion. Protein Sci. 2019, 28, 111–122. [Google Scholar] [CrossRef]
- Robinson, W.E.; Bassegoda, A.; Reisner, E.; Hirst, J. Oxidation-State-Dependent Binding Properties of the Active Site in a Mo-Containing Formate Dehydrogenase. J. Am. Chem. Soc. 2017, 139, 9927–9936. [Google Scholar] [CrossRef]
- Meneghello, M.; Oliveira, A.R.; Jacq-Bailly, A.; Pereira, I.A.C.; Leger, C.; Fourmond, V. Formate Dehydrogenases Reduce CO2 Rather than HCO3−: An Electrochemical Demonstration. Angew. Chem. Int. Ed. 2021, 60, 9964–9967. [Google Scholar] [CrossRef]
- Tiberti, M.; Papaleo, E.; Russo, N.; De Gioia, L.; Zampella, G. Evidence for the Formation of a Mo-H Intermediate in the Catalytic Cycle of Formate Dehydrogenase. Inorg. Chem. 2012, 51, 8331–8339. [Google Scholar] [CrossRef]
- Khan, M.R. Immobilized Enzymes: A Comprehensive Review. Bull. Natl. Res. Cent. 2021, 45, 207. [Google Scholar] [CrossRef]
- Bie, J.; Sepodes, B.; Fernandes, P.C.B.; Ribeiro, M.H.L. Enzyme Immobilization and Co-Immobilization: Main Framework, Advances and Some Applications. Processes 2022, 10, 494. [Google Scholar] [CrossRef]
- Porcar, R.; Lavandera, I.; Lozano, P.; Altava, B.; Luis, S.V.; Gotor-Fernandez, V.; Garcia-Verdugo, E. Supported Ionic Liquid-like Phases as Efficient Solid Ionic Solvents for the Immobilisation of Alcohol Dehydrogenases towards the Development of Stereoselective Bioreductions. Green Chem. 2021, 23, 5609–5617. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Basso, A.; Brady, D. New Frontiers in Enzyme Immobilisation: Robust Biocatalysts for a Circular Bio-Based Economy. Chem. Soc. Rev. 2021, 50, 5850–5862. [Google Scholar] [CrossRef] [PubMed]
- Lozano, P.; Garcia-Verdugo, E.; Bernal, J.M.; Izquierdo, D.F.; Isabel Burguete, M.; Sanchez-Gomez, G.; Luis, S.V. Immobilised Lipase on Structured Supports Containing Covalently Attached Ionic Liquids for the Continuous Synthesis of Biodiesel in ScCO2. ChemSusChem 2012, 5, 790–798. [Google Scholar] [CrossRef]
- Ren, S.; Jiang, S.; Yan, X.; Chen, R.; Cui, H. Challenges and Opportunities: Porous Supports in Carbonic Anhydrase Immobilization. J. CO2 Util. 2020, 42, 101305. [Google Scholar] [CrossRef]
- Molina-Fernandez, C.; Luis, P. Immobilization of Carbonic Anhydrase for CO2 Capture and Its Industrial Implementation: A Review. J. CO2 Util. 2021, 47, 101475. [Google Scholar] [CrossRef]
- Rasouli, H.; Nguyen, K.; Iliuta, M.C. Recent Advancements in Carbonic Anhydrase Immobilization and Its Implementation in CO2 Capture Technologies: A Review. Sep. Purif. Technol. 2022, 296, 121299. [Google Scholar] [CrossRef]
- Yuan, Y.; Wang, F.; Li, H.; Su, S.; Gao, H.; Han, X.; Ren, S. Potential Application of the Immobilization of Carbonic Anhydrase Based on Metal Organic Framework Supports. Process Biochem. 2022, 122, 214–223. [Google Scholar] [CrossRef]
- Molina-Fernandez, C.; Peters, A.; Debecker, D.P.; Luis, P. Immobilization of Carbonic Anhydrase in a Hydrophobic Poly(Ionic Liquid): A New Functional Solid for CO2 Capture. Biochem. Eng. J. 2022, 187, 108639. [Google Scholar] [CrossRef]
- Rouf, S.; Greish, Y.E.; Al-Zuhair, S. Immobilization of Formate Dehydrogenase in Metal Organic Frameworks for Enhanced Conversion of Carbon Dioxide to Formate. Chemosphere 2021, 267, 128921. [Google Scholar] [CrossRef]
- Singh, R.K.R.; Tiwari, M.K.; Singh, R.K.R.; Lee, J.-K. From Protein Engineering to Immobilization: Promising Strategies for the Upgrade of Industrial Enzymes. Molecules 2013, 18, 1232–1277. [Google Scholar] [CrossRef]
- Di Spiridione, C.; Aresta, M.; Dibenedetto, A. Improving the Enzymatic Cascade of Reactions for the Reduction of CO2 to CH3OH in Water: From Enzymes Immobilization Strategies to Cofactor Regeneration and Cofactor Suppression. Molecules 2022, 27, 4913. [Google Scholar] [CrossRef]
- Zhu, X.; Du, C.; Gao, B.; He, B. Strategies to Improve the Mass Transfer in the CO2 Capture Process Using Immobilized Carbonic Anhydrase. J. Environ. Manag. 2023, 332, 117370. [Google Scholar] [CrossRef]
- Ren, S.; Chen, R.; Wu, Z.; Su, S.; Hou, J.; Yuan, Y. Enzymatic Characteristics of Immobilized Carbonic Anhydrase and Its Applications in CO2 Conversion. Colloid. Surface B 2021, 204, 111779. [Google Scholar] [CrossRef]
- Shen, J.; Salmon, S. Biocatalytic Membranes for Carbon Capture and Utilization. Membranes 2023, 13, 367. [Google Scholar] [CrossRef] [PubMed]
- Wanjari, S.; Prabhu, C.; Satyanarayana, T.; Vinu, A.; Rayalu, S. Immobilization of Carbonic Anhydrase on Mesoporous Aluminosilicate for Carbonation Reaction. Micropor. Mesopor. Mat. 2012, 160, 151–158. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, B.; Qi, W.; Li, X.; Shin, Y.; Lei, C.; Liu, J. Enzymatic Conversion of CO2 to Bicarbonate in Functionalized Mesoporous Silica. Micropor. Mesopor. Mat. 2012, 153, 166–170. [Google Scholar] [CrossRef]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.; Nam, S.C. Carbonic Anhydrase Conjugated to Nanosilver Immobilized onto Mesoporous SBA-15 for Sequestration of CO2. J. Mol. Catal. B Enzym. 2012, 75, 60–67. [Google Scholar] [CrossRef]
- Mao, M.; Zhai, T.; Meng, L.; Meng, Z.; Liu, W. Controllable Preparation of Mesoporous Silica and Its Application in Enzyme-Catalyzed CO2 Reduction. Chem. Eng. J. 2022, 437, 135479. [Google Scholar] [CrossRef]
- Azari, F.N.-G.M. Reversible Denaturation of Carbonic Anhydrase Provides a Method for Its Adsorptive Immobilization. Biotechnol. Bioeng. 1999, 62, 193–199. [Google Scholar] [CrossRef]
- Kim, J.K.; Abdelhamid, M.A.A.; Pack, S.P. Direct Immobilization and Recovery of Recombinant Proteins from Cell Lysates by Using EctP1-Peptide as a Short Fusion Tag for Silica and Titania Supports. Int. J. Biol. Macromol. 2019, 135, 969–977. [Google Scholar] [CrossRef]
- Yong, J.K.J.; Stevens, G.W.; Caruso, F.; Kentish, S.E. In Situ Layer-by-Layer Assembled Carbonic Anhydrase-Coated Hollow Fiber Membrane Contactor for Rapid CO2 Absorption. J. Memb. Sci. 2016, 514, 556–565. [Google Scholar] [CrossRef]
- Ivanovski, V.; Shapovalova, O.E.; Drozdov, A.S. Structural Rearrangements of Carbonic Anhydrase Entrapped in Sol-Gel Magnetite Determined by ATR-FTIR Spectroscopy. Int. J. Mol. Sci. 2022, 23, 5975. [Google Scholar] [CrossRef]
- Moon, H.; Kim, S.; Jo, B.H.; Cha, H.J. Immobilization of Genetically Engineered Whole-Cell Biocatalysts with Periplasmic Carbonic Anhydrase in Polyurethane Foam for Enzymatic CO2 Capture and Utilization. J. CO2 Util. 2020, 39, 101172. [Google Scholar] [CrossRef]
- Jiao, M.; He, J.; Sun, S.; Vriesekoop, F.; Yuan, Q.; Liu, Y.; Liang, H. Fast Immobilization of Human Carbonic Anhydrase II on Ni-Based Metal-Organic Framework Nanorods with High Catalytic Performance. Catalysts 2020, 10, 401. [Google Scholar] [CrossRef]
- Abdelrahim, M.Y.; Martins, C.F.; Neves, L.A.; Capasso, C.; Supuran, C.T.; Coelhoso, I.M.; Crespo, J.G.; Barboiu, M. Supported Ionic Liquid Membranes Immobilized with Carbonic Anhydrases for CO2 Transport at High Temperatures. J. Memb. Sci. 2017, 528, 225–230. [Google Scholar] [CrossRef]
- Martins, C.F.; Neves, L.A.M.; Estevão, L.A.; Rosatella, A.; Alves, V.D.; Afonso, C.A.M.; Crespo, J.G.; Coelhoso, I.M. Effect of Water Activity on Carbon Dioxide Transport in Cholinium-Based Ionic Liquids with Carbonic Anhydrase. Sep. Purif. Technol. 2016, 168, 74–82. [Google Scholar] [CrossRef]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.; Nam, S.C. Capture and Sequestration of CO2 by Human Carbonic Anhydrase Covalently Immobilized onto Amine-Functionalized SBA-15. J. Phys. Chem. C 2011, 115, 20209–20216. [Google Scholar] [CrossRef]
- Shen, J.; Yuan, Y.; Salmon, S. Durable and Versatile Immobilized Carbonic Anhydrase on Textile Structured Packing for CO2 Capture. Catalysts 2022, 12, 1108. [Google Scholar] [CrossRef]
- Xv, J.; Zhang, Z.; Pang, S.; Jia, J.; Geng, Z.; Wang, R.; Li, P.; Bilal, M.; Cui, J.; Jia, S. Accelerated CO2 Capture Using Immobilized Carbonic Anhydrase on Polyethyleneimine/Dopamine Co-Deposited MOFs. Biochem. Eng. J. 2022, 189, 108719. [Google Scholar] [CrossRef]
- Shamna, I.; Jeong, S.K.; Margandan, B. Covalent Immobilization of Carbonic Anhydrase on Amine Functionalized Alumino-Siloxane Aerogel Beads for Biomimetic Sequestration of CO2. J. Ind. Eng. Chem. 2021, 100, 288–295. [Google Scholar] [CrossRef]
- Iliuta, I.; Rasouli, H.; Iliuta, M.C. Intensified CO2 Capture in Wall-Coated Microreactors with Immobilized Carbonic Anhydrase: Experimental and Modeling. Sep. Purif. Technol. 2023, 307, 122590. [Google Scholar] [CrossRef]
- Kimmel, J.D.; Arazawa, D.T.; Ye, S.-H.; Shankarraman, V.; Wagner, W.R.; Federspiel, W.J. Carbonic Anhydrase Immobilized on Hollow Fiber Membranes Using Glutaraldehyde Activated Chitosan for Artificial Lung Applications. J. Mater. Sci. Mater. Med. 2013, 24, 2611–2621. [Google Scholar] [CrossRef]
- Peirce, S.; Russo, M.E.; Isticato, R.; Lafuente, R.F.; Salatino, P.; Marzocchella, A. Structure and Activity of Magnetic Cross-Linked Enzyme Aggregates of Bovine Carbonic Anhydrase as Promoters of Enzymatic CO2 Capture. Biochem. Eng. J. 2017, 127, 188–195. [Google Scholar] [CrossRef]
- Chang, S.; He, Y.; Li, Y.; Cui, X. Study on the Immobilization of Carbonic Anhydrases on Geopolymer Microspheres for CO2 Capture. J. Clean. Prod. 2021, 316, 128163. [Google Scholar] [CrossRef]
- Zhang, X.; Shao, W.; Chen, B.; Wang, M. Cross-Linking of Carbonic Anhydrase and Formate Dehydrogenase Based on Amino Acid Specific Recognition: Conversion of Carbon Dioxide to Formic Acid. Enzym. Microb. Technol. 2021, 146, 109763. [Google Scholar] [CrossRef]
- Nelson, J.M.; Griffin, E.G. Adsorption of Invertase. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef]
- Jesionowski, T.; Zdarta, J.; Krajewska, B. Enzyme immobilization by adsorption: A review. Adsorption 2014, 20, 801–821. [Google Scholar] [CrossRef]
- Nabavi Zadeh, P.S.; Åkerman, B. Immobilization of Enzymes in Mesoporous Silica Particles: Protein Concentration and Rotational Mobility in the Pores. J. Phys. Chem. B 2017, 121, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Crumbliss, A.L.; Perine, S.C.; Stonehuerner, J.; Tubergen, K.R.; Zhao, J.; Henkens, R.W.; O’Daly, J.P. Colloidal Gold as a Biocompatible Immobilization Matrix Suitable for the Fabrication of Enzyme Electrodes by Electrodeposition. Biotechnol. Bioeng. 1992, 40, 483–490. [Google Scholar] [CrossRef]
- Suvannasara, P.; Juntapram, K.; Praphairaksit, N.; Siralertmukul, K.; Muangsin, N. Mucoadhesive 4-Carboxybenzenesulfonamide-Chitosan with Antibacterial Properties. Carbohydr. Polym. 2013, 94, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-J.; Yin, L.; Wang, Z.-F.; Jing, Y.-C.; Jiang, Z.-L.; Ding, Y.; Wang, H.-S. Enzyme-Immobilized Metal-Organic Frameworks: From Preparation to Application. Chem. Asian J. 2022, 17, e202200751. [Google Scholar] [CrossRef]
- Zhou, H.-C.; Long, J.R.; Yaghi, O.M. Introduction to Metal–Organic Frameworks. Chem. Rev. 2012, 112, 673–674. [Google Scholar] [CrossRef]
- Zhang, W.; Taheri-Ledari, R.; Saeidirad, M.; Qazi, F.S.; Kashtiaray, A.; Ganjali, F.; Tian, Y.; Maleki, A. Regulation of Porosity in MOFs: A Review on Tunable Scaffolds and Related Effects and Advances in Different Applications. J. Environ. Chem. Eng. 2022, 10, 108836. [Google Scholar] [CrossRef]
- Silva, A.R.M.; Alexandre, J.Y.N.H.; Souza, J.E.S.; Lima Neto, J.G.; de Sousa Junior, P.G.; Rocha, M.V.P.; dos Santos, J.C.S. The Chemistry and Applications of Metal-Organic Frameworks (MOFs) as Industrial Enzyme Immobilization Systems. Molecules 2022, 27, 4529. [Google Scholar] [CrossRef]
- Liu, Q.; Bai, X.; Pham, H.; Hu, J.; Dinu, C.Z. Active Nanointerfaces Based on Enzyme Carbonic Anhydrase and Metal-Organic Framework for Carbon Dioxide Reduction. Nanomaterials 2021, 11, 1008. [Google Scholar] [CrossRef]
- Huang, Y.; Zhang, S.; Chen, H.; Zhao, L.; Zhang, Z.; Cheng, P.; Chen, Y. A Zinc Coordination Complex Mimicking Carbonic Anhydrase for CO2 Hydrolysis and Sequestration. Inorg. Chem. 2019, 58, 9916–9921. [Google Scholar] [CrossRef]
- Bien, C.E.; Chen, K.K.; Chien, S.-C.; Reiner, B.R.; Lin, L.-C.; Wade, C.R.; Ho, W.S.W. Bioinspired Metal-Organic Framework for Trace CO2 Capture. J. Am. Chem. Soc. 2018, 140, 12662–12666. [Google Scholar] [CrossRef]
- Han, L.; Pham, T.; Zhuo, M.; Forrest, K.A.; Suepaul, S.; Space, B.; Zaworotko, M.J.; Shi, W.; Chen, Y.; Cheng, P.; et al. Molecular Sieving and Direct Visualization of CO2 in Binding Pockets of an Ultramicroporous Lanthanide Metal-Organic Framework Platform. ACS Appl. Mater. Interfaces 2019, 11, 23192–23197. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Nan, Y.; Zhao, Y.; Wang, X.; Huang, S.; Shi, J. Immobilization of Carbonic Anhydrase for Facilitated CO2 Capture and Separation. Chin. J. Chem. Eng. 2020, 28, 2817–2831. [Google Scholar] [CrossRef]
- Neves, L.A.; Afonso, C.; Coelhoso, I.M.; Crespo, J.G. Integrated CO2 Capture and Enzymatic Bioconversion in Supported Ionic Liquid Membranes. Sep. Purif. Technol. 2012, 97, 34–41. [Google Scholar] [CrossRef]
- Bednár, A.; Nemestóthy, N.; Bakonyi, P.; Fülöp, L.; Zhen, G.; Lu, X.; Kobayashi, T.; Kumar, G.; Xu, K.; Bélafi-Bakó, K. Enzymatically-Boosted Ionic Liquid Gas Separation Membranes Using Carbonic Anhydrase of Biomass Origin. Chem. Eng. J. 2016, 303, 621–626. [Google Scholar] [CrossRef]
- de Castro, A.M.; Neves, L.A.; Corvo, M.C.; Cabrita, E.J.; Crespo, J.G. Effect of Carbonic Anhydrase on CO2 Absorption Promoted by Choline Hydroxide Using Supported Liquid Membranes. Sep. Purif. Technol. 2022, 280, 119921. [Google Scholar] [CrossRef]
- Migliardini, F.; De Luca, V.; Carginale, V.; Rossi, M.; Corbo, P.; Supuran, C.T.; Capasso, C. Biomimetic CO2 Capture Using a Highly Thermostable Bacterial α-Carbonic Anhydrase Immobilized on a Polyurethane Foam. J. Enzym. Inhib. Med. Chem. 2014, 29, 146–150. [Google Scholar] [CrossRef]
- Zaidi, S.; Srivastava, N.; Khare, S.K. Microbial Carbonic Anhydrase Mediated Carbon Capture, Sequestration & Utilization: A Sustainable Approach to Delivering Bio-Renewables. Bioresour. Technol. 2022, 365, 128174. [Google Scholar] [CrossRef]
- Rasouli, H.; Iliuta, I.; Bougie, F.; Garnier, A.; Iliuta, M.C. Enhanced CO2 Capture in Packed-Bed Column Bioreactors with Immobilized Carbonic Anhydrase. Chem. Eng. J. 2022, 432, 134029. [Google Scholar] [CrossRef]
- Vinoba, M.; Bhagiyalakshmi, M.; Jeong, S.K.; Yoon, Y.I.I.; Nam, S.C. Immobilization of Carbonic Anhydrase on Spherical SBA-15 for Hydration and Sequestration of CO2. Colloids Surf. B Biointerfaces 2012, 90, 91–96. [Google Scholar] [CrossRef]
- Uygun, M.; Singh, V.V.; Kaufmann, K.; Uygun, D.A.; De Oliveira, S.D.S.; Wang, J. Micromotor-Based Biomimetic Carbon Dioxide Sequestration: Towards Mobile Microscrubbers. Angew. Chem.-Int. Ed. 2015, 54, 12900–12904. [Google Scholar] [CrossRef]
- Xu, X.; Kentish, S.E.; Martin, G.J.O. Direct Air Capture of CO2 by Microalgae with Buoyant Beads Encapsulating Carbonic Anhydrase. ACS Sustain. Chem. Eng. 2021, 9, 9698–9706. [Google Scholar] [CrossRef]
- Sifat, N.S.; Haseli, Y. A Critical Review of CO2 Capture Technologies and Prospects for Clean Power Generation. Energies 2019, 12, 4143. [Google Scholar] [CrossRef]
- Fu, L.; Ren, Z.; Si, W.; Ma, Q.; Huang, W.; Liao, K.; Huang, Z.; Wang, Y.; Li, J.; Xu, P. Research Progress on CO2 Capture and Utilization Technology. J. CO2 Util. 2022, 66, 102260. [Google Scholar] [CrossRef]
- Fagorite, I.V.; Chijioke, C.F.; Opara, I.A.; Onyekuru, S.O.; Oguzie, E.E. Environmental and Safety Issues Associated with Geological Carbon Storage: A Review. Euro-Mediterr. J. Environ. Integr. 2022, 7, 445–461. [Google Scholar] [CrossRef]
- Farrukh, S.; Wu, D.; Al-Dadah, R.; Gao, W.; Wang, Z. A Review of Integrated Cryogenic Energy Assisted Power Generation Systems and Desalination Technologies. Appl. Therm. Eng. 2023, 221, 119836. [Google Scholar] [CrossRef]
- International Energy Agency. CO2 Emissions in 2022. Available online: https://iea.blob.core.windows.net/assets/3c8fa115-35c4-4474-b237-1b00424c8844/CO2Emissionsin2022.pdf (accessed on 30 June 2023).
- International Energy Agency. Global Energy-Related CO2 Emissions by Sector. Available online: https://www.iea.org/data-and-statistics/charts/global-energy-related-co2-emissions-by-sector (accessed on 30 June 2023).
- Hepburn, C.; Adlen, E.; Beddington, J.; Carter, E.A.; Fuss, S.; Mac Dowell, N.; Minx, J.C.; Smith, P.; Williams, C.K. The Technological and Economic Prospects for CO2 Utilization and Removal. Nature 2019, 575, 87–97. [Google Scholar] [CrossRef]
- Gundersen, M.T.; Von Solms, N.; Woodley, J.M. Enzymatically Assisted CO2 Removal from Flue-Gas. In Energy Procedia; Department of Chemical and Biochemical Engineering, Technical University of Denmark: Lyngby, Denmark, 2014; Volume 63, pp. 624–632. [Google Scholar] [CrossRef]
- Zhang, S.; Du, M.; Shao, P.; Wang, L.; Ye, J.; Chen, J.; Chen, J. Carbonic Anhydrase Enzyme-MOFs Composite with a Superior Catalytic Performance to Promote CO2 Absorption into Tertiary Amine Solution. Environ. Sci. Technol. 2018, 52, 12708–12716. [Google Scholar] [CrossRef]
- Gladis, A.; Lomholdt, N.F.; Fosbøl, P.L.; Woodley, J.M.; von Solms, N. Pilot Scale Absorption Experiments with Carbonic Anhydrase-Enhanced MDEA-Benchmarking with 30 wt% MEA. Int. J. Greenh. Gas Control. 2019, 82, 69–85. [Google Scholar] [CrossRef]
- Kim, T.-J.; Lang, A.; Chikukwa, A.; Sheridan, E.; Dahl, P.I.; Leimbrink, M.; Skiborowski, M.; Roubroeks, J. Enzyme Carbonic Anhydrase Accelerated CO2 Absorption in Membrane Contactor. In Energy Procedia; SINTEF Materials and Chemistry: Trondheim, Norway, 2017; Volume 114, pp. 17–24. [Google Scholar] [CrossRef]
- Thee, H.; Smith, K.H.; Da Silva, G.; Kentish, S.E.; Stevens, G.W. Carbonic Anhydrase Promoted Absorption of CO2 into Potassium Carbonate Solutions. Greenh. Gases Sci. Technol. 2015, 5, 108–114. [Google Scholar] [CrossRef]
- Hu, G.; Nicholas, N.J.; Smith, K.H.; Mumford, K.A.; Kentish, S.E.; Stevens, G.W. Carbon Dioxide Absorption into Promoted Potassium Carbonate Solutions: A Review. Int. J. Greenh. Gas Control 2016, 53, 28–40. [Google Scholar] [CrossRef]
- Jin, P.; Zhang, S.; Liu, Y.; Zhang, W.; Wang, R. Application of Bacillus mucilaginosus in the Carbonation of Steel Slag. Appl. Microbiol. Biotechnol. 2021, 105, 8663–8674. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.H.; Seo, J.H.; Yang, Y.J.; Baek, K.; Choi, Y.S.; Pack, S.P.; Oh, S.H.; Cha, H.J. Bioinspired Silica Nanocomposite with Autoencapsulated Carbonic Anhydrase as a Robust Biocatalyst for CO2 Sequestration. ACS Catal. 2014, 4, 4332–4340. [Google Scholar] [CrossRef]
- Yang, G.; Li, L.; Li, F.; Zhang, C.; Lyu, J. Mechanism of Carbonate Mineralization Induced by Microbes: Taking Curvibacter lanceolatus Strain HJ-1 as an Example. Micron 2021, 140, 102980. [Google Scholar] [CrossRef]
- Bose, H.; Satyanarayana, T. Suitability of the Alkalistable Carbonic Anhydrase from a Polyextremophilic Bacterium Aeribacillus pallidus TSHB1 in Biomimetic Carbon Sequestration. Bioprocess Biosyst. Eng. 2016, 39, 1515–1525. [Google Scholar] [CrossRef]
- De Luca, V.; Vullo, D.; Scozzafava, A.; Carginale, V.; Rossi, M.; Supuran, C.T.; Capasso, C. An Alpha-Carbonic Anhydrase from the Thermophilic Bacterium Sulphurihydrogenibium azorense Is the Fastest Enzyme Known for the CO2 Hydration Reaction. Bioorg. Med. Chem. 2013, 21, 1465–1469. [Google Scholar] [CrossRef]
- Di Lorenzo, F.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Gil-San Millán, R.; Navarro, J.A.R.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. The Carbonation of Wollastonite: A Model Reaction to Test Natural and Biomimetic Catalysts for Enhanced CO2 Sequestration. Minerals 2018, 8, 209. [Google Scholar] [CrossRef]
- Wang, M.; Lawal, A.; Stephenson, P.; Sidders, J.; Ramshaw, C. Post-Combustion CO2 Capture with Chemical Absorption: A State-of-the-Art Review. Chem. Eng. Res. Des. 2011, 89, 1609–1624. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Koech, P.K.; Glezakou, V.-A.; Rousseau, R.; Malhotra, D.; Cantu, D.C. Water-Lean Solvents for Post-Combustion CO2 Capture: Fundamentals, Uncertainties, Opportunities, and Outlook. Chem. Rev. 2017, 117, 9594–9624. [Google Scholar] [CrossRef]
- Sharif, M.; Zhang, T.; Wu, X.; Yu, Y.; Zhang, Z. Evaluation of CO2 Absorption Performance by Molecular Dynamic Simulation for Mixed Secondary and Tertiary Amines. Int. J. Greenh. Gas. Control 2020, 97, 103059. [Google Scholar] [CrossRef]
- Bernhardsen, I.M.; Krokvik, I.R.T.; Jens, K.-J.; Knuutila, H.K. Performance of MAPA Promoted Tertiary Amine Systems for CO2 Absorption: Influence of Alkyl Chain Length and Hydroxyl Groups. Energy Procedia 2017, 114, 1682–1688. [Google Scholar] [CrossRef]
- Liu, B.; Cui, Z.; Tian, W. The Kinetics Investigation of CO2 Absorption into TEA and DEEA Amine Solutions Containing Carbonic Anhydrase. Processes 2021, 9, 2140. [Google Scholar] [CrossRef]
- Bui, M.; Adjiman, C.S.; Bardow, A.; Anthony, E.J.; Boston, A.; Brown, S.; Fennell, P.S.; Fuss, S.; Galindo, A.; Hackett, L.A.; et al. Carbon Capture and Storage (CCS): The Way Forward. Energy Environ. Sci. 2018, 11, 1062–1176. [Google Scholar] [CrossRef]
- Power, I.M.; Harrison, A.L.; Dipple, G.M. Accelerating Mineral Carbonation Using Carbonic Anhydrase. Environ. Sci. Technol. 2016, 50, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, H.; Bailey, E.; Wysokowski, M.; Jesionowski, T. Forced Biomineralization: A Review. Biomimetics 2021, 6, 46. [Google Scholar] [CrossRef]
- Bose, H.; Satyanarayana, T. Microbial Carbonic Anhydrases in Biomimetic Carbon Sequestration for Mitigating Global Warming: Prospects and Perspectives. Front. Microbiol. 2017, 8, 1615. [Google Scholar] [CrossRef]
- Wang, F.; Dreisinger, D.B.; Jarvis, M.; Hitchins, T. The Technology of CO2 Sequestration by Mineral Carbonation: Current Status and Future Prospects. Can. Metall. Q. 2018, 57, 46–58. [Google Scholar] [CrossRef]
- Hills, C.D.; Tripathi, N.; Carey, P.J. Mineralization Technology for Carbon Capture, Utilization, and Storage. Front. Energy Res. 2020, 8, 142. [Google Scholar] [CrossRef]
- Bhatia, S.K.; Bhatia, R.K.; Jeon, J.-M.; Kumar, G.; Yang, Y.-H. Carbon Dioxide Capture and Bioenergy Production Using Biological System–A Review. Renew. Sust. Energy Rev. 2019, 110, 143–158. [Google Scholar] [CrossRef]
- Zajac, M.; Krol, M.; Bullerjahn, F.; Deja, J. Effect of Temperature on Carbon Dioxide Mineralisation in Recycled Cement Paste. Adv. Cem. Res. 2023, 35, 1–12. [Google Scholar] [CrossRef]
- Yin, B.; Xu, H.; Fan, F.; Qi, D.; Hua, X.; Xu, T.; Liu, C.; Hou, D. Superhydrophobic Coatings Based on Bionic Mineralization for Improving the Durability of Marine Concrete. Constr. Build. Mater. 2023, 362, 129705. [Google Scholar] [CrossRef]
- Heldebrant, D.J.; Kothandaraman, J.; Mac Dowell, N.; Brickett, L. Next Steps for Solvent-Based CO2 Capture; Integration of Capture, Conversion, and Mineralisation. Chem. Sci. 2022, 13, 6445–6456. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Cizer, O.; Kudlacz, K.; Ibanez-Velasco, A.; Ruiz-Agudo, C.; Elert, K.; Burgos-Cara, A.; Ruiz-Agudo, E. The Multiple Roles of Carbonic Anhydrase in Calcium Carbonate Mineralization. CrystEngComm 2019, 21, 7407–7423. [Google Scholar] [CrossRef]
- Fernández, M.S.; Montt, B.; Ortiz, L.; Neira-Carrillo, A.; Arias, J.L. Effect of Carbonic Anhydrase Immobilized on Eggshell Membranes on Calcium Carbonate Crystallization In Vitro. In Biomineralization; Endo, K., Kogure, T., Nagasawa, H., Eds.; Springer: Singapore, 2018; pp. 31–37. [Google Scholar]
- Jin, P.; Wang, R.; Zhang, S.; Chen, Y. Effect of Carbonic Anhydrase Bacteria on the Carbonation Process of γ-C2S. Adv. Cem. Res. 2021, 34, 15–27. [Google Scholar] [CrossRef]
- Sharma, V.K.; Hutchison, J.M.; Allgeier, A.M. Redox Biocatalysis: Quantitative Comparisons of Nicotinamide Cofactor Regeneration Methods. ChemSusChem 2022, 15, e2022008. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Jaen, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and Challenges in the Selective Reduction of Carbon Dioxide to Methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Corrado, M.L.; Knaus, T.; Schwaneberg, U.; Mutti, F.G. High-Yield Synthesis of Enantiopure 1,2-Amino Alcohols from L-Phenylalanine via Linear and Divergent Enzymatic Cascades. Org. Process Res. Dev. 2022, 26, 2085–2095. [Google Scholar] [CrossRef]
- Singh, P.; Srivastava, R. Utilization of Bio-Inspired Catalyst for CO2 Reduction into Green Fuels: Recent Advancement and Future Perspectives. J. CO2 Util. 2021, 53, 101748. [Google Scholar] [CrossRef]
- Armstrong, F.A.; Cheng, B.; Herold, R.A.; Megarity, C.F.; Siritanaratkul, B. From Protein Film Electrochemistry to Nanoconfined Enzyme Cascades and the Electrochemical Leaf. Chem. Rev. 2023, 123, 5421–5458. [Google Scholar] [CrossRef]
- Meneghello, M.; Leger, C.; Fourmond, V. Electrochemical Studies of CO2-Reducing Metalloenzymes. Chem. Eur. J. 2021, 27, 17542–17553. [Google Scholar] [CrossRef]
- Rasheed, T.; Shafi, S.; Anwar, M.T.; Rizwan, K.; Ahmad, T.; Bilal, M. Revisiting Photo and Electro-Catalytic Modalities for Sustainable Conversion of CO2. Appl. Catal. A-Gen. 2021, 623, 118248. [Google Scholar] [CrossRef]
- Cadoux, C.; Milton, R.D. Recent Enzymatic Electrochemistry for Reductive Reactions. ChemElectroChem 2020, 7, 1974–1986. [Google Scholar] [CrossRef]
- Xing, X.; Liu, Y.; Lin, R.-D.; Zhang, Y.; Wu, Z.-L.; Yu, X.-Q.; Li, K.; Wang, N. Development of an Integrated System for Highly Selective Photoenzymatic Synthesis of Formic Acid from CO2. ChemSusChem 2023, 16, e2022019. [Google Scholar] [CrossRef]
- Wang, Q.; Pan, Z. Advances and Challenges in Developing Cocatalysts for Photocatalytic Conversion of Carbon Dioxide to Fuels. Nano Res. 2022, 15, 10090–10109. [Google Scholar] [CrossRef]
- Fang, X.; Kalathil, S.; Reisner, E. Semi-Biological Approaches to Solar-to-Chemical Conversion. Chem. Soc. Rev. 2020, 49, 4926–4952. [Google Scholar] [CrossRef]
- Ozgen, F.F.; Runda, M.E.; Schmidt, S. Photo-Biocatalytic Cascades: Combining Chemical and Enzymatic Transformations Fueled by Light. ChemBioChem 2021, 22, 790–806. [Google Scholar] [CrossRef]
- Chen, H.; Huang, Y.; Sha, C.; Moradian, J.M.; Yong, Y.-C.; Fang, Z. Enzymatic Carbon Dioxide to Formate: Mechanisms, Challenges and Opportunities. Renew. Sust. Energy Rev. 2023, 178, 113271. [Google Scholar] [CrossRef]
- Immanuel, S.; Sivasubramanian, R.; Gul, R.; Dar, M.A. Recent Progress and Perspectives on Electrochemical Regeneration of Reduced Nicotinamide Adenine Dinucleotide (NADH). Chem. Asian J. 2020, 15, 4256–4270. [Google Scholar] [CrossRef]
- Lee, Y.S.; Gerulskis, R.; Minteer, S.D. Advances in Electrochemical Cofactor Regeneration: Enzymatic and Non-Enzymatic Approaches. Curr. Opin. Biotechnol. 2022, 73, 14–21. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, X.; Ji, X. Developing and Regenerating Cofactors for Sustainable Enzymatic CO2 Conversion. Processes 2022, 10, 230. [Google Scholar] [CrossRef]
- Bachosz, K.; Zdarta, J.; Bilal, M.; Meyer, A.S.; Jesionowski, T. Enzymatic Cofactor Regeneration Systems: A New Perspective on Efficiency Assessment. Sci. Total Environ. 2023, 868, 161630. [Google Scholar] [CrossRef]
- Findrik, Z.; Vasic-Racki, D. Overview on Reactions with Multi-Enzyme Systems. Chem. Biochem. Eng. Q. 2009, 23, 545–553. [Google Scholar]
- Neves, L.A.; Afonso, C.A.M.; Coelhoso, L.M.; Crespo, J.G. CO2 Capture by Enzymatic Bioconversion in a Membrane Contactor with Task Specific Ionic Liquids. In Procedia Engineering; Universidade Nova de Lisboa: Lisbon, Portugal, 2012; Volume 44, pp. 557–558. [Google Scholar] [CrossRef]
- Qadir, M.I.; Dupont, J. Thermo- and Photocatalytic Activation of CO2 in Ionic Liquids. Angew. Chem. Int. Ed. 2023, e202301497. [Google Scholar] [CrossRef]
- Chenault, H.K.; Whitesides, G.M. Regeneration of Nicotinamide Cofactors for Use in Organic Synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef] [PubMed]
- Chenault, H.K.; Simon, E.S.; Whitesides, G.M. Cofactor Regeneration for Enzyme-Catalysed Synthesis. Biotechnol. Genet. Eng. Rev. 1988, 6, 221–270. [Google Scholar] [CrossRef]
- Yu, X.; Niks, D.; Ge, X.; Liu, H.; Hille, R.; Mulchandani, A. Synthesis of Formate from CO2 Gas Catalyzed by an O2-Tolerant NAD-Dependent Formate Dehydrogenase and Glucose Dehydrogenase. Biochemistry 2019, 58, 1861–1868. [Google Scholar] [CrossRef]
- Liao, Q.; Liu, W.; Meng, Z. Strategies for Overcoming the Limitations of Enzymatic Carbon Dioxide Reduction. Biotechnol. Adv. 2022, 60, 108024. [Google Scholar] [CrossRef]
- El-Zahab, B.; Donnelly, D.; Wang, P. Particle-Tethered NADH for Production of Methanol from CO2 Catalyzed by Coimmobilized Enzymes. Biotechnol. Bioeng. 2008, 99, 508–514. [Google Scholar] [CrossRef]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Tethering of Nicotinamide Adenine Dinucleotide inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. ACS Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef]
- Ren, S.; Wang, Z.; Bilal, M.; Feng, Y.; Jiang, Y.; Jia, S.; Cui, J. Co-Immobilization Multienzyme Nanoreactor with Co-Factor Regeneration for Conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef]
- Shukia, S.K.; Khokarale, S.G.; Bui, T.Q.; Mikkola, J.-P.T. Ionic Liquids: Potential Materials for Carbon Dioxide Capture and Utilization. Front. Mater. 2019, 6, 42. [Google Scholar] [CrossRef]
- Zeng, S.; Zhang, X.X.; Bai, L.; Zhang, X.X.; Wang, H.; Wang, J.; Bao, D.; Li, M.; Liu, X.; Zhang, S. Ionic-Liquid-Based CO2 Capture Systems: Structure, Interaction and Process. Chem. Rev. 2017, 117, 9625–9673. [Google Scholar] [CrossRef]
- Zhang, Z.; Muschiol, J.; Huang, Y.; Sigurdardottir, S.B.; von Solms, N.; Daugaard, A.E.; Wei, J.; Luo, J.; Xu, B.-H.; Zhang, S.; et al. Efficient Ionic Liquid-Based Platform for Multi-Enzymatic Conversion of Carbon Dioxide to Methanol. Green Chem. 2018, 20, 4339–4348. [Google Scholar] [CrossRef]
- The Methanol Institute. Available online: https://www.methanol.org/applications/ (accessed on 7 July 2023).
- Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. Reduction of CO2 to Methanol by a Polyenzymatic System Encapsulated in Phospholipids-Silica Nanocapsules. New J. Chem. 2013, 37, 3721–3730. [Google Scholar] [CrossRef]
- Singh, R.K.R.; Singh, R.K.R.; Sivakumar, D.; Kondaveeti, S.; Kim, T.; Li, J.; Sung, B.H.; Cho, B.-K.; Kim, D.R.; Kim, S.C.; et al. Insights into Cell-Free Conversion of CO2 to Chemicals by a Multienzyme Cascade Reaction. ACS Catal. 2018, 8, 11085–11093. [Google Scholar] [CrossRef]
- Dave, B.C.; Rao, M.S.; Burt, M.C. Converting Carbon Dioxide to Methanol Comprises Serial Reduction of the Carbon Dioxide to Methanol By Dehydrogenase Enzymes in the Presence of Reduced Nicotinamide Adenine Dinucleotide. US20070042479(A1), 18 August 2005. [Google Scholar]
- Alvarez-Malmagro, J.; Oliveira, A.R.; Gutierrez-Sanchez, C.; Villajos, B.; Pereira, I.A.C.; Velez, M.; Pita, M.; De Lacey, A.L. Bioelectrocatalytic Activity of W-Formate Dehydrogenase Covalently Immobilized on Functionalized Gold and Graphite Electrodes. ACS Appl. Mater. Interfaces 2021, 13, 11891–11900. [Google Scholar] [CrossRef]
- Barin, R.; Rashid-Nadimi, S.; Biria, D.; Asadollahi, M.A. Direct Electrochemical Regeneration of 1,4-NADH at the Copper Foam and Bimetallic Copper Foam. Electrochim. Acta 2017, 247, 1095–1102. [Google Scholar] [CrossRef]
- Song, H.; Ma, C.; Liu, P.; You, C.; Lin, J.; Zhu, Z. A Hybrid CO2 Electroreduction System Mediated by Enzyme-Cofactor Conjugates Coupled with Cu Nanoparticle-Catalyzed Cofactor Regeneration. J. CO2 Util. 2019, 34, 568–575. [Google Scholar] [CrossRef]
- Addo, P.K.; Arechederra, R.L.; Waheed, A.; Shoemaker, J.D.; Sly, W.S.; Minteer, S.D. Methanol Production via Bioelectrocatalytic Reduction of Carbon Dioxide: Role of Carbonic Anhydrase in Improving Electrode Performance. Electrochem. Solid-State Lett. 2011, 14, E9–E13. [Google Scholar] [CrossRef]
- Yuan, M.; Sahin, S.; Cai, R.; Abdellaoui, S.; Hickey, D.P.; Minteer, S.D.; Milton, R.D. Creating a Low-Potential Redox Polymer for Efficient Electroenzymatic CO2 Reduction. Angew. Chem. Int. Ed. 2018, 57, 6582–6586. [Google Scholar] [CrossRef]
- Kim, S.-H.; Chung, G.-Y.; Kim, S.-H.; Vinothkumar, G.; Yoon, S.-H.; Jung, K.-D. Electrochemical NADH Regeneration and Electroenzymatic CO2 Reduction on Cu Nanorods/Glassy Carbon Electrode Prepared by Cyclic Deposition. Electrochim. Acta 2016, 210, 837–845. [Google Scholar] [CrossRef]
- Chen, Y.; Li, P.; Zhou, J.; Buru, C.T.; Dordevic, L.; Li, P.; Zhang, X.; Cetin, M.M.; Stoddart, J.F.; Stupp, S.I.; et al. Integration of Enzymes and Photosensitizers in a Hierarchical Mesoporous Metal–Organic Framework for Light-Driven CO2 Reduction. J. Am. Chem. Soc. 2020, 142, 1768–1773. [Google Scholar] [CrossRef]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 Reduction to Formate by Formate Dehydrogenase from Candida Boidinii Coupling with Direct Electrochemical Regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Ji, M.; Liu, Y.; Wang, N.; Zhang, X.; Zhang, S.; Ji, X. Encapsulation of Multiple Enzymes in a Metal-Organic Framework with Enhanced Electro-Enzymatic Reduction of CO2 to Methanol. Green Chem. 2021, 23, 2362–2371. [Google Scholar] [CrossRef]
- Zhang, T.R.; Lu, S.Y. Sacrificial Agents for Photocatalytic Hydrogen Production: Effects, Cost, and Development. Chem. Catalysis 2022, 2, 1502–1505. [Google Scholar] [CrossRef]
- Hernandez-Ibanez, N.; Gomis-Berenguer, A.; Montiel, V.; Ania, C.O.; Iniesta, J. Fabrication of a Biocathode for Formic Acid Production upon the Immobilization of Formate Dehydrogenase from Candida boidinii on a Nanoporous Carbon. Chemosphere 2022, 291, 133117. [Google Scholar] [CrossRef]
- Cheng, Y.; Shi, J.; Wu, Y.; Wang, X.; Sun, Y.; Cai, Z.; Chen, Y.; Jiang, Z. Intensifying Electron Utilization by Surface-Anchored Rh Complex for Enhanced Nicotinamide Cofactor Regeneration and Photoenzymatic CO2 Reduction. Research 2021, 2021, 8175709. [Google Scholar] [CrossRef]
- Ottone, C.; Pugliese, D.; Laurenti, M.; Hernandez, S.; Cauda, V.; Grez, P.; Wilson, L. ZnO Materials as Effective Anodes for the Photoelectrochemical Regeneration of Enzymatically Active NAD+. ACS Appl. Mater. Interfaces 2021, 13, 10719–10727. [Google Scholar] [CrossRef]
- Tensi, L.; Macchioni, A. Extremely Fast NADH-Regeneration Using Phosphonic Acid as Hydride Source and Iridium-Pyridine-2-Sulfonamidate Catalysts. ACS Catal. 2020, 10, 7945–7949. [Google Scholar] [CrossRef]
- Liao, Q.; Guo, M.; Mao, M.; Gao, R.; Meng, Z.; Fan, X.; Liu, W. Construction and Optimization of a Photo-Enzyme Coupled System for Sustainable CO2 Conversion to Methanol. Process Biochem. 2023, 129, 44–55. [Google Scholar] [CrossRef]
- Guo, M.; Gu, F.; Meng, L.; Liao, Q.; Meng, Z.; Liu, W. Synthesis of Formaldehyde from CO2 Catalyzed by the Coupled Photo-Enzyme System. Sep. Purif. Technol. 2022, 286, 120480. [Google Scholar] [CrossRef]
- Kim, J.A.; Kim, S.; Lee, J.; Baeg, J.-O.; Kim, J.A. Photochemical Production of NADH Using Cobaloxime Catalysts and Visible-Light Energy. Inorg. Chem. 2012, 51, 8057–8063. [Google Scholar] [CrossRef] [PubMed]
- Laun, K.; Duffus, B.R.; Kumar, H.; Oudsen, J.-P.H.; Karafoulidi-Retsou, C.; Waffo, A.T.; Hildebrandt, P.; Ly, K.H.; Leimkuehler, S.; Katz, S.; et al. A Minimal Light-Driven System to Study the Enzymatic CO2 Reduction of Formate Dehydrogenase. ChemCatChem 2022, 14, e202201067. [Google Scholar] [CrossRef]
- Ji, X.; Wang, J.; Kang, Y.; Mei, L.; Su, Z.; Wang, S.; Ma, G.; Shi, J.; Zhang, S. Enhanced Solar Energy Harvest and Electron Transfer through Intra- and Intermolecular Dual Channels in Chlorosome-Mimicking Supramolecular Self-Assemblies. ACS Catal. 2018, 8, 10732–10745. [Google Scholar] [CrossRef]
- Lin, G.; Zhang, Y.; Hua, Y.; Zhang, C.; Jia, C.; Ju, D.; Yu, C.; Li, P.; Liu, J. Bioinspired Metalation of the Metal-Organic Framework MIL-125-NH2 for Photocatalytic NADH Regeneration and Gas-Liquid-Solid Three-Phase Enzymatic CO2 Reduction. Angew. Chem. Int. Ed. 2022, 61, e202206283. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-J.; Tan, L.-L.; Ng, Y.H.; Yong, S.-T.; Chai, S.-P. Graphitic Carbon Nitride (g-C3N4)-Based Photocatalysts for Artificial Photosynthesis and Environmental Remediation: Are We a Step Closer To Achieving Sustainability? Chem. Rev. 2016, 116, 7159–7329. [Google Scholar] [CrossRef]
- Zeng, P.; Ji, X.; Su, Z.; Zhang, S. WS2/g-C3N4 Composite as an Efficient Heterojunction Photocatalyst for Biocatalyzed Artificial Photosynthesis. RSC Adv. 2018, 8, 20557–20567. [Google Scholar] [CrossRef]
- Meng, J.; Tian, Y.; Li, C.; Lin, X.; Wang, Z.; Sun, L.; Zhou, Y.; Li, J.; Yang, N.; Zong, Y.; et al. A Thiophene-Modified Doubleshell Hollow g- C3N4 Nanosphere Boosts NADH Regeneration via Synergistic Enhancement of Charge Excitation and Separation. Catal. Sci. Technol. 2019, 9, 1911–1921. [Google Scholar] [CrossRef]
- Gao, Y.; Li, W.; Sun, X.; Zhao, Y.; Ji, H.; Luo, H.; Wu, G.; Wan, L.; Zhang, L. Boosting the Performance of Formate Dehydrogenase by Silver Nanoclusters for Photoreduction of CO2 to Formate. ACS Sustain. Chem. Eng. 2022, 10, 14888–14896. [Google Scholar] [CrossRef]
- Gupta, P.; Verma, N. Conversion of CO2 to Formate Using Activated Carbon Fiber-Supported g-C3N4-NiCoWO4 Photoanode in a Microbial Electrosynthesis System. Chem. Eng. J. 2022, 446, 137029. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, Y.; Zong, Y.; Li, J.; Yang, N.; Zhang, M.; Guo, Z.; Song, H. Construction of Functionally Compartmental Inorganic Photocatalyst-Enzyme System via Imitating Chloroplast for Efficient Photoreduction of CO2 to Formic Acid. ACS Appl. Mater. Interfaces 2020, 12, 34795–34805. [Google Scholar] [CrossRef]
- Yu, S.; Lv, P.; Xue, P.; Wang, K.; Yang, Q.; Zhou, J.; Wang, M.; Wang, L.; Chen, B.; Tan, T. Light-Driven Enzymatic Nanosystem for Highly Selective Production of Formic Acid from CO2. Chem. Eng. J. 2021, 420, 127649. [Google Scholar] [CrossRef]
- Ji, X.; Kang, Y.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Graphene Oxide and Polyelectrolyte Composed One-Way Expressway for Guiding Electron Transfer of Integrated Artificial Photosynthesis. ACS Sustain. Chem. Eng. 2018, 6, 3060–3069. [Google Scholar] [CrossRef]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Integration of Artificial Photosynthesis System for Enhanced Electronic Energy-Transfer Efficacy: A Case Study for Solar-Energy Driven Bioconversion of Carbon Dioxide to Methanol. Small 2016, 12, 4753–4762. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Anjong, T.F.; Jang, H.Y.; Kim, J.-Y.; Lee, C.; Park, S.; Lee, H.J.; Yoon, J.; Kim, J. Artificial Photocatalytic System Using Polydiacetylene-(-NH-Phen)Ru(Bpy)2 for Cofactor Regeneration and CO2 Reduction. J. Phys. Chem. C 2016, 120, 28407–28414. [Google Scholar] [CrossRef]
- Sokol, K.P.; Robinson, W.E.; Oliveira, A.R.; Warnan, J.; Nowaczyk, M.M.; Ruff, A.; Pereira, I.A.C.; Reisner, E. Photoreduction of CO2 with a Formate Dehydrogenase Driven by Photosystem II Using a Semi-Artificial Z-Scheme Architecture. J. Am. Chem. Soc. 2018, 140, 16418–16422. [Google Scholar] [CrossRef] [PubMed]
- Schuler, E.; Ermolich, P.A.; Shiju, N.R.; Gruter, G.-J. Monomers from CO2: Superbases as Catalysts for Formate-to-Oxalate Coupling. ChemSusChem 2021, 14, 1517–1523. [Google Scholar] [CrossRef]
- Murcia-Valderrama, M.A.; van Putten, R.-J.; Gruter, G.-J.M. The Potential of Oxalic—And Glycolic Acid Based Polyesters (Review). Towards CO2 as a Feedstock (Carbon Capture and Utilization–CCU). Eur. Polym. J. 2019, 119, 445–468. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Mueller, V. Whole-Cell Biocatalysis for Hydrogen Storage and Syngas Conversion to Formate Using a Thermophilic Acetogen. Biotechnol. Biofuels 2020, 13, 32. [Google Scholar] [CrossRef]
- Luxuan, L.; Zhijun, X.; Huang, X. Whole-Cell-Based Photosynthetic Biohybrid Systems for Energy and Environmental Applications. ChemPlusChem 2021, 86, 1021–1036. [Google Scholar]
- Hwang, H.; Yeon, Y.; Lee, S.; Choe, H.; Cho, D.; Park, S.; Kim, Y. Electro-Biocatalytic Production of Formate from Carbon Dioxide using an Oxygen-Stable Whole-Cell Biocatalyst. Bioresour. Technol. 2015, 185, 35–39. [Google Scholar] [CrossRef]
- Le, T.Q.A. Recent Applications and Strategies to Enhance Performance of Electrochemical Reduction of CO2 Gas into Value-Added Chemicals Catalyzed by Whole-Cell Biocatalysts. Processes 2023, 11, 766. [Google Scholar] [CrossRef]
- Alissandratos, A.; Kim, H.-K.; Easton, C.J. Formate Production through Carbon Dioxide Hydrogenation with Recombinant Whole Cell Biocatalysts. Bioresour. Technol. 2014, 164, 7–11. [Google Scholar] [CrossRef]
- Roger, M.; Brown, F.; Gabrielli, W.; Sargent, F. Efficient Hydrogen-Dependent Carbon Dioxide Rreduction by Escherichia coli. Curr. Biol. 2018, 28, 140–145.e2. [Google Scholar] [CrossRef]
- Leo, F.; Schwarz, F.M.; Schuchmann, K.; Mueller, V. Capture of Carbon Dioxide and Hydrogen by Engineered Escherichia coli: Hydrogen-Dependent CO2 Reduction to Formate. Appl. Microbiol. Biotechnol. 2021, 105, 5861–5872. [Google Scholar] [CrossRef]
- Schwarz, F.M.; Oswald, F.; Müller, V. Acetogenic Conversion of H2 and CO2 into Formic Acid and Vice Versa in a Fed-Batch-Operated Stirred-Tank Bioreactor. ACS Sustain. Chem. Eng. 2021, 9, 6810–6820. [Google Scholar] [CrossRef]
- Le, Q.A.T.; Kim, H.G.; Kim, Y.H. Electrochemical Synthesis of Formic Acid from CO2 Catalyzed by Shewanella oneidensis MR-1 Whole-Cell Biocatalyst. Enzym. Microb. Technol. 2018, 116, 1–5. [Google Scholar] [CrossRef]
- Singh, S.; Noori, M.T.; Verma, N. Efficient Bio-Electroreduction of CO2 to Formate on a Iron Phthalocyanine-Dispersed CDC in Microbial Electrolysis System. Electrochim. Acta 2020, 338, 135887. [Google Scholar] [CrossRef]
- Phan, U.T.; Jeon, B.W.; Kim, Y.H. Microbial Engineering of Methylorubrum extorquens AM1 to Enhance CO2 Conversion into Formate. Enzym. Microb. Technol. 2023, 168, 110264. [Google Scholar] [CrossRef]
- Chen, H.; Li, J.; Fan, Q.; Zheng, T.; Zhang, Y.; Yong, Y.-C.; Fang, Z. A feasible strategy for microbial electrocatalytic CO2 reduction via whole-cell-packed and exogenous-mediator-free rGO/Shewanella biohydrogel. Chem. Eng. J. 2023, 460, 141863. [Google Scholar] [CrossRef]
- Guntermann, N.; Mengers, H.G.; Franciò, G.; Blank, L.M.; Leitner, W. Bio-energy Conversion with Carbon Capture and Utilization (BECCU): Integrated Biomass Fermentation and Chemo-Catalytic CO2 Hydrogenation for Bioethanol and Formic Acid Coproduction. Green Chem. 2021, 23, 9860–9864. [Google Scholar] [CrossRef]
- Rowe, S.F.; Le Gall, G.; Ainsworth, E.V.; Davies, J.A.; Lockwood, C.W.J.; Shi, L.; Elliston, A.; Roberts, I.N.; Waldron, K.W.; Richardson, D.J.; et al. Light-Driven H2 Evolution and C=C or C=O Bond Hydrogenation by Shewanella oneidensis: A Versatile Strategy for Photocatalysis by Nonphotosynthetic Microorganisms. ACS Catal. 2017, 7, 7558–7566. [Google Scholar] [CrossRef]
- Tuyishime, P.; Sinumvayo, J.P. Novel Outlook in Engineering Synthetic Methylotrophs and Formatotrophs: A Course for Advancing C1-Based Chemicals Production. World J. Microbiol. Biotechnol. 2020, 36, 118. [Google Scholar] [CrossRef] [PubMed]
- Yishai, O.; Lindner, S.N.; Gonzalez de la Cruz, J.; Tenenboim, H.; Bar-Even, A. The formate bio-economy. Curr. Opin. Chem. Biol. 2016, 35, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shengyuan Guo, S.; Asset, T.; Atanassov, P. Catalytic Hybrid Electrocatalytic/Biocatalytic Cascades for Carbon Dioxide Reduction and Valorization. ACS Catalysis 2021, 11, 5172–5188. [Google Scholar] [CrossRef]
- Liang, B.; Zhao, Y.; Yang, J. Recent Advances in Developing Artificial Autotrophic Microorganism for Reinforcing CO2 Fixation. Front. Microbiol. 2020, 11, 592631. [Google Scholar] [CrossRef]
- Collas, F.; Dronsella, B.B.; Kubis, A.; Schann, K.; Binder, S.; Arto, N.; Claassens, N.J.; Kensy, F.; Orsi, E. Engineering the Biological Conversion of Formate into Crotonate in Cupriavidus necator. Metab. Eng. 2023, in press. [Google Scholar] [CrossRef]
- Cotton, C.A.; Claassens, N.J.; Benito-Vaquerizo, S.; Bar-Even, A. Renewable Methanol and Formate as Microbial Feedstocks. Curr. Opin. Biotechnol. 2020, 62, 168–180. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Organism | MW (kDa) | Activity (WAU/mg) | Activity (k−1) | Metal Coordination | Ref. |
|---|---|---|---|---|---|---|
| α | Bovine | 29.8 | 2540 | Zn(II), 3 His, H2O | [53] | |
| Homo sapiens (HCAI) | 28.7 | 920 | 2.0 × 105 | [54] | ||
| Homo sapiens (HCAII) | 29.1 | 8000 | 1.4 × 106 | [54] | ||
| Persephonella marina | 26.9 | 1748 | [55] | |||
| Thermosulfurimonas dismutans | 27.9 | 2032 | [53] | |||
| Thermovibrio ammonificans | 25.3 | 1016 | [53] | |||
| Bacillus halodurans | 37.0 | 3425 | [56] | |||
| Sulphurihydrogenibium yellowstonense | 26.0 | 7254 | [57] | |||
| β | Bacillus subtilis | 37.0 | 714 | Zn(II), His, 2 Cys | [58] | |
| Acetobacterium woodii | 22.0 | 1814 | [55] | |||
| Methanobacterium thermoautotrophicum | 19.9 | 580 | [55] | |||
| Aspergillus fumigatus | 23.0 | 20 | [59] | |||
| γ | Geobacillus kaustophilus | 22.0 | 179 | Zn(II) or Fe(II), 3 His, H2O | [60] | |
| Thermus thermophilus HB8 | 24.3 a | 0.9 | [61] | |||
| Methanosarcina thermophila | 40.0 | 4872 | [62] | |||
| Burkholderia pseudomallei | 28.2 | 5.3 × 105 | [63] | |||
| Vibrio cholerae | 26.3 | 7.39 × 105 | [63] | |||
| Porphyromonasgingivalis | 26.2 | 4.1 × 105 | [63] | |||
| δ | Thalassiosira weissflogii, TWCA1 | 27.0 | 1.3 × 105 | Zn(II), 3 His, H2O | [64] | |
| δ | Emiliania huxleyi | 18.3 | 1.3 × 106 (b) | Zn(II), 3 His, H2O | [65] | |
| ζ | Thalassiosira weissflogii, CDCA1 | 69 a | 1.5 × 106 | Cd(II) or Zn(II), His, 2 Cys | [42,66] | |
| η | Plasmodium falciparum | 26.2 | 1.4 × 105 | Zn(II), 2 His, Gln, H2O | [43,67] | |
| θ | Thalassiosirapseudonana | 26.0 | 122 | Cd(II) or Zn(II), His, 2 Cys | [38] | |
| Phaeodactylum tricornutum | 31.1 | 30.9 | [47] | |||
| ι | Burkholderia territorii | 28.2 | 3.0 × 105 | Cd(II) or Zn(II), His, 2 Cys | [46] | |
| Anabaena sp. PCC 7120 c | 19.3 | 16.7 | [52] | |||
| Bigelowiella natansd | 55.3 | 85.8 | [52] |
| A. NAD-Dependent FDHs. | ||||||||||||
| Formate Oxidation | CO2 reduction | |||||||||||
| Organism | MW (kDa) a | pH | kcat (s−1) (U mg−1) b | KM (mM) | kcat/KM (mM−1 s−1) | pH | kcat (s−1) (U mg−1) | KM (mM) | kcat/KM (mM−1 s−1) | ref | ||
| Myceliophthora thermophila | 42 | 10.5 | 0.32 | 7.2 | 0.04 | 7.0 | 0.10 | 0.43 | 0.23 | [97] | ||
| Ancylobacter aquaticus | 45 | 6.0 | (21.6) | 6.0 | (23) | 4.5 | [98] | |||||
| Candida boidinii | 41 | 7.0 | 1.081 (6.1) | 8.55 | 0.13 | 5.5 | 0.015 | 2.6 | 0.006 | [98] | ||
| Thiobacillus sp. KNK65MA | 45 | 6.5 | 1.769 (10.9) | 16.24 | 0.11 | 5.5 | 0.32 | 0.95 | 0.34 | [98] | ||
| Candida methylica | 42 | 8.0 | 1.31 (13.2) | 7.01 | 0.19 | 8.0 | 0.008 | 0.078 | 0.01 | [97] | ||
| Chaetomium thermophilum | 45 | 5.0 | 2.04(3.1) | 3.30 | 0.62 | 5.0 | 0.023 | 3.29 | 0.069 | [93,99] | ||
| Ceriporiopsis subvermispora | 40 | 6.5 | (1.3) | 6.0 | (0.8) | [98] | ||||||
| Moraxella sp. C-1 | 45 | 5.5 | (14.3) | 5.5 | (2.8) | [98] | ||||||
| Paracoccus sp. 12-A | 45 | 5.5 | (12.2) | 5.5 | (6.5) | [98] | ||||||
| B. Metal-Dependent FDHs | ||||||||||||
| Formate Oxidation | CO2 reduction | |||||||||||
| Organism | MW a (kDa) | Subunits b | Cofactors | pH | kcat(s−1) (U mg−1)a | KM(mM) | kcat/KM (mM−1 s−1) | pH | kcat(s−1) (U mg−1) a | KM(mM) | kcat/KM (mM−1 s−1) | ref |
| Syntrophobacter fumaroxidans FDH-1 | 175 | (αβγ)2 | W, SeCys 4 [Fe2S2] [Fe4S4] | 7.0 | (700) | 0.04 | - | 282 (900) | - | - | [100] | |
| Syntrophobacter fumaroxidans FDH-2 | 125 | (αβ)2 | W, SeCys 2 [Fe2S2] | 7.0 | (2700) | 0.01 | - | 282 (89) | - | - | [100] | |
| Desulfovibrio desulfuricans | 135 | (αβγ)3 | Mo, SeCys 2 MGDs 4 c-heme 2 [Fe4S4] | 8.0 | 543 | 0.0571 | 9526 | 7.0 | 46.6 | 0.0157 | 2968 | [101] |
| Escherichia coli FDH-H | 79 | αβ | Mo, SeCys 2 MGDs 1 [Fe4S4] | 7.5 | 2800 | 26 | 107.7 | 7.5 | 1.0 | 8.3 | 0.12 | [102,103] |
| Desulfovibrio vulgaris Hildenborough | 97.4 | (αβχ)3 | Mo, SeCys 4 [Fe4S4] 4 c-heme | 7.6 | 1310 (77) | 0.017 | 77.06 | 315(1.0) | 0.42 | 750 | [104,105] | |
| Acetobacterium woodii | 169 | (αβ)3 | Mo SeCys [4Fe-4S]) | 7.0 | (600) | 1.0 | - | 7.0 | 372 (132) | 3.8 | 97.9 | [106] |
| Cupriavidus necator | 178 | (αβγ)3 | Mo, 4 [Fe4S4] FMN 3 [Fe2S2] | 7.0 | 140 | 0.082 | 1707 | 7.0 | 11 | 2.7 | 4.07 | [107] |
| Rhodobacter capsulatus | 180 | (αβγ)2 | Mo, 4 Fe4S4, 1Fe2S2, 2 MGD, FMN | 5.0 | 36.5 | 281 | 0.13 | 7.7 | 1.48 | - | - | [108,109] |
| Pseudomonas oxalaticus | 315 | - | Mo, 2 FMN | - | 0.135 | - | 6.2 | 3.0 | 40 | 0.075 | [110,111] | |
| Clostridium Ijungdahlii | 80 | - | W Cys 2 MGD [Fe4S4] | 9.0 | 14.77 | 1.40 | 10.55 | 7.0 | 0.73 | 7.27 | 0.17 | [112,113] |
| Clostridium autoethanogenum | 74 | W 2 MGD [Fe4S4] | 9.0 | 1.04 | 4.51 | 0.231 | 7.0 | 4.00 | 23.15 | 0.17 | [113] | |
| Clostridium coskatii | 62 | W 2 MGD [Fe4S4] | 9.0 | 0.62 | 5.57 | 0.111 | 7.0 | 5.62 | 59.65 | 0.094 | [113] | |
| Clostridium ragsdalei | 74 | W | 9.0 | 11.88 | 44.83 | 0.265 | 7.0 | 3.28 | 31.20 | 0.11 | [113] | |
| Desulfovibrio gigas | 121 | (αβ)2 | W, SCys 4 [Fe4S4] | 8.0 | (34.1) | - | - | - | - | - | - | [114] |
| Moorella thermoacetica | (αβ)2 | W, SeCys 4 [Fe4S4] | 7.5 | (1100) | - | - | - | - | - | - | [115] | |
| Method | Support | Immobilization Conditions | Main Results | Ref |
|---|---|---|---|---|
| Physical adsorption | Mesoporous aluminosilicate | Phosphate buffer 100 mM, pH 7, 0.2 mL of enzyme (1 mg/mL) in 10 mg of support for 6 hr at 120 rpm. | CA loading up to 3 mg/mL optimal for CA activity. Carbonation activity was 55% (8 cycles of reuse | [140] |
| Carboxylic acid group-functionalized mesoporous silica (FMS) | 0.6–2.0 mg FMS with functional groups incubated with 70–1000 IL of 2.0 mg/mL BCA II in water (pH 6.5) for 2 h at 21 °C under 1200 rpm shaking. | High protein loading density 0.5 mg of protein/mg. High stability and activity (95.6%) of the immobilized enzyme | [141] | |
| Silver nanoparticles amine-functionalized mesoporous SBA-15 | 10 mg dispersed separately in 2 mL free HCA in buffer (3 mg/mL HCA in 100 mM sodium phosphate, pH 6.4) followed by incubation at 25 °C with shaking for 4 h. | The activity of the materials was ∼25-fold higher than the activity of the free HCA for converting CO2 to CaCO3 after 30 cycles. | [142] | |
| Mesoporous silica nanoparticles with polydopamine (PDA) and polyethyleneimine (PEI) | 5 mg FDH/1.6 mg CA added to 2 mL of phosphate buffer (0.05 M, pH 6). PDA/PEI-mSiO2 (0.05 g) and 5 μL GA aqueous solution (20 wt%) were added (30 °C for 24 h). | Optimal FDH and CA concentration was 2.5 and 0.8 mg/mL respectively, (specific activity of 0.045 mM/h/mg). 10 reuse cycles with 86.7% of activity. | [143] | |
| Palmityl-substituted sepharose 4B | Denatured Bovine CA solution by incubation for 15–120 min (58–65 °C), Tris-sulfate buffer, pH 7.5 (0.0125–0.5 mg/mL). | Denaturation and renaturation processes of the enzyme in the matrix. After renaturation at 60 °C, 85% of immobilization was achieved (70% of the original activity after 1 h). | [144] | |
| Silica or titania particles | The clarified lysates expressed BCA-peptide (0.088 mg protein) were incubated with 30 mg silica or 20 mg titania for 10 min with shaking at room temperature in 25 mM sodium phosphate buffer (pH 6.5). | After 10 days, 90 ± 4% and 95 ± 3% of silica and titania particles’ original activity remained. Immobilized BCA-peptide fusion protein shows ∼95% of its residual activity with up to 5 cycles of reuse. | [145] | |
| Porous polypropylene and a non-porous polydimethoysilane hollow fiber membrane | Application of CA in situ to the shell side surface of each fiber. | CO2 absorption into K2CO3 increased approximately three-fold when CA is adsorbed onto the Porous polypropylene or non-porous polydimethoxysilane PDMS membrane surface | [146] | |
| Entrapment or encapsulation | Magnetic nanoparticles (sol-gel ferria hydrosol) | 200 mL of ferria hydrosol mixed with CAB solution (10 g/L, 0.05 M Tris, pH 7.4) and dried at 20 °C. | Large thermal stability of CA immobilized, active up to 95 °C | [147] |
| Polyurethane foam (PUF) | 1 mL of CA (1 g/L) or whole-cell solution in 20 mM Tris-sulfate buffer (pH 8.3) was added to a 50 mL containing 1 g of prepolymer. The swelling of PUF continued for 30 min, and then an additional 10 min was allowed for curing. | An immobilization efficiency of 3.4%, 16-fold higher than that for free enzymes. The reusability of the immobilized whole-cell catalyst shows no apparent decrease in activity after 9 reuses. The rate of CO2 capture was accelerated by 80%. | [148] | |
| Ni-based MOFs (Ni-BTC) nanorods, | 100 mL cell lysate of His-HCA II (0.4 mg protein) and 900 mL Milli-Q water were incubated with Ni-based MOFs. | His-HCA II from cell lysate obtained an activity recovery of 99%. After storing for 10 days, the immobilized His-HCA II maintained 40% activity (free enzyme lost 91% activity). Immobilized His-HCA II retained 65% (8 cycles). | [149] | |
| Supported ionic-liquid membranes (SILMs) | 0.25 mg CA/g IL. | SILMs resent permeability (PCO2 = 733.73 barrier) at high temperatures (up to 373 K) and a good transport selectivity towards CO2 against N2. | [150] | |
| Cholinium-based ionic liquids | 0.1 mg CA/g IL | CA samples promote an enhancement of 63% in the carbon dioxide transport rate. | [151] | |
| Covalent binding and cross-linking | Mesoporous SBA-15 surfaces covalently functionalized with amines | HCA immobilization was achieved by mixing 10 mg of amine-functionalized SBA-15 with a 0.1% glutaraldehyde (GA) solution (50 mM sodium phosphate, pH 8.0, 1 h). The product was treated with free HCA (3 mg/mL HCA, pH 7.0), incubated and shaking (1 h, 25 °C). | Immobilized HCA retained activities after long-term storage, exposure to high temperatures, and reuse (40 cycles). CO2 capture efficiency of immobilized HCA was 36 times higher than that of free HCA, 75% of the enzymatic activity was retained after 40 cycles. | [152] |
| Chitosan | CA liquid was slowly added into the pH 5 chitosan solution with continuous stirring. Ratios of 1:0.05–1:2 chitosan:CA (g:mL) | Textile packing with covalently attached enzyme aggregates retained 100% of the initial 66.7% CO2 capture efficiency over 71 days and retained 85% of the initial capture efficiency after 1-year of ambient dry storage. | [153] | |
| Polyethyleneimine and polydopamine in MOF 808 | PBS buffer (pH 8, 10 mM), PEI/PDA-MOF-808 or PDA-MOF-808 was disseminated. 200–400 μL of CA solution (1 mg/mL, Milli-Q) and 10 μL of aqueous solution of GA (0–25 wt%) were added and shaken for a while at 28 C. | CaCO3 produced by CA@PEI/PDA-MOF-808 was 11.0-fold and 2.5-fold higher than free CA and PEI/PDA-MOF-808, respectively. After 8 consecutive rounds, the total production of CaCO3 by CA@PEI/PDA-MOF-808 was 92-fold higher than free CA. | [154] | |
| Alumino-siloxane hybrid aerogel beads | 100 mg of Al/Si-NH2 beads were treated with 0.5% GA for 1 h. 4 mL of free BCA in the buffer (1 mg/mL 100 mM phosphate buffer) was added to GA-treated Al/Si-NH2 beads and stirred (3 h, 25 °C) for BCA immobilization. | Free BCA retained 70% of its maximum activity (immobilized BCA, 88%). Free BCA and BCA-Al/Si-NH2 remained 80% of the activities, after ten days. BCA-Al/Si-NH2 retained 89% of their enzyme activity up to 10 cycles | [155] | |
| Amine-functionalized by co-deposition of polydopamine (PDA) and polyethyleneimine (PEI) | Co-deposition of PDA and PEI and CA covalently anchored on the surface via GA. Surface was treated with a mixture of PDA (2 mg/mL) and PEI, pH 8,5. Amine-functionalized micro-reactor surface was contacted with a mixture of CA and GA (concentration 2.0% (v/v)). | A steady CO2 absorption rate for several hours and good reusability of the immobilized enzyme which maintains its original absorption performance after 10 cycles of operation. | [156] | |
| Polypropylene hollow fiber membranes using GA-activated chitosan | Aminated knitted hollow fiber membranes mats (204 cm2 fiber surface area) were incubated in 5% GA in 100 mM phosphate buffer, pH 8.5 for 1 h under constant rocking at room temperature. | Chitosan/CA coated fibers exhibited accelerated CO2 removal in scaled-down gas exchange devices in buffer and blood (115% enhancement vs. control, 37% enhancement vs. control, respectively). | [157] | |
| Magnetic Cross-Linked Enzyme Aggregates (CLEAs) to bovine carbonic anhydrase (BCA) and magnetic nanoparticle | - Adsorption: the NPs suspension (5 and 2 mg solids) added to 1 mL of 10 mM PBS (pH 7.4), 10 g/L BCA. - Precipitation: NP-enzyme suspension added dropwise to 9 mL containing the precipitating agent (pH 7.4), 1 h or 0.5 h under mixing. - Cross-linking: 25% vol GA added. The system kept under mixing for 3, 16 and 22 h. - CLEAs separated by MF. | BCA-CLEAs can increase the CO2 absorption rate concerning the one observed in the same reactor filled with only alkaline solvent. Biocatalyst reusability analysis showed that BCA-CLEA retained 95% of its initial activity after five CO2 absorption tests at 1000 mg BCA CLEA/L and as many liquid-solid separation steps by membrane filtration. | [158] | |
| Geopolymer micro-spheres (GMS) and covalent attachment by GA | GMS were introduced into a Tris-HCl buffer solution (0.05 M, pH 8.0) with CAs solution (1 mg/mL) in a 50 mL centrifuge tube. After shaking and reacting, the GA was added for the cross-linking between the GMS and CAs. | Kcat/Km values were 61.50 and 12.36 M−1s−1 for the immobilized and free CAs, respectively. At 60 °C, free CAs were inactivated (immobilized CAs kept 34.8% activity). Immobilized CAs maintain 68.73% of activity (8 cycles). | [159] | |
| Microbial transgluta-minase (MTG) acts as a “cross-linking medium” | Iso-peptide bond between glutamine and the primary amine group of a lysine in artificial peptide tags. Equimolar amounts F-CA and M-FDH were added, and a metal constant temperature oscillator was used for the cross-linking reaction (reaction time 12 h, 25 ℃, 400 rpm. The amount of MTG used was 1 U/mL. | The remaining CA activity was more than 93%, and the remaining FDH activity was more than 84%. The efficiency of the cross-linked enzyme is increased by 5.8 times compared with free enzymes. FDH thermal stability at different temperatures is improved the optimal found CA/FDH ratio was 1:2. | [160] |
| Process | Idea of the Study | CO2 Uptake Conditions | Relevant Conclusions | CA Activities | Ref. |
|---|---|---|---|---|---|
| Chemical absorption | To examine the stability and activity of the enzyme under different pH values and amine solvents. Compare its performance to other common solvents. | CA tested for pH [7,8,9,10,11]. Temperature stability for up to 100 h of incubation. CA stability tested for 7 capture solvents (1 M or 3 M, 150 days, 40 °C). | CA stable at 60 °C, pH range [7,8,9,10,11]: residual activity at pH 5 or 12, ranging from 12 to 91%. The enzyme enhances reaction rates. NaCl, K2CO3, AMP, and MDEA show additive effects. | After 100 h (25 °C), CA activity was kept higher than 75% for 1M NaCl, AMP and MDEA and 125% in 1M K2CO3 | [191] |
| To develop the CA-MOFs composite, with superior catalytic performance and high stability to promote CO2 absorption into a tertiary amine solution. | 0.05 g L−1 and 40 °C, PCO2 15 kPa. | CA loading in ZIF-L-1 increased with the added enzyme amount. CA/ZIF-L-1 composite has higher catalytic activity and stability than the free CA. The immobilization of CA on ZIF-L-1 improves the CA conformational stability | CO2 absorption rate of CA/ZIF-L-1 in 1 M MEA and MDEA at 40 °C: 3.0 × 106 kmol s−1m−2. CA loading reached up to 87 mg g−1. The highest immobilized CA activity was 1.5 times that of the free CA | [192] | |
| Pilot-scale experiments with CA-enhanced MDEA for CC, bench-marking its mass transfer performance against the industrial standard 30 wt% MEA. | Experiments were done using 30 wt% MEA and MDEA solvents varying CA concentrations (0, 0.85, and 3.5 g/L) at different column L/G ratios. | Enzyme-enhanced MDEA solutions exhibit 80% of the mass transfer performance at 30 wt% MEA and have the potential to reduce the size and cost of absorbents in carbon capture. | CO2 capture efficiency higher than 90% for high L/G ratios. The mass transfer increased 20-fold for 25 and 50 wt% MDEA solutions by adding 0.2 g/L CA. | [193] | |
| Use of columns with membranes contractors with polyionic liquids (PIL), amines and CA for CO2 uptake. | PIL blend (F9:1(M10)) with immobilized CA in 30 wt% MDEA at low feed gas (15% CO2 in N2.) pressure (1.3 bar) | Addition of the enzyme to the MDEA solution with PILs significantly improves CO2 uptake rate and reduced the equilibration time. | CA addition to MDEA solution improved the CO2 uptake rate by 1.7 times. CA and membrane improved the uptake rate by twice. | [194] | |
| Carbonation | To present the effects of adding small quantities of immobilized CA on the absorption of CO2 into potassium carbonate. | CO2 absorption (partial pressure 90 kPa) in a 30% K2CO3 solution. A wet-walled column (40–80 °C) with 38 g/L CA. | CA addition improves the CO2 absorption in K2CO3 solvents. The rate of CA-catalyzed CO2 hydration increased with the CA concentration | Increasing CA concentration from 0.4 to 1.8 μM increases the absorption rate by 34% (40 °C) | [195] |
| To explore the potential of using enzymes to catalyze the conversion of CO2 into bicarbonate and on the carbonation rate of brucite, Mg(OH)2. | Gas flow (CO2/N2 10%/90%) at 10 psig. Experimental durations were 11, 7, and 3 days (low, medium, and high flow experiments). | CA accelerates the carbonation of brucite. Higher CO2 gas flow rates results in faster carbonation. Mineralogical compositions depend on the CO2 flow rate. | The carbonation rate of brucite using BCA was accelerated by up to 240% compared to controls. | [196] | |
| To propose a novel method based on microbially induced calcium precipitation to improve the cementitious properties of steel slag. | Bacillus mucilaginosus placed in a sealed container, the air pressure was reduced to −0.05 MPa. CO2 (99.99%) pumped to maintain the gas pressure (0.25 MPa, 30 °C, 32 h). | Ca-silicate promotes the growth of bacteria, enhances the production of polymers and improves bacterial adhesion. Ca-silicate-containing medium (CSCM) can be used as a microbial carrier. | Maximum bacterial growth rate in CSCM was 1.5 times higher than that in the control medium (CM). Bacterial activity in CSCM was 40% of that in CM (30 h). At a dosage of bacterial powders of 1 wt.%, carbonation reached the maximum. | [197] | |
| To develop and characterize a highly efficient and stable biocatalyst for CO2 sequestration using silica nanocomposite with auto-encapsulated CA. (CA)-based biocatalyst encapsulated in a biosilica with a peptide R5. | CaCO3 precipitation was carried out at 30 °C and monitored turbidimetrically at 600 nm. The final pH of the buffer was approximately 9.3 | The encapsulated CA was not leached from the silica matrix. Encapsulation in silica effectively improved the thermostability and activity of the enzyme. | Encapsulation efficiency greater than 95%. Residual activity of the self-encapsulated CA was more than 50% (30 min, 80 °C). Activity of ngCA-R5@silica decreased only after 2.5 days at 60 °C. Encapsulated CA: 98% and 80% of its initial activity (1 day and 5 days of incubation, 50 °C). CaCO3 precipitation is reduced by 5.5-fold when ngCA-R5@silica (30 μg/mL) is present compared to the uncatalyzed reaction. | [198] | |
| Mineralizaton | Mineralization experiments were performed using Curvibacter lanceolatus strain HJ-1, including its secreted extracellular polymeric substances (EPS) and CA (CA). | Three types of mineralization experiments: with CA (duration 96 h), with EPS (96 h), and with bacteria (50 days). | Strain HJ-1, EPS, and CA promote carbonate precipitation. HJ-1 and EPS1 experiments contained calcite and aragonite. CA formed calcite only. HJ-1 and EPS are favorable for aragonite precipitation. | The mass of precipitate (inorganic plus organic substances) increases until ca. 55 mg. The maximum degree of calcification was approximately 6.6%. In the control groups without CA no precipitate was formed. In the absence of HCO3−, the optimized calcification rate followed the order: HJ-1 (49.5%) > CA(6.6%) > EPS2(4.1%). | [199] |
| Production of CA from the bacterium Aeribacillus pallidus TSHB1, a thermostable and alkaline-stable bacterium, highly effective in the formation of CaCO3 from aqueous CO2. | Tris buffer (15 mL, pH 7.4) containing 0.9 g CaCl2-2H2O, CA 0.05 mg (37 °C). | A 3.8-fold higher CA production by A. pallidus than that under unoptimized conditions. Enzyme thermostable that retains activity at alkaline pH: it is useful in carbon sequestration. | The partially purified enzyme produces precipitation of 42.5-mg CO32− mg−1 protein. CO2 sequestration is efficient. | [200] | |
| Study of the biochemical properties, thermostability, and inhibition of CA from Sulfurihydrogenibium azorense, SazCA. | Hepes buffer 10 mM, NaBF4 20 mM, pH 7.5. Phenol red (0.2 mM) as an indicator. | SazCA is highly thermostable and can survive incubation at 90–100 °C. | kcat value 4.40 × 106 s−1, KM value 12.5 mM, kcat/KM 3.5 × 108 M−1 s−1, 5-fold faster than the second CA. SazCA is the second faster enzyme, after superoxide dismutase (SOD). | [201] | |
| To explore wollastonite (calcium silicate) carbonation for the removal of anthropogenic CO2 and evaluate the effectiveness of different natural (CA) and CA biomimetic catalysts (Zr-based MOFs) to enhance CO2 capture. | Water 170 mL, the catalyst (30 ppm), adjusted at pH 4 (25 °C). Wollastonite crystals (100 mg) added to the solution. CA is immobilized on the MOFs UiO-66 and MOF-808@Mg(OH)2 by an impregnation process. | CA accelerates carbonate precipitation but hinders carbonation of wollastonite. Thick carbonate coatings formed on the most reactive surfaces of wollastonite act as passivating layers leading to a reduction in the dissolution and carbonation rates. | The passivating effect explains why the conversion of wollastonite into calcite was so limited (up to 14 mol%). Zr-based MOFs accelerate the dissolution of wollastonite. | [202] |
| Reduction | Organism | Significant Assay Features/Conditions | Formate Production | Ref |
|---|---|---|---|---|
| Hydrogenation | Overexpressed genes of FDH from E. coli, Clostridium carboxidi-vorans, Pyrococcus furiosus and Methanobacterium thermos-formicicum in E. coli JM109(DE3) | H2 atmosphere. Wet cell pellet (0.5 g wet cells/mL) resuspended in 50 mM sodium phosphate buffer, pH 7.0, containing 0.25 M sodium bicarbonate as a source of CO2. Incubation 37 °C. | The highest formic yield (FDH from P. furiosus) was more than 1 g L−1 h−1 | [292] |
| Hydrogenation | Escherichia coli | CO2 and H2 placed under pressure (up to 10 bar). First, pressurized the system to rapidly convert 100% conversion of gaseous CO2 to formic acid. Next, NaOH addition to the E. coli cell suspension (pH > 8). | Formate concentration to 150 (4 bar) and 200 (6 bar) mmol L−1. Increasing the pressure to 10 bar (122.88 mmol L−1 CO2 and 3.61 mmol L−1 H2): > 0.5 mol L−1 formate at 23 h of reaction. | [293] |
| Hydrogenation | Acetobacterium woodii and Thermoanaerobacter kivui | Cells grown with 28 mM glucose or 0.1 M pyruvate (50 mM imidazole, 20 mM KCl, 20 mM MgSO4, 2 mM DTE, 4 μM resazurin, pH 7.0). 1 mg/mL in an anoxic medium. Shaking for 10 min (60 °C), with additional 300 mM KHCO3. The experiment started by replacing the gas phase with H2 + CO2 (80:20%, 2 × 105 Pa). | Optimal formate production rates of 234 mmol g−1protein h−1 | [270] |
| Hydrogenation | H2-dependent CO2 reductase from Acetobacterium woodie expressed in E. coli JM109 | Whole-cell E. coli in presence of formate (10 mM) and methylviologen (10 mM) as electron acceptor. E. coli cells incubated with H2 + CO2 (80:20%, 1.1 × 105 Pa). | 6 mM formic acid in 60 h. Addition of 2.5 mM HCO3− increased 4-fold the formate generation. | [294] |
| Hydrogenation | Acetobacterium woodii and Thermoanaerobacter kivui | 50 mM imidazole, 20 mM MgSO4, 20 mM KCl, 20 mM NaCl, 2 mM DTE, pH 7.0; or 50 mM K-phosphate, 20 mM KCl, 20 mM NaCl, 2 mM DTE, pH 7.0) was maintained at 30 and 60 °C for A. woodii and T. kivui, respectively. Gas flow rate was maintained at a value of 25 mL/min | 60 mM and 80 formic acid generation after 5 h of reaction for A. woodii and T. kivui, respectively. | [295] |
| Electrochemical | Methylobacterium extorquens AM1, | System: 1 mM H2SO4 with a platinum electrode placed in the anode; protons supplied to the cathode through a proton-exchange membrane. 1.9 g wet cells, 10 mM methyl viologen (MV) added to the cathode reactor as an electron mediator | Formate concentrations of up to 60 mM, 80 h (1.9 g wet cells, 10 mM MV, pH 7.0) | [290] |
| Electrochemical | Shewanella oneidensis MR-1 | The electrochemical cell with two compartments divided by a proton-exchange memberane. Copper plates and Ag/AgCl electrodes. Whole-cell S. oneidensis MR-1 (wet-cell, 0.5 g) and MV, 10 mM). RT, anaerobic conditions. | Formic acid at 0.59 mM h−1 for 24 h. Medium supplemented with fumarate and nitrate: 1.9 mM h−1 for 72 h. LB supplemented with 40 mM fumarate, 1mM nitrate and 20 mM DL-lactate: 136.84 mM formic acid at 72 h. (3.8 mM h−1 g−1wet-cell) | [296] |
| Electrochemical | E. coli | NaHCO3 electrolyte saturated with N2 or CO2 media at three different poised potentials, i.e., 0.4, 0.8 and 1.0 V vs. Ag/AgCl. E. coli-immobilized FePc-CDC/ACF and ACF electrodes in the presence of the NR mediator (FePc: iron pfthalocyanine; CDC: carbide-derived carbon; ACF: activated carbon fiber; NR: Neutral Red). | Maximum formate production rate of ~30 mg/L-h under CO2 flow (120 mg/L-h) with NR mediator | [297] |
| Electrochemical | Methylorubrum extorquens AM1 | Nafion 115 (proton permeable) membrane. Cathode: 0.6 g cell pellet (potassium phosphate 200 mM, pH 7.0) as catholyte. Electron mediator: ethyl viologen 10 mM. 0.6 g cell pellet suspended into catholyte (200 mM-potassium phosphate at pH 7) saturated by CO2 purging (30 min). Water splitting reaction in 100 mM-H2SO4. | Formate production: 6 mM h−1 | [298] |
| Electrochemical | Shewanella loihica PV-4 | Cathode: biohydrogel formed by Shewanella loihica PV-4 immobilized in graphene oxide. | High Faradaic efficiency (~99.5%) and 46-fold increase of formate titer without exogenous mediator (4.2 mM formate. 36 h) | [299] |
| Hydrogen/car-bohydrate fermentation | Saccharomyces cerevisiae | Two phases: Glucose fermentation for generating CO2 CO2 Ru catalysis hydrogenation | 26% of the CO2 was hydrogenated. Addition of His 150 mM: 128 mM in formic acid at 48 h. | [300] |
| Photocatalytic hydrogenation | Shewanella oneidensis MR-1 | Anaerobic conditions: N2-filled chamber and samples irradiated from outside the chamber by a KL5125 Cold 150W light source. Irradiation into MV, with triethanolamine (TEOA) as sacrificial agent in 50 mM HEPES, 50 mM NaCl, pH 7, 23 °C. | Formate (∼1500 nmol) was produced when MR-1 was incubated with CO2 (∼5000 nmol) after 48 h incubation | [301] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Villa, R.; Nieto, S.; Donaire, A.; Lozano, P. Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals. Molecules 2023, 28, 5520. https://doi.org/10.3390/molecules28145520
Villa R, Nieto S, Donaire A, Lozano P. Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals. Molecules. 2023; 28(14):5520. https://doi.org/10.3390/molecules28145520
Chicago/Turabian StyleVilla, Rocio, Susana Nieto, Antonio Donaire, and Pedro Lozano. 2023. "Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals" Molecules 28, no. 14: 5520. https://doi.org/10.3390/molecules28145520
APA StyleVilla, R., Nieto, S., Donaire, A., & Lozano, P. (2023). Direct Biocatalytic Processes for CO2 Capture as a Green Tool to Produce Value-Added Chemicals. Molecules, 28(14), 5520. https://doi.org/10.3390/molecules28145520